
Abstract
N6-methyladenosine (m6A) is the most common internal modification in eukaryotic mRNA. It is dynamically installed and removed, and acts as a new layer of mRNA metabolism, regulating biological processes including stem cell pluripotency, cell differentiation, and energy homeostasis. m6A is recognized by selective binding proteins; YTHDF1 and YTHDF3 work in concert to affect the translation of m6A-containing mRNAs, YTHDF2 expedites mRNA decay, and YTHDC1 affects the nuclear processing of its targets. The biological function of YTHDC2, the final member of the YTH protein family, remains unknown. We report that YTHDC2 selectively binds m6A at its consensus motif. YTHDC2 enhances the translation efficiency of its targets and also decreases their mRNA abundance. Ythdc2 knockout mice are infertile; males have significantly smaller testes and females have significantly smaller ovaries compared to those of littermates. The germ cells of Ythdc2 knockout mice do not develop past the zygotene stage and accordingly, Ythdc2 is upregulated in the testes as meiosis begins. Thus, YTHDC2 is an m6A-binding protein that plays critical roles during spermatogenesis.
Similar content being viewed by others


Introduction
Cell growth and differentiation depend upon the tight regulation of gene expression through controlled transcription and translation. In addition, post-transcriptional modifications of eukaryotic mRNA permit greater dynamic control of protein expression, faster responses to a variety of stimuli, and the fine tuning of protein localization1. N6-methyladenosine (m6A), the most abundant chemical modification in mRNA, has recently emerged as a critical post-transcriptional mRNA regulator2,3. There are on average about three m6A modifications on any mRNA within the consensus motif, DRACH (D=A,G,U; R=A,G; H=A,C,U) and these modifications are essential in mammals4,5,6. In mammalian cells, the m6A modification is installed by a methyltransferase “writer” complex composed of METTL3 and METTL14, and it can be removed by the “erasers” ALKBH5 and FTO7,8,9,10,11,12. METTL3 plays important roles in coordinating naive and primed stem cell pluripotency; signaling for stem cell exit from self-renewal toward differentiation; and control of the circadian period13,14,15. Deficiency in the “erasers” FTO and ALKBH5 influences cell growth, fertility in mice, and acute myeloid leukemia11,16,17.
m6A exerts many of its functions through “reader” proteins in the cytoplasm and nucleus that selectively bind to m6A-containing transcripts6,18,19,20,21,22,23,24,25. In particular, the YT521-B homology (YTH) family of proteins possess the evolutionarily conserved YTH domain that selectively recognizes m6A21. In the cytosol, YTH domain family 1 (YTHDF1) and YTHDF3 act in concert to affect the translation of their targets by facilitating ribosome loading in HeLa cells20,26,27 whereas YTHDF2 decreases mRNA stability by recruiting the CCR4-NOT deadenylase complex18,28. In the nucleus, YTH domain containing 1 (YTHDC1) influences mRNA splicing22. Working in concert, YTHDF1, YTHDF2, YTHDF3, and YTHDC1 may provide a fast track to quickly process mRNAs in the nucleus, control their translation, and then degrade the translated mRNAs.
The fifth member of the YTH protein family, YTHDC2, has not yet been shown to bind m6A, and its function remains unclear. Although it contains the highly conserved YTH domain found in the other YTH family proteins, YTHDC2 possesses multiple helicase domains that may also act on RNA (Supplementary information, Figure S1). Ythdc2 is highly expressed in mouse testes, and was found to interact with the meiosis-specific protein MEIOC, which is required for meiotic prophase I29,30,31. Here, we report that Ythdc2 is an m6A-binding protein with critical functions in spermatogenesis. We report one of the first mammalian knockout models for YTH family proteins, showing that Ythdc2 facilitates proper spermatocyte development.
Results
YTHDC2 preferentially binds m6A
To determine whether YTHDC2 binds m6A, we performed an in vitro pulldown assay using lysate from HeLa cells stably overexpressing FLAG-tagged YTHDC2. YTHDC2 bound preferentially to an RNA probe containing m6A compared to a control RNA probe containing unmodified A (approximately 3.2-fold difference) (Figure 1A). Moreover, YTHDC2 bound preferentially to an RNA probe containing an m6A consensus motif (GGACU) compared to a control RNA probe containing a random sequence (ACAGA). YTHDC2 preferred binding to m6A rather than A regardless of the sequence of the probe (Figure 1A and Supplementary information, Figure S2B). We further confirmed that YTHDC2 binds selectively to m6A-containing RNA using gel shift (Supplementary information, Figure S2A). As YTHDC2 appears to play a role in spermatogenesis, we repeated our experiments using a mouse testes lysate, where we saw that Ythdc2 also bound preferentially to m6A within the GGACU motif (Supplementary information, Figure S2C). To further confirm the consensus motif, we performed crosslink immunoprecipitation sequencing (CLIP-seq) in adult mouse testes, and observed that Ythdc2-bound fractions enrich a consensus motif of GGACU (Figure 1B and Supplementary information, Figure S2D, Table S1). Moreover, the binding sites of YTHDC2 cluster around the stop codon, mirroring the locations of m6A along the mRNA transcript (Supplementary information, Figure S2E). These results indicate that YTHDC2 can preferentially recognize m6A-containing transcripts.

YTHDC2 binds preferentially to m6A-marked RNA transcripts. (A) In vitro probe pulldown assay in HeLa cells overexpressing FLAG-YTHDC2 showing YTHDC2 binds preferentially to probe with m6A on GGACU. GGYCU Probe sequence: 5′-CGUGGYCUGGCU-B-3′ (Y = m6A or A, B = biotin) ACYGA Probe sequence: 5′-GAUACYGAGAAG-B-3′ (Y = m6A or A, B = biotin). Labels: relative protein expression. (B) Consensus motif of Ythdc2 binding identified by HOMER of CLIP-seq of Ythdc2 in mouse testes. (C) Overlap of Ythdc2 RIP-seq genes and genes containing m6A in young mouse testes. Targets of YTHDC2 are defined as genes enriched in the RIP: Log2(RIP/input) ≥ 1. Non-targets are defined as genes depleted in the RIP: Log2(RIP/input) ≤ −1. Genes with m6A are defined as genes enriched in the m6A IP: Log2(m6A IP/input) ≥ 2.
Because previous work indicated that Ythdc2 may play a role in early spermatogenesis, we performed RNA immunoprecipitation sequencing (RIP-seq) of Ythdc2 in the testes of mice at 16.5 days post-partum (d.p.p.), which are enriched in meiotic spermatocytes, to identify gene targets of Ythdc230. Two biological replicates of RIP-seq assays identified 522 target genes of Ythdc2 (Supplementary information, Table S2). Of these target genes, 477 (91.4%) contain m6A, compared to 50.3% of genes depleted in the IP fraction (“Ythdc2 non-targets”) (Figure 1C and Supplementary information, Figure S2F-S2G). These results strengthen our conclusion that Ythdc2 is an m6A “reader” protein.
Ythdc2 affects mouse spermatogenesis
Gene ontology (GO) analysis of Ythdc2 RIP-seq targets in mouse testes identified meiosis-related categories among the most enriched pathways (Supplementary information, Figure S2H and Table S3). In agreement with a previous report, we also identified that Ythdc2 is highly expressed in mouse testes (Figure 2A). Our results pointed us toward examining the function of Ythdc2 in spermatogenesis.

