- RESEARCH HIGHLIGHT
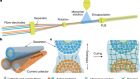
Novel chemistry for covalent inhibitors
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature Reviews Drug Discovery 19, 754 (2020)
doi: https://doi.org/10.1038/d41573-020-00161-6
References
Huang, H. et al. Covalent inhibition of NSD1 histone methyltransferase. Nat. Chem. Biol. https://doi.org/10.1038/s41589-020-0626-6 (2020)
Brighty, G.J. et al. Using sulfuramidimidoyl fluorides that undergo sulfur(vi) fluoride exchange for inverse drug discovery. Nat. Chem. https://doi.org/10.1038/s41557-020-0530-4 (2020)