
Abstract
Immunotherapy using dendritic cell (DC)-based vaccination is an approved approach for harnessing the potential of a patient's own immune system to eliminate tumor cells in metastatic hormone-refractory cancer. Overall, although many DC vaccines have been tested in the clinic and proven to be immunogenic, and in some cases associated with clinical outcome, there remains no consensus on how to manufacture DC vaccines. In this review we will discuss what has been learned thus far about human DC biology from clinical studies, and how current approaches to apply DC vaccines in the clinic could be improved to enhance anti-tumor immunity.
Similar content being viewed by others

Introduction
Originally described by Ralph Steinman and Zanvil Cohn in 1973, dendritic cells (DCs) have a critical role in mediating innate immune response and inducing adaptive immune response. Since then, DCs, often referred to as “Nature's adjuvant” have been recognized as the most potent antigen-presenting cells (APCs), capable of activating both naive and memory immune responses. DCs have a superior capacity for acquiring and processing antigens for presentation to T cells and express high levels of costimulatory or coinhibitory molecules that determine immune activation or anergy1. Almost 40 years later, the importance of DCs was recognized when the Nobel Prize for Medicine or Physiology was awarded to Ralph Steinman in 2011 for his discovery of these critical innate immune cells.
DC immunology
DC ontogeny and characteristics
DCs are a sparsely distributed, heterogenous group of specialized APCs that originate in the bone marrow from CD34+ stem cells. Data obtained from in vitro human and mouse studies and in vivo animal studies, and limited in vivo human studies, provide support for our understanding of DC differentiation. The most recently accepted model proposes that monocytes and DCs originate from the same common progenitors called the monocyte and DC progenitors (MDPs). These two cell types diverge in the bone marrow when the MDPs give rise to monocytes and the committed DC progenitors (CDPs). The CDPs give rise to pre-DCs that migrate out of the bone marrow to produce the two major sub-populations of DCs2. Stromal cell culture systems comprising hematopoietic stem cells cultured with mouse bone marrow stromal cells and stem cell factor (SCF), granulocyte-macrophage colony-stimulating factor (GM-CSF) and Flt3L have identified a definitive DC precursor population that gives rise to DC subsets found in the blood3,4,5. Although this model has largely been studied in mouse, studies in humans have confirmed these findings as well. Human granulocyte monocyte DC precursors sequentially develop into monocyte DC precursors, which subsequently give rise to common DC progenitors that are restricted to produce the three major subsets of DCs: CD1c+ DCs and CD141+ DCs (which are together considered conventional DCs or cDCs), and plasmacytoid DCs (pDCs). A migratory phenotype (hpre-cDC) has also been identified in human cord blood, bone marrow, blood, and peripheral lymphoid organs, which sustains the cDC pools through differentiation. Furthermore, Flt3L given systemically to humans has been shown to increase the pre-cDC pool4,6.
Phenotypically, human DCs lack lineage (Lin) markers (CD3, CD19, CD14, CD20, CD56, and glycophorin A), but constitutively express major histocompatibility complex (MHC) class II7,8. cDCs and pDCs represent the two major types of DCs in the blood and lymphoid tissue. cDCs are MHC-II+CD11c+ and are further subdivided into CD1c+ and CD141+ subsets. Three DC populations can also be distinguished by molecular signatures: CD1c+ DCs express IRF4, Notch2, Rbpj, and Klf4; CD141+ DCs express IRF8, batf3, Bc16, and Flt3; and pDCs express IRF8, Bcl11a, Spi-B, E2-2, Runx1, and IL-3RA9,10. CD1c+ DCs are the predominant subset, whereas the CD141+ DCs are a minor population, at least in the blood. CD141+ DCs are believed to be the human equivalent of mouse CD8+ DCs, which have the ability to cross present cell-associated antigens to CD8+ T cells. CD1c+ DCs express toll-like receptor (TLR) 1-8 and when stimulated, can secrete interleukin-12 (IL-12), tumor necrosis factor- (TNFα), IL-8, and IL-10. CD141+ DCs express TLR3 and 8 and secrete high levels of type I interferon upon stimulation with synthetic dsRNA poly-ICLC11. This DC subset is also known for producing high levels of IL-29 or type III interferon in response to TLR3 activation12. CD141+ DCs exclusively express Clec9A (DC NK lectin group receptor-1), an endocytic receptor that renders cells more capable of taking up and presenting antigens derived from necrotic cells13,14, and also XCR1. Although both CD1c+ and CD141+ DCs can cross-present antigens to CD4+ and CD8+ T cells, CD141+ DCs may be more efficient, although this may depend on the type and form of antigens and how they are accessed15.
pDCs are defined as Lin−MHC-II+CD303+CD304+ cells. They are major effector cells in immune responses due to their ability to produce up to 1000-fold more type I interferons (IFN-α/β) in response to viral infections than other cell types16. pDCs can also acquire antigens through, e.g., receptor-mediated endocytosis or uptake of dying cells, although not as efficiently as cDCs17. Moreover, pDCs can rapidly cross-present antigens, including components of influenza virus, to CD8+ T cells18, after processing in endosomal-type vesicles. pDCs express high levels of TLR7 and TLR9, which enable them to recognize viral and self nucleic acids19. Although cDCs can be found in almost every peripheral tissue as well as in lymphoid organs, pDCs have a more restricted distribution. They are found mostly in the T cell area of lymphoid organs such as the lymph nodes, tonsils, spleen, thymus, BM, and Peyer's patches, the blood, and some peripheral tissues including the liver and nasal mucosa. pDCs can activate melanoma-specific CD8+ T cell responses20, but they may also inhibit anti-tumor immune responses. pDCs with diminished capacity to produce IFNα have been found in many tumors20,21 and the accumulation of pDCs expressing indoleamine 2,3-dioxygenase (IDO), which is responsible for the degradation of tryptophan (an amino acid essential for T cell proliferation) and implicated in the generation of regulatory T cells, has been observed in tumor-draining lymph nodes22,23,24,25. Therefore, modulating pDCs may be an important step in generating effective anti-tumor immunity.