Ythdc2-deficient mice display defects in testes. (A) RT-PCR analysis of Ythdc2 mRNA levels in various organs of adult mice. (B) Schematic diagram of sgRNAs targeting at Ythdc2 locus. Two founders were generated by micro-injection of sgRNA/Cas9 mRNA into one-cell embryos. PAM sequences are underlined and highlighted in green. sgRNA targeting sites are in red. Mutant sequence are in blue. (C) Testes and epididymis from adult Ythdc2−/− mice are smaller in size than wild type. (D) The testis/body weight ratio was calculated for adult Ythdc2+/+ and Ythdc2−/− mice. Error bars, mean ± sd, n = 4, biological replicates ***P < 0.001 (Student's t-test) (E) Periodic Acid Schiff (PAS) staining of adult testes and epididymis. Scale bar, 50 μm.
To investigate the biological functions of Ythdc2, we generated two Ythdc2−/− founder mice by microinjection of CRISPR/Cas9 targeting exon 5 of the Ythdc2 gene (Figure 2B and Supplementary information, Figure S3A-S3B). Ythdc2−/− mice with a 37-bp deletion were viable and reached adulthood. However, both male and female mice were infertile; male mice had smaller testes, and female mice had smaller ovaries and showed progressive loss of germ cells (Figure 2C-2D and Supplementary information, Figure S4A-S4C). Comparative histological staining of the testes, caput epididymis, and cauda epididymis of adult mice showed that Ythdc2−/− mice lacked spermatozoa in the seminiferous tubules and epididymis (Figure 2E). Ythdc2−/− mice with a 4-bp deletion showed the same phenotype as those with the 37-bp deletion (Supplementary information, Figure S3C), eliminating the possibility of off-target effects induced by CRISPR/Cas9.
We examined expression levels of Ythdc2 mRNA and protein over time in testes, and found that Ythdc2 expression rises between 7 and 12 d.p.p. and remains steady through adulthood (Figure 3A and Supplementary information, Figure S5). As meiosis in male mice is initiated around 8 d.p.p., we hypothesized that meiotic prophase I may be affected in Ythdc2−/− mice32. Immunofluorescence staining showed abnormalities in meiotic prophase I in Ythdc2−/− mice. Spermatocytes in Ythdc2−/− mice seemed to undergo leptotene normally (Supplementary information, Figure S6A). However, unlike control spermatocytes, the Ythdc2−/− spermatocytes failed to undergo pachytene and instead reached a terminal zygotene-like stage, displaying fragmented axial elements and partially synapsed lateral elements (Figure 3B and Supplementary information, Figure S6A, data not shown). We also observed a similar failure to reach pachytene in oocytes of Ythdc2−/− mice (Supplementary information, Figure S6B-S6C). We reasoned that the abnormal spermatocytes may undergo apoptosis, and thus performed TUNEL assays on testes sections from mice at the age of 8.5–10.5 d.p.p., around the time of meiotic initiation. We found substantially more apoptosis in the testes of Ythdc2−/− mice than those of the wild-type after 9.5–10.5 d.p.p. (Figure 3C-3D). Cleaved Caspase-3 staining of young mouse testes also revealed that spermatocytes in Ythdc2−/− mice undergo apoptosis at 9.5–10.5 d.p.p. (Figure 3E). The number of spermatocytes in Ythdc2−/− mice is drastically reduced after 9.5 d.p.p. (Supplementary information, Figure S7). The defect in spermatogenesis in Ythdc2−/− mice appeared to be limited to the spermatocyte stage, as Ythdc2−/− mice displayed normal spermatogonia and sertoli cells (Supplementary information, Figures S8 and S9). Thus, Ythdc2 is required for proper spermatocyte development.

Ythdc2-deficient mice display defects in meiotic prophase I. (A) RT-qPCR analysis of Ythdc2 mRNA levels in wild type testes over time, normalized to Rn18s as an internal control. Error bars, mean ± sd, n = 3, technical replicates (Student's t-test). (B) Ythdc2+/+ and Ythdc2−/− spermatocyte chromosome spreads at 15.0 d.p.p. stained for SYCP1 (red) and SYCP3 (green). Scale bar, 10 μm. (C) TUNEL assay of testes sections from 8.5 d.p.p. to 10.5 d.p.p.. DNA was stained with Hoechst (blue). Scale bar, 20 μm. (D) Quantification of apoptotic cells in Ythdc2+/+ (black columns) and Ythdc2−/− (grey columns) testes at 8.5 to 10.5 d.p.p.. Error bars, mean ± sd, n = 3, biological replicates *P < 0.05, *P < 0.01 (Student's t-test). (E) Cleaved Caspase-3 staining of testes sections at 8.5–10.5 d.p.p.. Scale bar, 20 μm.
Ythdc2 affects translation efficiency and mRNA abundance of its targets
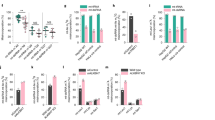
To investigate the function of Ythdc2 in mouse testes, we evaluated the expression of its targets in 8.5 d.p.p. mouse testes. We focused on Smc3, a top RIP-seq target of Ythdc2 involved in spermatogenesis. Our m6A-seq data showed that Smc3 contains m6A. We confirmed the presence of m6A in transcripts by qRT-PCR (Supplementary information, Table S4 and Figure S10B). Western blots revealed decreased expression of Smc3 in the 8.5 d.p.p. testes of Ythdc2−/− mice (Supplementary information, Figure S10A). In contrast, qRT-PCR of Smc3 in 8.5 d.p.p. Ythdc2−/− mouse testes showed an increase in its mRNA abundance (Figure 4A). The net outcome was a 37% decrease in translation efficiency in Ythdc2−/− mice (Figure 4B). We also saw similar changes of translation efficiency and mRNA abundance of Cep76, another top RIP-seq target of Ythdc2 that is involved in centriole reduplication (Supplementary information, Figure S10C-S10F). Thus, Ythdc2 may function by affecting the translation efficiency and mRNA abundance of its targets in mouse testes.

YTHDC2 affects the translation efficiency and mRNA abundance of its targets. (A) Relative mRNA expression of Smc3, normalized to Rn18s, in 8.5 d.p.p. Ythdc2+/+ and Ythdc2−/− testes. Error bars, mean ± sd, n = 3. **P < 0.01 (Student's t-test). (B) Translation efficiency of Smc3 in 8.5 d.p.p. Ythdc2+/+ and Ythdc2−/− testes, calculated as: translation efficiency = Smc3 protein level / Smc3 mRNA expression level. Smc3 protein level was determined by western blot and normalized to tubulin. Error bars, mean ± sd, n = 3. **P < 0.01 (Student's t-test). (C) Constructs in the tethering reporter assay. Effector: the YTH domain of YTHDC2 was replaced with λ peptide (N-YTHDC2-λ), which binds with high affinity to the BoxB RNA motif. Reporter: 5 BoxB domains were fused to the 3′ UTR of firefly luciferase mRNA (F-Luc-5BoxB). Renilla luciferase (R-Luc) mRNA was used as an internal control to normalize luciferase signals from different samples. (D) mRNA level of F-Luc normalized to R-Luc 4 h post F-Luc induction. Error bars, mean ± sd, n = 4. **P < 0.01 (Student's t-test). (E) Translation efficiency of F-Luc normalized to R-Luc 4 h post F-Luc induction. Error bars, mean ± sd, n = 4. ****P < 0.0001; (Student's t-test). (F) Cumulative frequency of log2-fold changes of RNA expression upon Ythdc2 depletion, in RIP targets and non-targets. P value was calculated using two-sided Mann-Whitney U test. (G) Top GO terms in GO analysis of components of protein complex containing YTHDC2 obtained from tandem affinity purification followed by mass spectrometry.
