
- NEWS AND VIEWS
Backbone of RNA viruses uncovered
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 556, 182-183 (2018)
doi: https://doi.org/10.1038/d41586-018-03923-w
References
Wolfe, N. D., Dunavan, C. P. & Diamond, J. Nature 447, 279–283 (2007).
Woolhouse, M. E. J. & Brierley, L. Sci. Data 5, 180017 (2018).
Olival, K. J. et al. Nature 546, 646–650 (2017).
Holmes, E. C. J. Virol. 85, 5247–5251 (2011).
Koonin, E. V., Senkevich, T. G. & Dolja, V. V. Biol. Direct 1, 29 (2006).
Shi, M. et al. Nature 556, 197–202 (2018).
Tristem, M., Herniou, E., Summers, K. & Cook, J. J. Virol. 70, 4864–4870 (1996).
Reuter, G. et al. J. Gen. Virol. 96, 2607–2613 (2015).
Ip, H. S., Lorch, J. M. & Blehert, D. S. Emerg. Microbes Infect. 5, e97 (2016).
Holmes, E. C., Dudas, G., Rambaut, A. & Andersen, K. G. Nature 538, 193–200 (2016).
Faria, N. R., Suchard, M. A., Rambaut, A., Streicker, D. G. & Lemey, P. Phil. Trans. R. Soc. B 368, 20120196 (2013).
Geoghegan, J. L. & Holmes, E. C. Open Biol. 7, 170189 (2017).
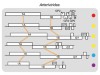 Read the paper: The evolutionary history of vertebrate RNA viruses
Read the paper: The evolutionary history of vertebrate RNA viruses
 Viral strategies at sea
Viral strategies at sea
 Zapping viral RNAs
Zapping viral RNAs