There are five major types of DCs found in the skin: Langerhans cells (LCs), CD14+ DCs, CD1a+ DCs, CD1c+ DCs, and CD141+ DCs. LCs are the major APCs found in the epidermis of the skin. They are positive for CD45, MHC-II, epithelial cell adhesion molecule, Langerin, and contain large granules called Birbeck granules. LCs acquire antigens in the epidermis and transport them to the regional lymph nodes where they present the antigens to T cells to initiate immune responses. During inflammation, pDCs and inflammatory DCs are also recruited to the skin. Inflammatory DCs are CD11c++MHC++CD40+CD80+CD86+ and are differentiated from the circulating monocytes in the blood only during inflammation/infection to infiltrate the site of inflammation/infection26. Dermal DCs are thought to comprise a heterogeneous mixture of CD14+ cells, the less characterized CD1a+ DCs, CD1c+ DCs as well as CD141+ DCs, but most recently, dermal CD14+ cells have been shown to be tissue-resident monocyte-derived macrophages27.
Lymphoid tissues have CD1c+ and CD141+ DCs; some of the secondary lymphoid tissues have specialized resident DC subsets. For example, CD103+ DCs have been found in the mesenteric lymph nodes (MLNs)28 where they have a crucial role in the induction of tolerance to commensal bacteria and food antigens. In addition, these cells may be similar to the CD103+ DCs found in murine MLNs that have an enhanced ability to metabolize vitamin A to generate retinoic acid, which drives the differentiation of gut-homing regulatory T cells29.
The concept of maturation
In their resting state, DCs are considered to be immature but primed to acquire antigens in situ through a variety of receptors and mechanisms. Upon exposure to “activating stimuli,” DCs undergo a complicated series of phenotypic and functional changes referred here to as “activation” and “maturation”, respectively30. The process of DC activation is an intricate and tightly controlled differentiation process that is closely associated with antigen acquisition. Activation is characterized by the upregulation of chemokine receptors (e.g., CCR7), adhesion molecules, costimulatory molecules (CD54, CD80, and CD86), immunoproteosomes and MHC class I and II molecules, all essential for migration of the cells to the lymphoid tissues and optimal activation of the immune responses. Cytokines produced during this process influence the immune responses generated by subtypes of CD4+ T cells such as T helper 1 (Th1) cells (by IL-12), Th2 cells, and regulatory T cells. The role of DCs in priming Th9 or Th22 cells is still under investigation. In addition, DCs qualitatively and quantitatively orchestrate the type of immune responses that develop by activating naive and memory B cells31, natural killer (NK) cells (via the action of IL-12, IL-15, and type I IFNs)32, and NKT cells (through antigen presentation on the CD1 molecule)33. DC maturation is characterized by the reduction in phagocytic capacity, enhancement in antigen processing and presentation, improved migration to lymphoid tissues, and increase in the capacity to stimulate B and T cells. Maturation is induced by microbial products that trigger the activation of pattern recognition receptors such as TLRs34, or activation of introcellular sensors such as RIG-I35 or inflammasome36, or by the action of inflammatory molecules (TNFα, IL-1, IL-6, and IFNα) produced by the cells of the immune system or by damaged tissues37. Dead cells can also release factors that activate DCs (e.g., heat-shock proteins, RNA, and DNA)38.
Antigen acquisition and presentation
DCs take up antigens through phagocytosis, micro- or macro-pinocytosis, and endocytosis using Fc receptors (Fcγ receptor type I or CD64 and Fcγ receptor type II or CD32), integrins (αvβ3 or αvβ5), C-type lectin receptors (CLRs, including mannose receptor and DEC205), apoptotic cell receptors, and scavenger receptors. Antigens are either processed into peptides via the endogenous pathway for presentation on MHC class I molecules to CD8+ T cells, or processed via the exogenous pathway for presentation on MHC class II molecules to the CD4+ T cells. Alternatively, DCs can also process antigens via two cross presentation pathways39: the cytosolic pathway and the vacuolar pathway40. In the cytosolic pathway, antigens are transferred to the cytoplasm, where they are processed in the proteasome before being loaded onto the newly formed MHC class I molecules; and this process may involve the participation of the ER machinery. The vacuolar pathway is less defined but is thought to occur in the endocytic compartments because the pathway is resistant to proteasome inhibitors, but sensitive to inhibitors of lysosomal proteolysis, and depends on cathepsin S. Recently it has been found that TLR signaling influences the maturation of phagosomes and induces the accumulation of MHC class I molecules in the phagosomes for optimal cross presentation41. Although the underlying mechanisms of cross presentation are still being clarified, the ability of DCs to utilize this process to activate CD8+ T cells is in many cases well established42,43,44. DCs can present antigens in various forms besides conventional peptides. For example, phosphopeptides and citrullinated antigens have been shown to be presented on HLA molecules and recognized by T cells45,46. These antigens, which can be expressed on cancer cells, are novel targets of immunotherapy (i.e., NCT01846143). DCs can also process lipid antigens and present them on the CD1d molecule to activate NKT cells. Furthermore, they can recognize and acquire antigens containing carbohydrate structures via CLRs including MMR (macrophage mannose receptor), DEC205, and DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin). CLRs can function as endocytic receptors to internalize antigens for antigen processing and presentation, but some of the CLRs, e.g., MICL (myeloid C-type lectin-like receptor) and DICR (dendritic cell immunoreceptor), may be inhibitory47.