This led us to investigate the direct role of YTHDC2 in regulating translation efficiency and mRNA abundance using a luciferase-based tethered reporter assay in HeLa cells as a model19 (Figure 4C). We replaced the YTHDC2 C-terminal YTH domain, which engages in m6A binding in YTH family proteins33,34, with λ peptide (N-YTHDC2-λ) known to bind specifically and tightly to F-Luc-5BoxB (five Box B RNA sequences inserted into the 3′ UTR of a firefly luciferase reporter). We installed a Tet-Off inducible promotor, which prevents transcription in the presence of doxycycline (DOX), onto F-Luc-5BoxB, thus allowing inducible expression of the luciferase reporter. We compared the expression of firefly luciferase (normalized to renilla luciferase) with and without the presence of N-YTHDC2-λ, and saw a 30% increase in translation accompanied by a 15% decrease in mRNA abundance (Figure 4D and Supplementary information, Figure S10G). Overall, this resulted in a 52% increase in translation efficiency, indicating that YTHDC2 can enhance translation efficiency and decrease the mRNA abundance of its targets in HeLa cells (Figure 4E).
To investigate whether Ythdc2 also affects the mRNA abundance and m6A profile of its targets, we performed m6A-seq on the testes of 8.5 d.p.p. Ythdc2−/− mice (Supplementary information, Table S4). m6A was located on its usual consensus motif of GGACU in both control and Ythdc2−/− testes (Supplementary information, Figure S11A). The CDS and 3′ UTR harbored the majority of m6A, as expected, but showed no substantial changes in m6A upon Ythdc2 depletion (Supplementary information, Figure S11B). There was a slight decrease of m6A in the 5′ UTR of mRNAs from Ythdc2−/− testes compared to control mice (Supplementary information, Figure S11B). We then compared RNA expression levels of genes in Ythdc2+/+ and Ythdc2−/− testes. We observed a slight increase in the expression of genes with m6A in Ythdc2−/− testes (Supplementary information, Figure S11C). This difference was not detectable when comparing all genes in Ythdc2+/+ and Ythdc2−/− testes, regardless of whether m6A is present, (Supplementary information, Figure S11D). We also observed a modest increase in the expression of Ythdc2 targets in Ythdc2−/− mice compared to Ythdc2+/+ mice (Figure 4F and Supplementary information, Figure S11E). Together our results indicate a role of YTHDC2 in regulating target RNA stability.
Next, we asked whether translation efficiency and stability of targets of YTHDC2 are regulated through its binding of m6A. We knocked down the m6A methyltransferase METTL3 in HeLa cells and performed ribosome profiling7. Upon reduction of m6A, we observed an overall decrease in translation efficiency and increase in mRNA abundance of YTHDC2 target transcripts, as determined by PAR-CLIP in HeLa cells (Supplementary information, Figure S12A-S12C and Table S5). This data suggests that the roles of YTHDC2 in enhancing translation efficiency and decreasing mRNA stability could be affected through its binding of m6A, although this protein possesses multiple domains that can also bind RNA and potentially affect targets' fates.
To explore how YTHDC2 may affect translation efficiency and mRNA stability, we performed polysome profiling of HeLa cells, separating cellular fractions along a 10%-50% sucrose gradient. The expression of YTHDC2 in different fractions was detected with western blot, and we observed YTHDC2 in the 40-80S ribosome fraction, suggesting that YTHDC2 may interact with cellular machinery involved in translation initiation (Supplementary information, Figure S10H). We then immunoprecipitated the YTHDC2-containing protein complexes from HeLa cells using tandem affinity purification and subjected the isolated peptides to mass spectrometry (Supplementary information, Table S6 and Figure S13). GO analysis of the top binding partner proteins revealed functions related to translation and translation initiation (Figure 4G). Moreover, the third most significant GO term of top binding partner proteins of YTHDC2 was nonsense-mediated decay, a process coupled with translation that eliminates mRNAs with premature translation-termination codons35,36. YTHDC2 partner proteins with known roles in nonsense-mediated decay included the exoribonuclease XRN1 and helicases UPF1 and MOV1037,38. Thus, YTHDC2 may recruit decay machineries to accelerate the degradation of mRNA targets during or after translation in mammalian cells. Although the effect on stability observed in testes appears to be very modest, these experiments were carried out upon the heterogeneous cell population of entire testes. It is possible that future studies on purified spermatocytes could reveal more significant effects. Together, our results suggest that YTHDC2 may interact with the translation and decay machineries to enhance the translation efficiency and decrease the mRNA abundance of its targets.
Discussion
YTHDF1, YTHDF2, YTHDF3, and YTHDC1 are members of the YTH protein family with characterized roles in the cytoplasm and nucleus. The role of the final member of the YTH protein family, YTHDC2, remained uncertain. To address this we have here characterized the function of Ythdc2 and its role in spermatogenesis using a knockout mouse model that has yet to be reported for any other YTH family proteins. Our results show that YTHDC2 preferentially binds m6A within its consensus RNA motif (Figure 1A-1C). YTHDC2 may increase the translation efficiency of its targets, as we showed in a tethering reporter assay (Figure 4E), as well as decrease their mRNA abundance (Figure 4D, 4F). Ythdc2 appears to be critical for fertility because both male and female mice lacking Ythdc2 are infertile and do not contain germ cells able to develop beyond the zygotene stage of meiotic prophase I (Figure 2 and Supplementary information, Figure S6).
The YTH family proteins display prominent effects on m6A-containing mRNA transcripts. YTHDF1-3 function in the cytoplasm; YTHDF1 and YTHDF3 affect the translation efficiency of their targets, and YTHDF2 decreases the stability of its targets. YTHDC1 acts in the nucleus, affecting mRNA processing. Like its family members, YTHDC2 contains the highly evolutionarily conserved YTH domain, whose interaction with m6A has been described through recent crystallographic studies on YTHDF1, YTHDF2, and YTHDC121,33,39. Thus, it is very likely that the YTH domain confers m6A binding specificity to YTHDC2. However, YTHDC2 is otherwise distinct from its other family members. It is by far the largest protein (approximately160 kDa as compared to approximately 60-85 kDa), and it is the only family member to contain helicase domains (Supplementary information, Figure S1). These helicase domains may contribute to RNA binding, or may have effects on RNA secondary structure. YTHDC2 also contains two Ankyrin repeats between its helicase domains; these repeats mediate a diverse range of protein-protein interactions. As the YTHDC2-containing protein complex includes proteins involved in translation and translation initiation (Figure 3E), the Ankyrin repeats may be important for the recruitment or binding of other protein complex members.
Because YTHDC2 contains multiple RNA-binding domains, it is possible that it may be a protein with multiple roles. We have elucidated roles pertaining to the regulation of translation efficiency and stability, and our data suggest that these roles may depend on the binding of YTHDC2 to m6A (Supplementary information, Figure S12). We have recently proposed that m6A is deposited onto transcripts to mark them for coordinated translation and decay40. This coordination may allow efficient utilization of the transcriptome and RNA turnover in cells undergoing differentiation. As little transcription happens after germ cells enter meiotic prophase I41,42, transcripts that have been translated and are no longer necessary in a later meiotic stage may need to be cleared so that the translation machinery can more effectively target the remaining transcripts. Thus Ythdc2 might indeed work on transcripts to accelerate both translation and decay.
