- NEWS AND VIEWS
Reactive carbon species tamed for synthesis
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 554, 36-38 (2018)
doi: https://doi.org/10.1038/d41586-018-01308-7
References
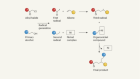
Wang, Z., Herraiz, A. G., del Hoyo, A. M. & Suero, M. G. Nature 554, 86–91 (2018).
Fischer, E. O., Ruhs, A. & Plabst, D. Z. Naturforsch. B 32B, 802–804 (1977).
Levy, O., Musa, S. & Bino, A. Dalton Trans. 42, 12248–12251 (2013).
Bino, A., Ardon, M. & Shirman, E. Science 308, 234–235 (2005).
Bogoslavsky, B. et al. Angew. Chem. Int. Edn 51, 90–94 (2012).
Ye, F. et al. J. Am. Chem. Soc. 137, 4435–4444 (2015).
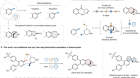
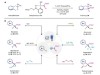 Read the paper: Generating carbyne equivalents with photoredox catalysis
Read the paper: Generating carbyne equivalents with photoredox catalysis
 Nickel steps towards selectivity
Nickel steps towards selectivity
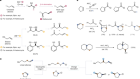
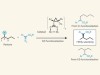 Precision pruning of molecules
Precision pruning of molecules