Induction of immunity
Upon maturation, DCs migrate to the secondary lymphoid tissues such as the lymph nodes (capturing antigen from the skin and solid organs), the spleen (capturing antigen from the blood), or the Peyer's patches (capturing antigen from the gut lumen) where they come in contact with T and B cells (reviewed in Benvenuti48). cDCs migrate through the afferent lymph from non-lymphoid tissues to the T cell-rich areas of lymph nodes. pDCs, which also migrate into T cell areas of secondary lymphoid tissues, do so through high endothelial venules of lymph nodes and the marginal zone of the spleen, likely using CCR7 and CD62-L49. Both activated blood cDCs and pDCs can migrate in response to lymph node homing chemokines (CCL19 and CCL21) through the expression of CCR7. CCR7 expression and CD141+ DC infiltration both have been shown to correlate with intratumoral T cell infiltration and better clinical outcome in melanoma50. Most recently, it was shown in the mouse that strategically localized DCs residing in the lymph nodes can induce T cell responses much sooner and independently of migratory DC51. These DCs reside in the lymphatic sinus endothelium, where they can “scan” lymph to capture soluble lymph-borne antigens to rapidly initiate immune responses without delay. It is conceivable certain antigen adjuvant platforms can be developed to access these DC subsets.
Through their TCRs, T cells specifically recognize antigens bound on MHC molecules on the surface of DCs. Peptides bound on MHC class I molecules are recognized by CD8+ T cells, whereas peptides bound on MHC class II molecules are recognized by CD4+ T cells. Activation of T cells is dependent on the intensity and duration of DC-T cell interactions, mediated through the immunological synapse (IS). IS forms as the result of cytoskeletal reorganization within the T cell leading to the dynamic clustering of T cell surface receptors and signaling molecules into supramolecular activation clusters, which provide an optimal environment for signaling molecules downstream of the TCR. The upregulation of costimulatory molecules (CD40, CD86) and MHC molecules during DC maturation is critical for making stable and long-lasting contacts with T cells through the IS, which is required for T cell expansion and differentiation into memory and effector T cells.
DCs can also activate naive and memory B cells31, primarily through their ability to stimulate CD4+ T cells (such as T follicular helper cells), which induce B-cell growth and antibody production. Antibody class switching is influenced through the production of factors that activate and induce B-cell proliferation (B-Lys and APRIL)52. The follicular DCs, which are present in the germinal centers of lymph nodes, support maintenance of B-cell memory via formation of multiple antigen-antibody complexes and continuous stimulation of B cells. DCs also activate NK cells via IL-12, IL-15, and type I IFNs32. IL-12 produced by DCs potentiates the cytolytic activity of NK cells. Conversely, the interaction with NK cells can induce further DC maturation. The production of XCL1 and XCL2 by NK cells, as well as CD8+ T cells, can recruit and initiate specific responses in XCR1-expressing DCs53,54. Finally, DCs activate NKT cells through the expression of invariant CD1 molecules and presentation of glycolipid molecules33. Therefore, DCs have important roles by mobilizing multiple arms of both the innate and adaptive immune responses.
In addition to inducing immunity, DCs mediate peripheral tolerance and prevent autoimmunity55,56. Immature DCs do not activate T cells due to their low expression levels of MHC and costimulatory molecules. DCs can also express IDO in response to certain stimuli, consequently driving T cells to undergo cell cycle arrest or apoptosis possibly due to a counter-regulatory effect. Moreover, the metabolites produced by tryptophan catabolism can exert direct cytotoxic effects on T cells57,58. Through production of IDO, DCs can induce the differentiation of regulatory T cells59,60, which can infiltrate several types of tumors61,62; they can also exert their effects through TGFβ, IL-10, and CTLA-4, among other mechanisms, to inhibit proliferating T cells56,63. Migratory DCs have a specialized role in mediating peripheral tolerance by inducing the generation of regulatory T cells64. As part of immune surveillance, immature DCs acquire self-antigens and in the absence of maturation stimuli, these DCs induce tolerance to these self-antigens. DCs can also mediate tolerance to self-antigens through the uptake of apoptotic cells via receptors including LOX-1, CD36, integrins (αvβ3 and αvβ5), and complement receptors (CR3 and CR4). Binding of apoptotic cells to CR3 on DCs leads to impaired maturation of the DCs in response to lipopolysaccharide (LPS) stimulation and an impaired priming and activation of the memory T cell responses65. Furthermore, binding of apoptotic microparticles to CD44 can inhibit human DCs66. Altogether, these properties of DCs make them ideal tools for inducing tolerance in the settings of transplantation and autoimmunity.
DC immunotherapy
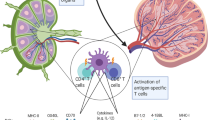
The role of the immune system in eliminating tumors has been established in several studies. First, incidences of spontaneous regression of metastatic melanoma have been proposed to be caused by the body's immunologic responses67. Second, the existence of immunogenic tumor-associated or tumor-specific antigens68 that are recognized by B and T cells has been reported. Third, knockout mice lacking components of the IFN signaling pathway, perforin, or recombination activating gene 1/2 have significantly higher risk of developing tumors69. Finally, adoptive transfer of tumor-infiltrating lymphocytes as well as engineered tumor antigen-specific T cells has led to the regression of tumors in melanoma patients68. Altogether, these studies prove a protective role of the immune system in eliminating tumors. The complex relationship between the immune system and tumors is now described as consisting of three phases of cancer immunoediting: elimination, equilibrium, and escape70. The first phase involves induction of the anti-tumor immune responses whereby both the innate and adaptive arms of the immune systems are activated to destroy the tumors. Equilibrium is the second phase when the immune system exerts a selection pressure on the tumor. Tumor cells that have become resistant to this selection pressure enter the third phase, escape. It is during this third phase that tumors grow and expand beyond control, and become malignant. Given the central role of DCs in initiating immune responses and surveillance, investigators have theorized that DCs would serve as an ideal tool for boosting endogenous anti-tumor responses that can lead to the effective eradication of tumors. As a proof of principle, we conducted a controlled study in healthy subjects and showed that, a single injection of DC vaccine comprising antigen-pulsed DCs was sufficient to induce antigen-specific immune responses in vivo71, whereas soluble antigen alone failed to induce immunity. This result indicates that DCs can be successfully loaded with antigens, and following injection, traffic to the lymphoid tissues to activate antigen-specific T cells. In support of this notion, early clinical trials using ex vivo-generated DCs pulsed with tumor antigens demonstrated that immune responses could indeed be induced thus setting the stage to further manipulate DCs to enhance immune responses in vivo (reviewed in Constantino et al.72).