It is quite possible that the various RNA-binding domains of YTHDC2 could have functions independent of its m6A-binding, which should be further studied in the future. Experiments introducing individual mutations to various domains could elucidate the roles of the individual domains and the mechanism underlying YTHDC2 function. Moreover, although we identified over 8 000 genes containing m6A in mouse testes, RIP-seq of Ythdc2 in mouse testes revealed binding only to a subset of these genes (Figure 1C). Future studies are necessary to determine the overarching role of m6A throughout spermatogenesis, as well as the selectivity of Ythdc2 to its targets.
Two recent articles have pointed toward a role of Ythdc2 in spermatogenesis. Abby et al. suggested that the meiosis-specific protein MEIOC interacts with YTHDC2 in an RNA-independent manner to stabilize transcripts during meiosis prophase I, allowing proper induction of the meiotic program30. Soh et al. also found that MEIOC interacts with Ythdc2, but propose that MEIOC may destabilize its targets and that it is required to sustain the prolonged length of meiotic prophase I31. Our results indicate that the role of YTHDC2 can be complex. Its interactions with XRN1, UPF1, and MOV10 could suggest a role in destabilizing its target RNAs. This receives support from our tethering reporter assays and sequencing data, which also reveal that YTHDC2 could destabilize its target RNAs. It is possible that MEIOC and YTHDC2 work in tandem to respectively influence the stability and translation efficiency of their targets. MEIOC may work to stabilize meiotic transcripts synthesized at the start of meiotic prophase I. YTHDC2 may enhance the translation of transcripts, and then destabilize the transcripts after translation is completed, allowing proper progression of meiotic prophase I.
The m6A demethylase Alkbh5 has also been shown to be most highly expressed in mouse testes, and its depletion leads to compromised spermatogenesis11. The m6A mark may thus be an important regulator of spermatogenesis. Interestingly, unlike the germ cells in Ythdc2−/− mice, the germ cells in Alkbh5−/− mice develop into spermatozoa, albeit at a dramatically decreased rate and with morphological abnormalities. Thus, the m6A mark may hold multiple roles throughout the various stages of spermatogenesis, and its misregulation may lead to defects in multiple pathways. Here we demonstrate defects in meiosis in Ythdc2−/− mice and show that Ythdc2 targets meiosis-related genes in mouse testes. However, we cannot exclude the possibility that the effect of Ythdc2 on meiosis may be indirect and that the loss of Ythdc2 leads to other defects that indirectly affect meiosis.
Our present studies are limited to functions of Ythdc2 in early spermatogenesis. It will be important to determine whether Ythdc2 functions beyond meiotic prophase I as will be possible through inducible knockout of Ythdc2. YTHDC2 may play important roles in other systems. Our results showed that Ythdc2 is also highly expressed in the spleen and brain (Figure 2A); YTHDC2 may thus have potential roles in the immune and nervous systems. YTHDC2 has also been identified as a frequently mutated gene in pancreatic adenocarcinoma patients, and plays a role in cancer metastasis by increasing the translation efficiency of HIF-1α43,44. As a protein with multiple domains, YTHDC2 could possess complex functions in different cellular contexts, potentially involving the regulation of both translation efficiency and transcript stability. The contributions of each domain to such differing functions requires more detailed future study.
Materials and Methods
Animals
All mice were housed under specific pathogen-free (SPF) conditions on a 12-h light-12-h dark cycle. All animal protocols were approved by the Animal Care and Use Committee of Nanjing Medical University.
In vitro transcription and microinjection of CRISPR/Cas9
Two sgRNAs were designed to target Exon 5 of the Ythdc2 gene. Oligos for the generation of sgRNA expression plasmids were annealed and cloned into the BsaI sites of pUC57-sgRNA (Addgene 51132)45. Oligo sequences are as follows: sgRNA1_up: TAGGAGTACAAAATATTCGGCAG; sgRNA1_down: AAACctgccgaatattttgtact; sgRNA2_up: TAGGacgactagcagcgattgctg; sgRNA2_down: AAACCAGCAATCGCTGCTAGTCGT. In vitro transcription and microinjection of CRISPR/Cas9 were performed as described45. Briefly, the Cas9 expression construct pST1374-Cas9-N-NLS-Flag-linker (Addgene 44758) was linearized with AgeI and transcribed using the the T7 Ultra Kit (Ambion), followed by purification using RNeasy Mini Kit (Qiagen). pUC57-sgRNA expression vectors were linearized by DraI and transcribed using the MEGAshortscript Kit in vitro (Ambion). sgRNAs were purified by MEGAclear Kit (Ambion). Mixture of Cas9 mRNA (20 ng/μl) and two sgRNAs (5 ng/μl each) were injected into the cytoplasm and male pronucleus of the zygote, obtained by CBF1 mating. Injected zygotes were transferred into pseudo-pregnant CD1 female mice. Founder mice were backcrossed to C57BL/6J. Mice used for experiments were of mixed C57BL/6J and CBA genetic backgrounds.
Primers used for genotyping of Ythdc2 mice: GATTCCTCAGTTCCTGTTAGA and GACCAATTCTTTCTCTTCTCTC.
Antibodies
The antibodies used in this study (listed in the format of name (catalogue; supplier; dilution) were as follows: Goat anti-YTHDC2 (Sc-249370; Santa-Cruz; IP/20μg; Western Blot/1:1 000); Rabbit anti-Ythdc2 (A303-026A; BETHYL; Western Blot/1:2 000); Rabbit anti-m6A (202003; Synaptic Systems; IP/2.5 μg); Rabbit anti-eIF3a (3411; Cell Signaling Technology; Western Blot/1:2 000); Mouse anti-FLAG M2-peroxidase HRP (A8592; Sigma; Western Blot/1:10 000); Goat anti-GAPDH HRP (A00192-100; GenScript; Western Blot/1:5 000); Mouse anti-DDX4 (ab27591; Abcam; IF/1:200); Mouse anti-γ-H2A.X (ab26350; Abcam; IF/1:100); Mouse anti-SYCP3 (ab97672; Abcam; IF/1:100); Rabbit anti-SYCP3 (ab15093; Abcam; IF/1:100); Rabbit anti SYCP1 (ab15090; Abcam; IF/1:100); Mouse anti-SMC3 (ab128919; Abcam; Western Blot/1:2 000, IF/1:200); Rabbit anti-cleaved caspase3 (9661; CST; IHC/1:200); Rabbit anti-Cep76 (gift from Dr Xueliang Zhu, Institute of Biochemistry and Cell Biology, SIBS, CAS, Western Blot/1:2 000); Mouse anti-PLZF (sc-28319; Santa-Cruz; IF/1:200); Rabbit anti-Lin28 (13456-1-AP; Proteintech; IHC/1:200); Rabbit anti-SOX9 (ab5535; Chemicon; IF/1:200); Rabbit anti-IMP3 (IGF2BP3) (A303-426A; Bethyl; Western Blot/1:1 000); Rabbit anti-YBX1 (ab76149; Abcam; Western Blot/1: 1 000); Rabbit anti-MOV10 (A301-571A; Bethyl; Western Blot/1:1 000); Rabbit anti-XRN1 (A300-443A; Bethyl; Western Blot/1:1 000); Mouse anti-Tubulin(AT819; Beyotime; Western Blot/1:10 000); Donkey anti-Rabbit IgG-HRP (sc-2313; Santa-Cruz; Western Blot/1:2 000); Donkey anti-Goat IgG-HRP (sc-2033; Santa-Cruz; Western Blot/1:2 000). Alexa Fluor® 555 donkey anti-mouse IgG (A31570; Life Technologies; IF/1:500); Alexa Fluor® 555 donkey anti-goat IgG (A21432; Life Technologies; IF/1:500); Alexa Fluor® 488 donkey anti-rabbit IgG (A21206; Life Technologies; IF/1:500); Alexa Fluor® 488 donkey anti-mouse IgG (A21202; Life Technologies; IF/1:500); Alexa Fluor® 555 donkey anti-rabbit IgG (A31572; Life Technologies; IF/1:500); and Alexa Fluor® 633 donkey anti-goat IgG (A21082; Life Technologies; IF 1:500).
