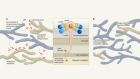
- NEWS AND VIEWS
Newly made mitochondrial DNA drives inflammation
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 560, 176-177 (2018)
doi: https://doi.org/10.1038/d41586-018-05764-z
References
Zhong, Z. et al. Nature 560, 198–203 (2018).
Bambouskova, M. et al. Nature 556, 501–504 (2018).
Mills, E. L. et al. Nature 556, 113–117 (2018).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. Nature 469, 221–225 (2011).
Mills, E. L. et al. Cell 167, 457–470.e13 (2016).
He, Y., Hara, H. & Núñez, G. Trends Biochem. Sci. 41, 1012–1021 (2016).
Schroder, K. & Tschopp, J. Cell 140, 821–832 (2010).
Shimada, K. et al. Immunity 36, 401–414 (2012).
Zhong, Z. et al. Cell 164, 896–910 (2016).
Nakahira, K. et al. Nature Immunol. 12, 222–230 (2011).
Murphy, M. P. et al. Cell Metab. 13, 361–366 (2011).
Bernardi, P. & Di Lisa, F. J. Mol. Cell. Cardiol. 78, 100–106 (2015).
Sugiura, A., McLelland, G.-L., Fon, E. A. & McBride, H. M. EMBO J. 33, 2142–2156 (2014).
Dhir, A. et al. Nature 560, 238–242 (2018).
 Read the paper: New mitochondrial DNA synthesis enables NLRP3 inflammasome activation
Read the paper: New mitochondrial DNA synthesis enables NLRP3 inflammasome activation
 Read the paper: Mitochondrial double-stranded RNA triggers antiviral signalling in humans
Read the paper: Mitochondrial double-stranded RNA triggers antiviral signalling in humans
 The inflammasome turns 15
The inflammasome turns 15
 Gut sensor halts viral attack
Gut sensor halts viral attack