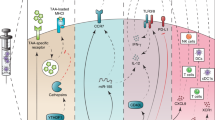
Despite many studies, however, the approach involving DCs remains to be optimized. Though the most commonly adopted method in clinical trials uses ex vivo-generated monocyte-derived DCs, it is currently unknown which primary DC subset is the best subset to use as an adjuvant or a target for vaccination. This is important to point out because monocyte-derived DCs are not comparable to the steady-state DC subsets present in the body; instead, they resemble more the cells that appear during an inflammatory response in vivo. With the plethora of maturation stimuli available, more studies are required to compare the performance of these cells. In addition, the number of DCs to inject (0.3 × 106 cells to 200 × 106 cells per injection), the vaccination schedule (once every 2 weeks vs 3-4 doses or even up to 10 injections given every 3-4 weeks) and the route of injection (subcutaneous, intradermal, intranodal, intravenous, or even intratumoral) to induce the most potent and long-lasting immune response have also not been determined. Below, we discuss current and future approaches to apply DC-based and targeted vaccines in the clinic (Figure 1).

Current and future approaches for dendritic cell (DC)-based vaccination. The current approaches for DC-based vaccination are mainly based on antigen loading on in vitro-generated DCs from monocytes or CD34+ cells, activating them with different TLR ligands, cytokine combinations, and injecting them back to the patients. The in vivo targeting approaches comprise administering specific cytokines (e.g., Flt3L, GM-CSF) and targeting the DCs with antibodies to C-type lectin receptors or agonistic antibodies (e.g., anti-CD40) that are conjugated with antigen of interest. Future approach may target DC subsets based on their specifically expressed C-type lectin receptors or chemokine receptors. Another potential approach is the generation of genetically engineered DCs from induced pluripotent stem cells and use of neoantigen-loaded DCs for inducing better clinical outcome.
Current approaches
Ex vivo approaches to DC vaccination
Current methods for generating DCs used in the clinical trials include differentiation from monocyte precursors or CD34+ hematopoietic precursors, in vivo expansion of circulating DCs, and most recently, isolation and enrichment of circulating blood DC subsets. Although no direct comparison of all these methods of DC generation has been made in clinical trials, analyses of the transcriptional profiles of the DCs generated ex vivo using various methods have revealed fundamental differences from the in vivo DC subsets73. Monocyte-derived DCs cluster with CD34+-derived DCs73 and are more closely related to macrophages74. Furthermore, immune system-related transcripts expressed by the ex vivo-generated DCs differ from those expressed by the in vivo DC subsets, suggestive of functional differences between them. Despite these differences, DCs derived using the most commonly used methods (monocyte-derived DCs and CD34-derived DCs) have been shown to stimulate antigen-specific T cell responses in both preclinical and clinical studies. The different methods of DC differentiation and their advantages are discussed below.
Monocyte-derived DCs Differentiation of DCs from monocytes in peripheral blood mononuclear cells (PBMCs) obtained from whole blood or leukapheresis is by far the most commonly used approach. DCs derived from this process are called monocyte-derived DCs (MDDCs). CD14+ monocytes are selected from PBMCs by plastic adherence or positive selection using immunomagnetic beads75,76,77,78,79 and are induced to differentiate into immature CD14−CD83− DCs in culture for several days in the presence of IL-4 and GM-CSF. The immature DCs are stimulated to mature by exposure to a maturation stimulus and simultaneously loaded with tumor antigen for an additional 1-2 days. The differentiated, matured, and antigen-loaded DCs are then harvested and cryopreserved in aliquots and are thawed at each scheduled vaccination date. This approach is both time-consuming and costly to undertake but is used in many treatment centers. The method has been adapted by some into a programmable closed culture system. Although autologous DC vaccination is the preferred approach, the use of allogeneic DCs has been explored for the treatment of renal cell carcinoma and demonstrated to be immunogenic80. This approach was also tested in acute myeloid leukemia (NCT01373515). Alternatively, umbilical cord blood-derived CD11c+ DCs can be a possible source of allogeneic DCs81. Allogeneic DCs may be an attractive source as they could be prepared from an unrelated healthy donor whose immune system has not been rendered dysfunctional as in cancer patients82. Finally, although DC production would still be expensive and time-consuming, the allogeneic approach would enable the off-the-shelf treatment that would be more viable and attractive for pharmaceutical companies. Partially HLA-matched allogeneic DCs may yet be a useful source for vaccination as (a) HLA-mismatched DCs can induce a strong immune response and (b) the use of different DC donors will still boost the immune response to the antigen of interest83,84. However, it is possible that this approach may not be feasible over a prolonged period due to antigen competition.
CD34+ precursors CD34+ precursors can be mobilized from the bone marrow prior to leukapheresis by the pre-treatment of granulocyte-colony stimulating factor. The harvested cells are further expanded in vitro for up to 12 days in the presence of GM-CSF, Flt3L, TNFα, TGFβ, and SCF, producing a mixture of MDDCs that are phenotypically similar to epidermal LCs, and a large proportion of myeloid cells at different stages of differentiation77. Compared with MDDCs and dermal-interstitial DCs, mature LCs stimulate more effective CD8+ T cell responses, a process requiring IL-15 produced by the LCs85. In a clinical trial aimed to compare melanoma peptide-loaded LCs and MDDCs, LCs were more potent stimulators of antigen-specific T cell responses than MDDCs, but MDDCs supplemented with IL-15 stimulated significantly more antigen-specific effector memory T cells86. Perhaps retroviral transduction of LCs, which enables sustained antigen expression, may enhance their efficacy in vivo87; however, this approach has been less enthusiastically accepted. Most recently, it was shown that CD34+ cells (derived from cord blood, bone marrow, or peripheral blood) cultured with MS5 stromal cells in the presence of Flt3L, SCF, and GM-CSF can develop into all three major DC subsets88. This new method has allowed a clear delineation of how the three subsets develop from their progenitors. The authors showed that granulocyte-monocyte-DC progenitors develop into monocyte-DC progenitors, which give rise to monocytes and common DC progenitors. These common DC progenitors differentiate into the three DC subsets. In addition, the authors identified an immediate DC precursor population circulating in the blood and showed these cells differentiate from the common DC progenitor cells in response to Flt3L administration4. Adaptation of this method has allowed investigators to obtain higher numbers of each DC subset; e.g., yields of pDCs and CD141+ DCs can be increased by 1.5- and 9-fold, respectively. The CD1c+ DCs in this culture system are not very well characterized and they may be more close to the MDDCs than the CD1c+ DCs circulating in the blood. More importantly, these investigations have made it possible to adjust the methodology for good manufacturing practice (GMP) production, thus allowing the potential comparison of all three DC subsets in the clinic for their relative immunogenicity. Significantly, this approach is also amenable to genetically manipulating DC subsets to improve their antigen presenting function.