Histological analysis and immunofluorescence
Testes were fixed in Modified Davidson's Fluid (MDF), embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E). For immunofluorescence, after dewaxing and hydration, paraffin sections were subjected to antigen retrieval by boiling in citrate buffer (1.8 mM citric acid, 8.2 mM sodium citrate, pH6.0), washing with PBS three times, blocking in 5% bovine serum albumin (BSA) for 2 h, and incubating with primary antibodies overnight at 4 °C. After washing with PBS three times, secondary antibodies were added to the samples for 2 h at room temperature (RT). The sections were then washed in PBS three times and incubated with Hoechst 33258 (Sigma) for 5 min at RT. Periodic acid-Schiff (PAS) staining of testis sections was performed according to the manufacturer's protocol (Sigma-Aldrich Cat. No. 395B-1KT).
For immunohistochemistry, after dewaxing and hydration, the sections were incubated with 10% hydrogen peroxide for 10 min to eliminate endogenous peroxidase activity at 37 °C. After antigen retrieval with citrate buffer and washing, the sections were blocked in 5% BSA and incubated with primary antibodies and secondary antibodies. DAB staining kit (ZSGB-BIO, ZLI-9018) was used to detect the signal according to the manufacturer's instructions. The sections were counterstained with hematoxylin. For spermatocyte chromosome spreads, testes and ovaries were dissected and placed in hypotonic extraction buffer (30 mM Tris-HCl pH 8.5, 50 mM sucrose, 17 mM citric acid, 5 mM EDTA, 2.5 mM DTT, 1 mM PMSF) for 45 min at RT. Tissues were separated and minced in 100 mM sucrose. Cell suspensions were pipetted onto slides and fixed (1% PFA, 0.15% TritonX-100, 10 mM sodium borate) for 3 h. After washing four times with TBS (150 mM NaCl, 20 mM Tris-HCl pH 7.6), slides were blocked in ADB (1% Normal donkey serum, 0.3% BSA, 0.05% TritonX-100) for 1 h at RT, incubated with primary antibodies overnight at 37 °C, blocked in ADB for 5 h, and finally incubated with secondary antibodies for 1.5 h at 37 °C and washed with TBS.
H&E images were taken using an Axioskop 2 plus (ZEISS). Immunofluorescence images were taken with a LSM710 laser scanning confocal microscope (ZEISS).
Plasmid construction and protein expression
Flag-tagged YTHDC2 was cloned from commercial cDNA clones (Dharmacon) into vector pFastBac (BamHI, XhoI; forward primer with coding sequence for Flag-tag, 5′-GCGGATCCATGGACTACAAAGACGATGACGACAAGTCCAGGCCGAGCAGCGTCTC-3′; reverse primer, 5′-GCCTCGAGTCAATCAGTTGTGTTTTTTTCTCCCAAG-3′). Flag-tagged YTHDC2 was purified using the Bac-to-Bac Baculovirus Expression System (Life Technologies) following the manufacturer's protocols.
Tether effector (N-YTHDC2-λ) was cloned from commercial cDNA clones into vector pcDNA3.0 (BamHI, XhoI; forward primer, 5′-GCGGATCCATGTCCAGGCCGAGCAG-3′; reverse primer with λ peptide transcript 5′-GCCTCGAGTCAGTTTGCAGCTTTCCATTGAGCTTGTTTCTC AGCGCGACGCTCACGTCGTCGTGTTTGTGCGTCCATAGGCATGTTTGGTCTTGG-3′). Tether control effector and reporter constructed have been previously described19.
Mammalian cell culture, shRNA knockdown, and plasmid transfection
The human HeLa cell line used in this study was purchased from ATCC (CCL-2) and grown in DMEM (Gibco, 11995) media supplemented with 10% FBS and 1% 100× Pen/Strep (Gibco). The HeLa Tet-off cell line was purchased from Clontech and grown in DMEM (Gibco) media supplemented with 10% FBS (Tet system approved, Clontech), 1% 100× Pen/Strep (Gibco), and 200 μg/ml G418 (Sigma).
YTHDC2 shRNA were purchased from GE Dharmacon (RHS4533-EG64848); YTHDC2 shRNA-3 target sequence: ATATAAGAGATGTGACGAGGG; YTHDC2 shRNA-5 target sequence: CTTTAGTCGAAGTTCTGACTA. Lentiviruses containing the YTHDC2 shRNAs were produced in 293T cells with packaging plasmids. After 48 h, virus was were collected and used to infect Hela cells. Stable YTHDC2 knock down cell lines were selected through 3 days of puromycin selection (2 μg/ml).
YTHDC2 with an N terminal FLAG tag and HA tag in tandem was stably overexpressed in HeLa cells by puromycin selection (2 μg/ml) with a modified pPB-CAG vector. The control cell line expressing only FLAG and HA peptides in tandem was created similarly.
Tethering reporter assay
The tethering reporter assay was performed as previously reported, with slight modifications20. 30 ng of the reporter plasmid (pmirGlo-Ptight-5BoxB) and 270 ng of the effector plasmid (N-YTHDC2-λ or FLAG-GGS-λ as a control in pcDNA3.0) were used to transfect HeLa Tet-Off Advanced Cells (613356, Clontech) in each well of a six-well plate at 60%-80% confluence under doxycycline (Dox, 100 ng/ml) inhibition. After six hours, the transfection mixture was replaced with fresh media containing DOX (100ng/ml). Eighteen hours later, each well was digested with trypsin, washed extensively with PBS by resuspending and centrifuging the cells three times. Cells were re-seeded into six wells of a 96-well plate (1:10 in each well) and one plate of a 12-well plate (4:10) in media without DOX to induce transcription of F-luc. Four hours later, cells in the 96-well plate were analyzed by Dual-Glo Luciferase Assay System (E2920, Promega), and F-luc activity was normalized to R-Luc to evaluate protein production from the reporter. Concurrently, the RNA was extracted from the cells in the 12-well plate with Direct-zol RNA Microprep Kit (Zymo Research), following the manufacturer's protocol and including DNase I digestion. mRNA expression of F-luc and R-luc were quantified by RT-qPCR, and the expression of F-luc was normalized to that of R-luc. Translation efficiency of F-luc mRNA was calculated as the ratio of normalized F-luc protein level to normalized F-luc mRNA level.
