- NEWS AND VIEWS
A non-tailed twist in the viral tale
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 554, 38-39 (2018)
doi: https://doi.org/10.1038/d41586-018-00923-8
References
Cobián Güemes, A. G. et al. Annu. Rev. Virol. 3, 197–214 (2016).
Suttle, C. A. Nature 437, 356–361 (2005).
Børsheim, K. Y., Bratbak, G. & Heldal, M. Appl. Environ. Microbiol. 56, 352–356 (1990).
Wommack, K. E., Hill, R. T., Kessel, M., Russek-Cohen, E. & Colwell, R. R.. Appl. Environ. Microbiol. 58, 2965–2970 (1992).
Brum, J. R., Schenck, R. O. & Sullivan, M. B. ISME J. 7, 1738–1751 (2013).
Fuhrman, J. A. Nature 399, 541–548 (1999).
Kauffman, K. M. et al. Nature 544, 118–122 (2018).
Holmfeldt, K. et al. Proc. Natl Acad. Sci. USA 110, 12798–12803 (2013).
Hug, L. A. et al. Nature Microbiol. 1, 16048 (2016).
Sullivan, M. B., Waterbury, J. B. & Chisholm, S. W. Nature 424, 1047–1051 (2003).
Krupovic, M. & Koonin, E. V. Proc Natl Acad. Sci. USA 114, E2401–E2410 (2017).
Kang, I., Oh, H.-M., Kang, D. & Cho, J.-C. Proc. Natl Acad. Sci. USA 110, 12343–12348 (2013).
Zhao, Y. et al. Nature 494, 357–360 (2013).
Steward, G. F. & Culley, A. I. in Manual of Aquatic Viral Ecology 154–165 (Assoc. Sci. Limnol. Oceanogr., 2010)
Comeau, A. M. & Krisch, H. M. Mol. Biol. Evol. 25, 1321–1332 (2008).
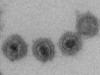 Read the paper: A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria
Read the paper: A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria
 Viral strategies at sea
Viral strategies at sea
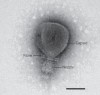 Arms race in a drop of sea water
Arms race in a drop of sea water