Blood DCs The first FDA-approved cell-based therapy for the treatment of hormone-refractory prostate cancer, Provenge (Sipuleucel-T), is a CD54-enriched peripheral blood vaccine89. Provenge consists of a mixture of DCs, B cells, monocytes, and NK cells that have been cultured ex vivo with a recombinant fusion protein containing prostatic acid phosphatase (PAP) and GM-CSF, before the mixture is infused intravenously back into patients within 48 h of the leukapheresis collection90. Patients receive three freshly processed products; products two and three contain primed T cells in addition to activated APCs, which include DCs. phase I and II clinical trials have demonstrated that Provenge is safe and patients develop immune responses to the fusion protein91. Reactivity to the native protein also develops but it is less strong. The increase in CD54 expression upon culture with the fusion protein correlated with APC activation and was used as a surrogate marker for potency of the therapy92. The phase III IMPACT trial showed a 4.1-month improvement in median overall survival at 36 months and the survival rate was 31.7% in treated patients vs 23.0% in placebo patients89. In addition to inducing circulating humoral (with antigen spreading as evidenced by responses against PSA93) and cellular immune responses94, Provenge has also been shown to induce T cell infiltration in the tumor microenvironment (TME)95. Provenge is currently being tested in combination with anti-PD1 (NCT01420965), anti-CTLA-4 (NCT01832870, NCT01804465), and IL-7 (NCT01881867) to see whether any of these combinations would further enhance its efficacy.
Administration of growth factor Flt3L induces the in vivo expansion of circulating DCs96. Daily administration of Flt3L for 10 days led to a 130-fold increase in CD1c+ DC number, 48-fold increase in CD141+ DC number, and 6- to 16-fold increase in pDC number from the mean baseline count. CD34+ cells also increased 23-fold from the mean baseline count in healthy human volunteers97. Flt3L administration may also increase the number of precursors to CD1c+ DCs and CD141+ DCs (the pre-cDCs)4, and the number of precursors for pDCs, when combined with thrombopoietin, at least in vitro98. Clinical trials using Flt3L are currently ongoing in melanoma in combination with DEC205/NY-ESO-1 fusion protein and poly-ICLC (NCT02129075) with preliminary results confirming the immunogenicity and safety of the combined product. The treatment is also being given intratumorally in low grade B-cell lymphoma in combination with poly-ICLC and radiation (NCT01976585), where the response rate is > 30% (J Brody, personal communication). Interestingly, intratumoral Flt3L not only mobilizes DCs into the TME but also achieves a systemic mobilization of DCs. Therefore, by increasing the DC numbers and localization to tumors and also providing a maturation stimulus, Flt3L may substantially improve anti-tumor T cell priming. The contribution of individual DC subsets in acquiring and presenting tumor-derived antigens in these settings remains to be determined. Nevertheless, such studies are enabling the analysis of DCs at the individual cell level by analyzing their phenotype and transcriptome through strategically obtained tumor biopsies. Flt3L has also been delivered as a single agent to patients with acute myeloid leukemia (NCT00006223) and colorectal cancer (NCT00003431); and has been given in combination with HLA-A2-restricted TAAs emulsified in Montanide ISA-51 adjuvant to patients with melanoma or renal cell cancer (NCT00019396).
CD1c+ DCs and pDCs Due to their paucity, blood DC subsets have not been rigorously compared and tested in clinical trials. In one study, however, autologous-activated pDCs loaded with tumor-associated peptides were evaluated in patients with melanoma. One group successfully isolated sufficient numbers of pDCs to test their immunogenicity in vivo. pDCs were isolated by leukapheresis using a CliniMACS isolation system with average purity of 75% and yields of 13 × 106 to 33 × 106 cells. Isolated pDCs were then cultured in the presence of IL-3 and loaded with HLA-A2-restricted gp100 and tyrosinase peptides. The study showed that despite the limited cell numbers, vaccination with autologous pDCs loaded with tumor-associated peptides was safe and induced antigen-specific CD4+ and CD8+ T cell responses in the patients99. This study was the first to show the feasibility of isolating primary DC subsets and using them for immunotherapy. A recent study by the same group demonstrated the successful isolation of autologous primary CD1c+ DCs from leukapheresis product using the CliniMACS isolation system from metastatic melanoma patients enrolled in the study100. Positively selected CD1c+ DCs had an average purity of 93% and yields of 27-96 × 106 cells. The isolated cells were cultured overnight in the presence of GM-CSF and loaded with HLA-A2-restricted gp100 and tyrosinase peptides. Patients received 3-10 × 106 DCs given intranodally every 2 weeks. The study demonstrated the feasibility and safety of isolation of primary CD1c+ DCs for vaccination. The vaccine induced peptide-specific CD8+ T cell responses in a small subset of patients. It remains to be seen whether these cells will elicit a superior anti-tumor response than pDCs or even CD141+ DCs, which also produce type I and III IFNs but have the capacity to migrate from tumor tissues to draining nodes and to cross-present antigens to T cells.