RIP-seq
RIP-seq was performed as previously described, with minor modifications18. Testes were isolated from 2 16.5 d.p.p. male mice per sample; tunica vaginalis was removed and discarded. The remaining seminiferous tubules were lysed in high salt lysis buffer (300 mM NaCl, 0.2% NP-40, 20 mM Tris-HCl pH 7.6, 0.5 mM DTT, 1:100 cOmplete EDTA-free protease inhibitor (Roche), 1:100 SUPERase In RNase Inhibitor (Ambion)) for 30 min with gentle rotary shaking at 4 °C, followed by centrifugation at 16 000g for 20 min. The supernatant was diluted 1:1 in 20 mM Tris-HCl pH 7.6, and filtered through a 0.22 μm filter. The sample was precleared with 33 μl Dynabeads Protein G beads (Life technologies) for 1 h at 4 °C. 100 μl Dynabeads Protein G beads were coated with 20 μg of Goat anti-YTHDC2 or normal goat IgG (Santa-Cruz) at 4 °C for 1 h following the manufacturer's instructions. 1/10 of the sample was saved as RNA input; the rest was incubated with the pre-coated beads for 4 h at 4 °C. The beads were washed six times with NT2 wash buffer (200 mM NaCl, 2 mM EDTA, 0.05% NP-40, 50 mM Tris-HCl pH 7.6, 0.5 μM DTT, 1:1 000 protease inhibitor, 1:1 000 SUPERase In RNAse Inhibitor) to remove excess RNA, and RNA was isolated with Direct-zol RNA Microprep Kit (Zymo Research). The sample immunoprecipitated using normal goat IgG did not contain any detectable RNA as detected by Nanodrop. rRNA was depleted in parallel from both the Input and IP samples using RiboMinus Eukaryote Kit v2 (Ambion). 5 ng of Input and IP RNA, 2 biological replicates each, were used to generate the library using TruSeq stranded mRNA sample preparation kit (Illumina).
CLIP-seq
CLIP-seq was performed as described for PAR-CLIP46, without using 4SU and with the following modifications. Testes were isolated from 5 adult male mice per sample; tunica vaginalis was removed and discarded. The remaining seminiferous tubules were separated to single cells using a tissue grinder (Fisher), filtered through a 40 μm cell strainer in 5 ml PBS, then crosslinked with 254 nm UV light three times using a Stratalinker 2 400 at 150 mJ/cm2.
Mild enzyme digestion47: The first round of T1 digestion was carried out using 0.2U ml-1 for 15 minutes. The second round of T1 digestion was conducted using 10 U/ml for 8 min.
Library construction: the final recovered RNA sample was further cleaned by RNA Clean & Concentrator (Zymo Research) before library construction using the Truseq small RNA sample preparation kit (Illumina).
PAR-CLIP
PAR-CLIP was performed as previously described18,46 with the following modifications. The preparation was made from 150-200 × 106 HeLa cells stably expressing Flag-HA-tagged YTHDC2. The YTHDC2-RNA complex was purified by SDS-PAGE selecting proteins migrating between 130-200 kDa. RNA fragments were extracted by proteinase K digestion of the gel slices followed by ethanol precipitation. The purified RNA pellet was dissolved in 12 μl RNase-free water, of which 6 μl was subjected to library construction by Truseq small RNA sample preparation kit (Illumina).
m6A-seq
m6A-seq was performed as previously described6. Briefly, 100 μg total cellular RNA was extracted from the seminiferous tubules of 8.5 d.p.p. mice using TRIzol following the manufacturer's protocol. PolyA mRNA was enriched using Dynabeads mRNA DIRECT Kit (Ambion) following the manufacturer's protocols. mRNA was sonicated to ∼100 nt, mixed with 2.5 mg affinity-purified anti-m6A polyclonal antibody (Synaptic Systems) in IP buffer (150 mM NaCl, 0.1% NP-40, and 10 mM Tris-HCl, pH 7.4), and incubated for 2 h at 4 °C. The antibody-RNA complex was isolated by incubation with protein A beads (Invitrogen) at 4 °C for 2 h. The beads were washed three times and eluted competitively with an m6A monophosphate solution. RNA in the eluate was isolated using RNA Clean and Concentrator (Zymo Research) and used for library preparation with TruSeq stranded mRNA sample preparation kit (Illumina).
Polysome profiling
Polysome profiling of HeLa Control or shYTHDC2 knockdown cells was performed as previously described26 with the following modifications: (1) Before collection, cycloheximide (CHX) was added to the media at 100 μg/ml for 7 min. (2) The lysis buffer was 20 mM HEPES, pH 7.6, 100 mM KCl, 5 mM MgCl2, 100 μg/ ml CHX, 1% Triton-X-100, 1:100 protease inhibitor (Roche), and 40 U/ml SUPERasin (Ambion).
Data analysis
Gene structure annotations were downloaded from UCSC mm10 RefSeq. Data analyses were performed as previously described6,19. Briefly, after removing adapters, sequencing reads were aligned to the reference genome (mm10) using TopHat (v2.0.14)48. For RIPSeq, RPKM were calculated by Cuffnorm49. RIP-seq targets were defined as genes enriched in the IP (log2(RIP/input) ≥ 1), and RIP-seq non-targets were defined as genes depleted in the IP (log2(RIP/input) ≤ −1). For m6A-seq, the longest isoform was used if multiple isoforms were detected. Aligned reads were extended to 100 nt (average fragment size) and converted from genome-based coordinates to isoform-based coordinates to eliminate interference from introns in peak calling. To call m6A peaks, the longest isoform of each human gene was scanned using a 100 nt sliding window with 10 nt steps. To reduce bias from potentially inaccurate gene structure annotation and the arbitrary usage of the longest isoform, windows with reads counts less than 1/20 of the top window in both m6A IP and input sample were excluded. For each gene, the reads count in each window was normalized by the median count of all windows of that gene. A negative binomial model was used to identify the differential windows between IP and input samples by using the edgeR package50. The window was called positive if FDR < 1% and log2(enrichment score) ≥ 1. Overlapping positive windows were merged. The following four numbers were calculated to obtain the enrichment score of each peak (or window): read count of the IP sample in the current peak/window (a), median read count of the IP sample in all 100 nt windows on the current mRNA (b), read count of the input sample in the current peak/window (c), and median read count of the input sample in all 100 nt windows on the current mRNA (d). The enrichment score of each window was calculated as (a × d)/(b × c). For CLIP-seq, we used a modified version of PARalyzer51 to allow for all mutations, rather than just T to C mutations, for peak calling, while filtering out all SNPs and potential sequencing errors. Consensus motif was determined using HOMER52. For PAR-CLIP, we analyzed data using PARalyzer with default settings. Nonparametric Mann-Whitney U-test (Wilcoxon rank-sum test, two sided, significance level = 0.05) was applied to RNA-seq and ribosome profiling data analysis, as previously reported19,53.
RT-qPCR
All RNA templates used in RT-qPCR were digested with DNase I during purification with Direct-zol RNA Microprep Kit (Zymo Research) to avoid genomic DNA contamination. All RT-qPCR primers covered exon-exon junctions when possible. 500ng of RNA was reverse-transcribed into cDNA with PrimeScript RT (Takara), then subjected to qPCR analysis with FastStart SYBR Green Master Mix (Roche) in a Roche LightCycler 96. Primer sequences are as follows: mYthdc2_F GGTCCGATCAATCATCTGT; mYthdc2_R GAAGTAACGAATAGGCATGT; m18s_F CATTCGAACGTCTGCCCTATC; m18s_R CCTGCTGCCTTCCTTGGA; mSmc3_F AACCAGGAGCTCAACGAGAC; mSmc3_R TGGAAACTCAGCAGCAAGAGA; mCep76_F CCTCGGTCACCAGCAATGAA; mCep76_R GCTTGTTGTTCGCTCAAACTCA. The primer sequences of F-luc and R-luc have been reported19.
