
- NEWS AND VIEWS
How the ubiquitous GPCR receptor family selectively activates signalling pathways
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 558, 529-530 (2018)
doi: https://doi.org/10.1038/d41586-018-05503-4
References
Santos, R. et al. Nature Rev. Drug Discov. 16, 19–13 (2017).
Koehl, A. et al. Nature 558, 547–552 (2018).
Draper-Joyce, C. J. et al. Nature 558, 559–563 (2018).
García-Nafría, J., Nehmé, R., Edwards, P. C. & Tate, C. G. Nature 558, 620–623 (2018).
Kang, Y. et al. Nature 558 , 553–558 (2018).
Milligan, G. & Kostenis, E. Br. J. Pharmacol. 147, S46–S55 (2006).
Rasmussen, S. G. F. et al. Nature 477, 549–555 (2011).
García-Nafría, J., Lee, Y., Bai, X., Carpenter, B. & Tate, C. G. eLife 7, e35946 (2018).
Mahoney, J. P. & Sunahara, R. K. Curr. Opin. Struct. Biol. 41, 247–254 (2016).
Andressen, K. W. et al. FASEB J. 32, 1059-1069 (2018).
Gregorio, G. G. et al. Nature 547, 68–73 (2017).
Wacker, D. et al. Cell 168, 377–389 (2017).
Lane, J. R., May, L. T., Parton, R. G., Sexton, P. M. & Christopoulos, A. Nature Chem. Biol. 13, 929–937 (2017).
Roth, B. L., Irwin, J. J. & Shoichet, B. K. Nature Chem. Biol. 13, 1143–1151 (2017).
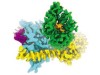 Read the paper: Structure of the μ-opioid receptor–Gi protein complex
Read the paper: Structure of the μ-opioid receptor–Gi protein complex
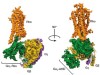 Read the paper: Cryo-EM structure of human rhodopsin bound to an inhibitory G protein
Read the paper: Cryo-EM structure of human rhodopsin bound to an inhibitory G protein
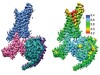 Read the paper: Structure of the adenosine-bound human adenosine A1 receptor–Gi complex
Read the paper: Structure of the adenosine-bound human adenosine A1 receptor–Gi complex
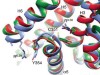 Read the paper: Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go
Read the paper: Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go
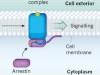 Activation mechanisms for a universal signalling protein
Activation mechanisms for a universal signalling protein
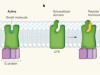 Signalling under the microscope
Signalling under the microscope
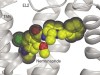 A new era of rationally designed antipsychotics
A new era of rationally designed antipsychotics





