- NEWS AND VIEWS
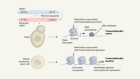
Viruses hijack a long non-coding RNA
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 552, 184-185 (2017)
doi: https://doi.org/10.1038/d41586-017-07692-w
References
Engreitz, J. M., Ollikainen, N. & Guttman, M. Nature Rev. Mol. Cell Biol. 17, 756–770 (2016).
Tycowski, K. T. et al. Genes Dev. 29, 567–584 (2015).
Wang, P., Xu, J., Wang, Y. & Cao, X. Science http://dx.doi.org/10.1126/science.aao0409 (2017).
Goodwin, C. M., Xu, S. & Munger, J. Trends Microbiol. 23, 789–798 (2015).
Heward, J. A. & Lindsay, M. A. L. Trends Immunol. 35, 408–419 (2014).
The ENCODE Project Consortium. Nature 489, 57–74 (2012).
 Virology: Ins and outs of picornaviruses
Virology: Ins and outs of picornaviruses
 HIV: Going for the watchman
HIV: Going for the watchman