Western blotting
Proteins were extracted with 8 M urea lysis buffer (8 M urea, 75 μM NaCl, 50 μM Tris-Cl pH 8.2) containing 1 mM PMSF, and quantified using the Bradford Protein Assay Kit (Beyotime). Protein samples were separated in a 7% SDS-PAGE gel and transferred to PVDF membranes (Millipore). Appropriate primary antibodies were incubated overnight at 4 °C after blocking in 5% non-fat milk. After washing three times in TBST, secondary antibodies were used at appropriate dilution for 2 h at RT. The signals were detected by the addition of Ncm ECL Ultra® (New Cell & Molecular Biotech Co., Ltd).
TUNEL assay
Detection of apoptotic cells was performed using TUNEL Apoptosis Detection Kit (Vazyme) according to the manufacturer's instructions. Images were taken with a LSM710 laser scanning confocal microscope (ZEISS).
In vitro probe pull-down
40 × 106 HeLa cells stably overexpressing FLAG-tagged YTHDC2 or the seminiferous tubules of three 16.5 d.p.p. mice were lysed at 4 °C for 30 minutes in 3.5 ml lysis buffer (250 mM NaCl, 0.5% NP40, 10% glycerol, 20mM Tris pH 7.5, 1:100 cOmplete EDTA-free protease inhibitor (Roche), 1:100 SUPERase In RNase Inhibitor (Ambion)) or high salt lysis buffer (300 mM NaCl, 0.2% NP-40, 20 mM Tris-HCl pH 7.6, 0.5 mM DTT, 1:100 cOmplete EDTA-free protease inhibitor (Roche), 1:100 SUPERase In RNase Inhibitor (Ambion)), respectively, followed by centrifugation at 16 000 g for 20 min. Supernatant of seminiferous tubules were diluted 1:1 in 20 mM Tris-HCl pH 7.6. Both were filtered through a 0.22 μm filter. 80 μl of the filtered supernatant was saved as input. The remaining supernatant was divided evenly among 4 tubes and incubated with 1.5 μg of the biotinylated RNA probes indicated in Figure 1. The lysate-probe mixture was incubated at 4 °C for 2 h with rotation. 15 μl Dynabeads MyOne Streptavidin C1 beads (Invitrogen) were washed 3× with lysis buffer, and added to each sample. The samples were then incubated at 4 °C for 1.5 h with rotation. The beads were isolated and washed four times with lysis buffer, and protein was eluted with 50 μl 2× Laemmli sample buffer (Bio-rad) at 95 °C for 5 min. Protein was subjected to FLAG or YTHDC2 blotting analysis.
EMSA (electrophoretic mobility shift assay/gel shift assay)
The RNA probe was synthesized with a sequence of 5′-ACCGGXCUGUUACCAACACCCACACCCC-FAM 3′ (X = m6A or A; FAM = 6-carboxyfluorescein). FLAG-YTHDC2 was diluted to concentration series of 24 μM, 12 μM, 6 μM, 3 μM, 1.5 μM, and 0.75 μM in binding buffer (10 mM HEPES, pH 8.0, 50 mM KCl, 1 mM EDTA, 0.05% Triton-X-100, 5% glycerol, 10 μg/ml salmon DNA, 1 mM DTT and 40 U/ml RNasein). 1 μl RNA probe (4 nM final concentration) and 1 μl protein (2 400 nM, 1 200 nM, 600 nM, 300 nM, 150 nM, 75 nM final concentration) were added to 8 μl binding buffer, and the solution was incubated on ice for 30 minutes. The entire 10 μl RNA-protein mixture was loaded onto a Novex 6% TBE gel and run at 4 °C for 90 minutes at 90V, and fluorescence was quantified. The Kd (dissociation constant) was calculated with nonlinear curve fitting (log(inhibitor) vs. response) using Prism 7 software, where X = Log(protein concentration) and Y is the ratio of [RNA–protein]/([free RNA]+[RNA-protein]).
Tandem affinity purification of YTHDF3 protein interactome
We followed the previously reported procedure26,54. Protein complex components were identified by mass spectrometry and validated by western blot.
Author Contributions
PH, YZ, HM, and YG contributed equally to this work. PH, HM, BS, and CH designed the experiments. PH, YZ, HM, XS, YL, MQ, YG, JW, and YC performed the experiments and analyzed the data. PH and ZL processed the high-throughput RNA sequencing data. HS and GL assisted with the experiments and provided valuable discussion. QD generated the RNA probes. PH, BS, and CH wrote the manuscript. All authors provided comments and suggestions to the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
Moore MJ . From birth to death: the complex lives of eukaryotic mRNAs. Science 2005; 309:1514–1518.
Desrosiers R, Friderici K, Rottman F . Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA 1974; 71:3971–3975.
Zhao BS, Roundtree IA, He C . Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 2016; 18:31–42.
Wei C, Moss B . Nucleotide Sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 1977; 16:1672–1676.
Wei C, Gershowitz A, Moss B . 5′-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry 1976; 15:397–401.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012; 485:201–206.
Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 2014; 10:93–95.
Wang X, Feng J, Xue Y, et al. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature 2016; 534:1–15.
Wang P, Doxtader KA, Nam Y . Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Molecular Cell 2016; 63:306–317
Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 2011; 7:885–887.
Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 2013; 49:18–29.
Ping X, Sun B, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 2014; 24:177–189.
Geula S, Moshitch-Moshkovitz S, Dominissini D, et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015; 347:1002–1006.
Fustin JM, Doi M, Yamaguchi Y, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 2013; 155:793–806.
Batista PJ, Molinie B, Wang J, et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014; 15:707–719.
Fischer J, Koch L, Emmerling C, et al. Inactivation of the Fto gene protects from obesity. Nature 2009; 458:894–898.
Li Z, Weng H, Su R, et al. FTO Plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine rna demethylase. Cancer Cell 2017; 31:127–141.
Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014; 505:117–120.
Wang X, Zhao BS, Roundtree IA, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015; 161:1388–1399.
Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res 2017; 27:315–328.
Xu C, Wang X, Liu K, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 2014; 10:927–929.
Xiao W, Adhikari S, Dahal U, et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell 2016; 61:507–519.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T . N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015; 518:560–564.
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB . A mRNA methylation directs translational control of heat shock response. Nature 2015; 526:591–594.
Zhao BS, Wang X, Beadell A V, et al. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 2017; 542:475–478.
Wang X, Zhao BS, Roundtree IA, et al. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015; 161:1388–1399.
Li A, Chen YS, Ping XL, et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res 2017; 27:444–447.
Du H, Zhao Y, He J, et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun 2016; 7:12626.
Morohashi K, Sahara H, Watashi K, et al. Cyclosporin A associated helicase-like protein facilitates the association of hepatitis C virus RNA polymerase with its cellular cyclophilin B. PLoS One 2011; 6:e18285.
Abby E, Tourpin S, Ribeiro J, et al. Implementation of meiosis prophase I programme requires a conserved retinoid-independent stabilizer of meiotic transcripts. Nat Commun 2016; 7:10324.
Soh YQS, Mikedis MM, Kojima M, Godfrey AK, Rooij G De, Page DC . Meioc maintains an extended meiotic prophase I in mice. PLoS Genet 2017; 13:e1006704.