Although most of clinical studies have cited the induction of immune responses by DC vaccine as a measure of their success, most T cell responses are evaluated after long in vitro stimulation cultures. And those who monitored the direct ex vivo response reported weak T cell reaction primarily involving the CD4+ cells, but not the CD8+ cells. Presumably the nature of antigen (tumor antigen to which patients may have already developed tolerance) and possibly the quality and amount of cells, as well as the lack of efficient migration to the lymph nodes may compromise the possibility of eliciting a potent response as was observed in healthy patients receiving DC vaccines when the antigens were technically neoantigens, e.g., keyhole limpet hemocyanin (KLH) or antigens derived from pathogens71. In an unpublished study from our group, our DC vaccine induced very weak ex vivo CD8+ T cell responses to HLA-A2-restricted melanoma peptides (Melan-A, gp100, tyrosinase, MAGE-A3, and NY-ESO-1) but very strong CD8+ T cell responses to KLH, consistent with this hypothesis. Indeed, DCs seem to be quite capable of eliciting CD8+ T cell responses to tumor-derived neoantigens, suggesting that the choice of antigen dictates the effectiveness in priming T cell responses101. Studies with Provenge using a fusion protein of PAP-GM-CSF also support this assertion.
Maturation of DCs
Mature DCs have enhanced expression of costimulatory molecules, produce cytokines and chemokines necessary for the efficient activation of T cell responses42, and can migrate to the lymphoid tissues102. In contrast, immature DCs fail to induce antigen-specific responses103 and may in fact induce the differentiation of regulatory T cells56,104. In clinical studies, only the mature, peptide-loaded DCs had the capacity to induce antigen-specific T cell responses in healthy subjects71 and patients with metastatic melanoma105.
Maturation stimuli DCs can be matured in various ways. In the laboratory, TLR agonists such as LPS (TLR4), poly IC (TLR3), and resiquimod (TLR7) are commonly used as DC activators. In the clinic, a commonly used maturation cocktail of proinflammatory cytokines TNFα, IL-1β, and IL-6 combined with prostaglandin E2 (PGE2) was initially established as the gold standard for maturation106. This cocktail induced the upregulation of MHC class I and II molecules, CD40, CD80, CD86, and CCR7 but failed to effectively induce IL-12p70106. However, when compared with other DC maturation stimuli (CD40L trimer, poly IC, and LPS), the cytokine cocktail induced the most uniform maturation in terms of upregulation of DC maturation markers, with the highest yield and recovery; it also stimulated the highest levels of allogeneic T cell proliferation and cytokine production, and it induced priming of Th1 responses106. Other studies have indicated that PGE2 may induce differentiation of regulatory T cells and Th2 responses107, IDO expression108, and is responsible for the lack of IL-12p70 production109. However, PGE2 has also been shown to have important roles in promoting the migration of DCs into lymphoid tissues through the upregulation of CCR7 on DCs110, and in enhancing T cell proliferation through the induction of OX40L, CD70, and 4-1BBL on DCs111.
Alternative methods of maturing DCs have also been explored. CD40 ligand (CD40L) is expressed primarily by activated T cells and B cells, and binds to its receptor CD40 on DCs. Engagement of CD40L with CD40 on DCs induces upregulation of costimulatory molecules and secretion of cytokines such as IL-12112. An irradiated CD40L-expressing K562 cell line together with IFNγ has been used to mature DCs for vaccination in melanoma patients. The DC vaccine produced IL-12p70, and the amount of IL-12p70 produced positively correlated with the induction of CD8+ T cell responses to HLA-A2-restricted gp100 as measured by tetramer binding assays113. TLR agonists have also been used to activate DCs. Activation of the TLRs on DCs can induce DC maturation, upregulation of costimulatory molecules, and production of cytokines and chemokines114. Moreover, simultaneous triggering of different TLRs on DCs mediates synergistic effects resulting in the “superinduction” of IL-12115,116. Consequently, TLR agonists have the potential of inducing optimal DCs to stimulate an effective immune response while concurrently conditioning the environment in vivo to favor the development of immune response. For MDDCs, the TLR3 agonist poly-ICLC combined with TNFα, IL-1β, IFNα, and IFNγ has been used in clinical trials to mature DCs. DCs matured with this cocktail are called α-type-1 polarized DCs (αDC1). They produce high levels of IL-12p70, migrate in response to CCR7 ligand, and induce cytotoxic T lymphocyte-mediated responses against tumor-associated antigen (TAA)117,118. Administration of αDC1 in patients with high-grade glioma proved to be safe and immunogenic and resulted in progression-free status in 9/22 patients lasting at least 12 months119. A clinical grade TLR4 agonist LPS has also been used to mature DCs for vaccination in ovarian cancer patients120. LPS-matured DCs produced IL-12p70 and induced tumor-specific T cell responses in melanoma patients. A modified, less toxic version of LPS called 3-O-deacylated monophosphoryl lipid A (MPLA), which is derived from Salmonella minnesota R595, has been approved for use in human and is used in combination with Cervarix. MPLA matures DCs and together with IFNγ was demonstrated to produce high levels of IL-12 and induce CD8+ T cell responses in vitro121. A new synthetic TLR4 agonist, glucopyranosyl lipid A, has been used in combination with anti-DEC205 that targets HIV gag p24 to DCs in mouse studies. It upregulates CD86 and CD40 and induces IL-12p70 production from DCs in vivo122. It is now being used in human studies as an immune modulator with and without anti-PD-1 (NCT02501473) to modify the TME in follicular non-Hodgkin's lymphoma.
Alternatively, a cocktail of commonly used preventative vaccines consisting of BCG (Bacille Calmette-Guerin)-SSI, Influvac, and Typhim, which contains agonists of TLRs, has been used to mature DCs in vitro. The DCs matured with this cocktail showed high expression of CD80, CD83, and CD86, was able to migrate, and produced IL-12p70123. This cocktail of preventative vaccines was tested in a clinical trial to mature DCs for vaccination in melanoma patients. The DC vaccine induced T cell responses that coincided with longer overall survival; however, local and systemic grade 2 and 3 adverse events were observed in a number of patients and were attributed to the presence of BCG in the maturation cocktail, precluding any future use of the cocktail124.
Another potential factor that can affect the quality of the DC vaccine is the timing of the maturation process. DC vaccines used in clinical trials are typically matured for 24-48 h and cytokine assessments are performed during that period125. However, most of the cytokines produced after DC maturation are produced within 24 h and they only regain the capacity to produce cytokines after the T cell encounter and CD40L ligation126. Thus, maturation protocols that are < 24 h may be more ideal so that the DC vaccines would still retain their ability to produce cytokines upon injection in vivo.
Therefore, the most ideal maturation method is one that induces the most effective APCs in order to induce the most successful immune response in vivo.
Antigen loading
DCs are typically loaded through incubation with peptides, proteins, RNA, or autologous/allogeneic tumor cells79. Peptides are loaded directly on the MHC molecules on the surface of the DCs, whereas the use of proteins and tumor cells requires processing into peptides before loading on the MHC molecules.
Short synthetic peptides (8-15 aa) that correspond to defined CD8+ T cell epitopes within the defined TAAs have been used in clinical trials. However, use of these short peptides necessitates the knowledge of patient's HLA haplotypes and the defined epitopes that would bind to these specific haplotypes. Most recently, the use of synthetic long peptides (SLPs) that are 28-35 aa long has gained some favor. Due to their length, SLPs are preferentially taken up, processed, and presented by DCs through cross presentation127 leading to the activation of CD8+ T cell responses as well as CD4+ T cell responses; however, the advantages of SLPs have been underappreciated until recently128. Clinical trials using SLPs covering the entire length of HPV-16 E6 and E7 sequences emulsified in Montanide ISA-51 adjuvant in patients with advanced HPV-16+ cervical cancer showed that the vaccine was safe and immunogenic129,130. In another study, 79% of patients vaccinated with the HPV-16 SLPs with Montanide ISA-51 adjuvant had objective responses after 12 months with no evidence of virus in the original lesions in five patients131. On the basis of the success of these studies, vaccine formulations using SLPs have been further explored in cervical132, ovarian133,134 and colorectal cancer135,136 and HIV infection137 with evidence of induction of CD4+ and CD8+ T cell responses. An study of DCs with SLPs from NY-ESO-1 and Melan-A/MART-1 is currently being conducted in melanoma. The study is a randomized two arm study comparing the immunogenicity of DC vaccine to Montanide ISA-51 (NCT02334735), and should determine whether DCs can cross-present long peptides to T cells as has been shown to be the case when Montanide is used as the adjuvant.
DCs loaded with proteins138 or lysates of autologous or allogeneic whole tumors or tumor cell lines76,139,140,141,142 have also been used in treating numerous cancers. The major advantages of this method are that (1) multiple epitopes can be presented on MHC molecules of different haplotypes, thus having the potential of inducing both CD4+ and CD8+ T cell responses to a wide spectrum of antigens, and (2) the necessary requirement for processing results in prolonged antigen presentation143. One way to enhance immunogenicity is to increase the exposure/release of damage-associated molecular patterns (DAMPs) derived from tumor cells144 to enhance DC maturation145. Coincidentally, the most common procedure used for killing of autologous or allogeneic whole-tumor cells or tumor cell lines relies on multiple cycles of freeze-thaw, which have been demonstrated to release endogenous DAMPs146. Potent CD4+ and CD8+ T cell responses were detected in ovarian cancer patients vaccinated with DCs activated with hypochlorous acid-oxidized autologous whole-tumor lysate generated via multiple cycles of freeze-thaw120. Most recently, a study in a mouse model of high-grade glioma showed that hypercidin-based photodynamic therapy-activated DC vaccines increased survival, which was accompanied by an immunostimulatory shift in the brain contexture from regulatory T cells to Th1/cytotoxic T cells/Th17 cells147. Alternatively, DCs have been fused to autologous tumor cells with polyethylene glycol. The fusion allows for the tumor to be accessible to both the endogenous and exogenous pathways for activation of both CD4+ and CD8+ T cell responses. This approach was tested in patients with multiple myeloma and shown to be safe and effective, resulting in both CD4+ and CD8+ T cell responses148. These various approaches of antigen loading are currently being explored in larger studies. A phase II multicenter study in multiple myeloma using DCs fused to tumor cells led by Dr David Avigan is set to open. A phase III study using DCs with autologous tumor lysates (DCVAX-L) of glioblastoma is currently ongoing (NCT00045968).
Bacterial or viral vectors have been evaluated to target DCs with tumor antigens. For example, vectors derived from bacteria such as BCG, Listeria monocytogenes, Salmonella, and Shigella, and viruses including Canarypox virus, Newcastle disease virus, vaccinia virus, Sindbis virus, yellow fever virus, human papillomavirus, adenovirus, adeno-associated virus, and lentiviruses have all been explored in this capacity75,149,150,151,152,153,154. These vectors have distinct advantages including the ability to insert genes encoding TAAs and to remove genes encoding virulence or replication factors for safety, and/or the ability to induce DC maturation. Human DCs infected with a killed but metabolically active (KBMA) L. monocytogenes strain encoding an epitope of Melan-A/Mart-1 induced the maturation of DCs, which were able to prime Mart-1-specific CD8+ T cells, resulting in the lysis of patient-derived melanoma cells in vitro. Furthermore, KBMA bacteria efficiently targeted APCs in vivo to induce protective antitumor responses in a mouse tumor model155. However, pre-existing immunity against a vector may ultimately limit its ability to induce immune responses in vivo. Lentiviral vectors have several properties that offer distinct advantages over current vectors used in clinical trials156. Although lentiviral vectors are typically less immunogenic, they can activate the immune system via endosomal or cytoplasmic sensors, i.e., TLRs, RIG-I, and PKR, etc. Use of lentiviral vectors can prime tumor-specific T cell responses in vitro157, activate CD8+ T cells, and inhibit the growth of pre-existing tumors in mouse models158,159. They have the additional capacity of transducing non-dividing cells such as MDDCs, which are usually propagated from CD14+ or CD34+ progenitors160,161,162,163. Lentiviral vectors have been successfully used in adoptive T cell therapy to transduce T cells to express chimeric antigen receptors164. Most recently, a new technology was developed involving transduction of CD14+ monocytes with lentiviral vectors co-expressing GM-CSF/IL-4 and a melanoma antigen to generate so called “SmartDCs” (self-differentiated myeloid-derived antigen-presenting cells reactive against tumors-DCs). In vitro studies have shown that the SmartDCs expressed high levels of CD80, CD86, and those that were transduced with the tyrosinase antigen (SmartDC-TRP2) stimulated antigen-specific T cell responses165. It is possible that viral vectors may augment DC vaccination in the context of virus-induced inflammation and modulate the immunosuppressive TME166 as has been observed with the use of oncolytic viruses.