Kerr JB, Loveland KL, O'Bryan MK, De Kretzker DM . Cytology of the testis and intrinsic control mechanisms. In: Knobil and Neill's Physiology of Reproduction. 2006, 25:827–947.
Zhu T, Roundtree IA, Wang P, et al. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res 2014; 24:1493–1496.
Wang X, He C . Dynamic RNA modifications in posttranscriptional regulation. Mol Cell 2014; 56:5–12.
Popp MWL, Maquat LE . Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Rev Genet 2013; 47:139–165.
Brogna S, Wen J . Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol 2009; 16:107–113.
Gregersen LH, Schueler M, Munschauer M, et al. MOV10 Is a 5′ to 3′ RNA Helicase Contributing to UPF1 mRNA target degradation by translocation along 3′ UTRs. Mol Cell 2014; 54:573–585.
Nagarajan VK, Jones CI, Newbury SF, Green PJ . XRN 5′→3′ exoribonucleases: Structure, mechanisms and functions. Biochim Biophys Acta - Gene Regul Mech 2013; 1829:590–603.
Li F, Zhao D, Wu J, Shi Y . Structure of the YTH domain of human YTHDF2 in complex with an m6A mononucleotide reveals an aromatic cage for m6A recognition. Cell Res 2014; 24:1490–1492.
Roundtree IA, Evans ME, Pan T, Chuan He . Dynamic RNA modifications in gene expression regulation. Cell 2017; 169:1187.
Monesi V . Ribonucleic acid synthesis during mitosis and meiosis in the mouse testis. J Cell Biol 1964; 22:521–532.
Page J, Fuente R de la, Manterola M, et al. Inactivation or non-reactivation: what accounts better for the silence of sex chromsoomes during mammalian male meiosis? Chromosoma 2012; 121:307–326.
Fanale D, Iovanna JL, Calvo EL, et al. Germline copy number variation in the YTHDC2 gene: does it have a role in finding a novel potential molecular target involved in pancreatic adenocarcinoma susceptibility? Expert Opin Ther Targets 2014; 18:841–850.
Tanabe A, Tanikawa K, Tsunetomi M, et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett 2016; 376:34–42.
Shen B, Zhang W, Zhang J, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 2014; 11:399–402.
Hafner M, Landthaler M, Burger L, et al. PAR-CliP — A method to identify transcriptome-wide the binding sites of rna binding proteins. J Vis Exp 2010 Jul 2. pii:2034. doi: 10.3791/2034
Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M . A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods 2011; 8:559–564.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL . TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14:R36.
Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012; 7:562–578.
Robinson MD, McCarthy DJ, Smyth GK . edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009; 26:139–140.
Corcoran DL, Georgiev S, Mukherjee N, et al. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol 2011; 12:R79.
Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol Cell 2010; 38:576–589.
Bazzini AA, Lee MT, Giraldez AJ . Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 2012; 336:233–237.
Shi Y, Sawada J, Sui G, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003; 422:735–738.
Acknowledgements
We thank Prof Tao Pan (The University of Chicago) for kindly providing the polysome profiling instrument. We thank Peter Chen and Phil Xie for technical assistance. We also thank the Institutes of Biomedical Sciences at Fudan University for the protein mass spectrometry experiments. This work was supported by the National Science Fund for Excellent Young Scholars (31622039 to BS), the National Key Research and Development Program of China (2016YFC1000600 to BS, 2016YFA0500902 to ML, 2017YFA0103803 to JS), the Science Foundation for Distinguished Young Scholars of Jiangsu Province (BK20160045 to BS), and National Institutes of Health grants HG008935 and GM113194 (to CH). CH is an investigator of the Howard Hughes Medical Institute (HHMI). HM is supported by the Postdoctoral International Exchange Program of the China Postdoctoral Council (CPC). PH is supported by the University of Chicago Medical Scientist Training Program (MSTP; NIH MSTP training grant T32GM007281).
Author information
Authors and Affiliations
Corresponding authors
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Domains of YTHDC2 (https://www.ncbi.nlm.nih.gov/gene/64848). (PDF 39 kb)
Supplementary information, Figure S2
(Supplement to Figure 1) YTHDC2 binds preferentially to m6A-marked RNA transcripts. (PDF 148 kb)
Supplementary information, Figure S3
Characterization of Ythdc2−/− mouse. (PDF 105 kb)
Supplementary information, Figure S4
Female Ythdc2−/− mice demonstrate defects in the ovaries. (PDF 152 kb)
Supplementary information, Figure S5
Expression of Ythdc2 protein in wild type testes over time. (PDF 110 kb)
Supplementary information, Figure S6
Meiotic defects in Ythdc2-deficient meiocytes. (PDF 195 kb)
Supplementary information, Figure S7
Comparative histological analysis of testes of young Ythdc2+/+ and Ythdc2−/− mice. (PDF 436 kb)
Supplementary information, Figure S8
Ythdc2-deficient mice show normal spermatogonia. (PDF 199 kb)
Supplementary information, Figure S9
Ythdc2-deficient mice show normal sertoli cells. (PDF 445 kb)
Supplementary information, Figure S10
(Supplement to Figure 4) YTHDC2 affects the translation efficiency and stability of its targets. (PDF 141 kb)
Supplementary information, Figure S11
m6A-seq of 8.5 d.p.p. Ythdc2+/+ and Ythdc2−/− testes. (PDF 107 kb)
Supplementary information, Figure S12
Effect of YTHDC2 on mRNA abundance and translation efficiency may be m6A dependent. (PDF 108 kb)
Supplementary information, Figure S13
Confirmation of components of protein complex containing YTHDC2 obtained from tandem affinity purification followed by western blot. (PDF 111 kb)
Supplementary information, Table S1
Target transcripts of Ythdc2 in adult mouse testes, determined by CLIP-seq (XLSX 1895 kb)
Supplementary information, Table S2
Target transcripts of Ythdc2 in 16 d.p.p. mouse testes, determined by RIP-seq (XLSX 1406 kb)
Supplementary information, Table S3
Top 40 GO terms of Ythdc2 RIP-seq targets in mouse testes (XLSX 48 kb)
Supplementary information, Table S4
m6A-seq of control and Ythdc2−/− mice (XLSX 8512 kb)
Supplementary information, Table S5
Targets of YTHDC2 in HeLa cells, determined by PAR-CLIP (XLSX 120 kb)
Supplementary information, Table S6
Binding partner proteins of YTHDC2 in HeLa cells, determined by Tandem Affinity Purification followed by mass spectrometry (XLSX 1107 kb)
Rights and permissions
About this article

Cite this article
Hsu, P., Zhu, Y., Ma, H. et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res 27, 1115–1127 (2017). https://doi.org/10.1038/cr.2017.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2017.99
Keywords
This article is cited by
-
The m6A methyltransferase METTL3 drives thyroid cancer progression and lymph node metastasis by targeting LINC00894
Cancer Cell International (2024)
-
Crosstalk between m6A modification and autophagy in cancer
Cell & Bioscience (2024)
-
RNA m6A methylation and regulatory proteins in pulmonary arterial hypertension
Hypertension Research (2024)
-
Emerging role of RNA modification and long noncoding RNA interaction in cancer
Cancer Gene Therapy (2024)
-
METTL16 promotes liver cancer stem cell self-renewal via controlling ribosome biogenesis and mRNA translation
Journal of Hematology & Oncology (2024)