DCs can also be transfected with mRNAs encoding TAAs. A number of studies have confirmed the ability of these DCs to induce potent tumor antigen-specific T cell responses in vivo167,168,169,170. Transfection of DCs with mRNA can be accomplished with the use of a cationic lipid, i.e., DOTAP, or electroporation. Electroporation has been demonstrated to be the most efficient method to introduce mRNA into DCs171 and has been successfully used in preclinical172,173,174 and clinical175 studies. Autologous DCs electroporated with RNAs encoding CD40L and HIV antigens were demonstrated to be safe and capable of inducing HIV-specific immune responses176. Results from a phase II study using DCs co-electroporated with amplified tumor RNA and synthetic CD40L RNA (AGS-003) in combination with Sunitinib in metastatic clear cell renal cell carcinoma showed that the vaccine was well tolerated and that 62% of vaccinated patients experienced clinical benefit. Furthermore, the magnitude of increases in effector/memory T cells correlated with the overall survival177. The phase III trial (NCT01582672) is currently ongoing and a phase II trial was recently opened for non-small cell lung cancer (NCT02662634).
DC vaccines have typically targeted viral antigens, cancer-testis antigens (CT antigens), overexpressed antigens, and differentiation antigens178. The development of deep sequencing technologies has made it possible to identify unique mutations within the protein-encoding parts of the tumor genomes and to predict potential antigens. These antigens are not expressed in the normal human genome and therefore serve as effective stimulators of naive antigen-specific T cells179. In support of this, whole-exome sequencing and RNA transcript analysis showed that tumors with high mutational burden had more CD8+ T cells180 and that the cytolytic activity of NK and CD8+ T cells correlated with the mutational burden181. Moreover, three studies have suggested that mutational burden in melanoma and lung cancer correlates with improved survival in response to checkpoint inhibitors such as anti-CTLA-4 and anti-PD-1 antibodies182,183,184. Mutational burden alone does not necessarily correlate with improved survival however, and the response rate can be influenced by immune suppressive factors in the TME185. Altogether these studies suggest that mutated antigens deserve to be tested in the clinic where they could prove to be far more immunogenic than conventionally used antigens, e.g., CT antigens, overexpressed antigens, and differentiation antigens.
Indeed, DCs matured with GMP-grade CD40L-expressing K562 cells, IFNγ, poly IC, and R848, and loaded with neoantigen peptides derived from missense mutations in the tumor primed or boosted HLA-A2-restricted CD8+ T cell responses in patients with melanoma186. Additional factors such as pretreatment with cyclophosphamide and checkpoint blockade antibodies to eliminate or reduce regulatory T cells, and administration of DCs via intravenous infusion to allow better trafficking to secondary lymphoid tissues, may have contributed to the success of this study. These very encouraging results indicate that DC vaccination can be used to stimulate neoantigen-specific T cell responses in patients. Given that only a subset of predicted neoantigens were shown to be immunostimulatory there is a need to further refine prediction tools to identify high affinity immunogenic neoantigens that can prime both CD4+ and CD8+ T cell responses.
As noted earlier, DCs have the capacity to present non-conventional antigens. Newer classes of antigens include phosphopeptides, which are synthesized as a result of deregulated phosphorylation, citrullinated peptides, and antigens derived from noncoding DNAs that are expressed in cancer cells. In the latter case these include antigens expressed by endogenous retroviral remnants. It remains to be seen where such antigens will fit into the realm of effective anti-tumor antigens in DC-based cancer vaccines.
Administration and migration of DCs
Migration to the lymph nodes is critical for inducing immune responses. DC vaccines have been administered intradermally, subcutaneously, intravenously, intranodally, or intratumorally, but the optimal route of administration has yet to be established. Large amount of intradermally administered 111-indium-labeled DC vaccine loaded with melanoma peptides remained at the injection site, lost viability, and was cleared within 48 h, with < 5% reaching the draining lymph nodes187. Intratumoral administration of DC vaccines showed retention at the injection site with little detected in the draining lymph nodes188,189, indicating failure of the vaccines to reach their targets.
Newer strategies include administration of DC vaccines via more than one route, i.e., intradermally plus intravenously to induce a systemic response, and administration directly into the lymph nodes (intranodally). Intranodally administered DC vaccine loaded with melanoma peptides redistributed to multiple lymph nodes within 30 min of injection; however, despite direct delivery of DCs into the lymph nodes, immunologic responses elicited were comparable to187 or not better189 than intradermally administered DC vaccines. Many factors likely contributed to this outcome. One possibility is the technical difficulty of properly injecting a vaccine to lymph nodes; improper injection could disrupt the lymph node architecture. Another possibility is that intranodal injection delivers all the vaccines into the lymph nodes; whereas with intradermal injection, only the viable, mature, and fully functional DCs migrate into the lymph nodes to stimulate T cell responses. It may not matter how many DCs reach the lymph nodes as one study has shown that reducing the number of cells injected improved migration of DCs190. Pre-conditioning the site of injection has been identified as a way to increase DC vaccine migration. First tested in mice by Martin-Fontecha et al., preconditioning with the cytokine TNF or activated DCs improved lymph node homing and efficacy of DC vaccines6. Similarly, topical application of TLR7 agonist imiquimod, which recruits T cells and DCs into the skin, also enhanced DC vaccine migration7. In a recently published study, Mitchell et al.191 showed that pre-conditioning the site of DC vaccine injection with tetanus/diphtheria toxoid (Td) vaccine improved DC migration in patients with glioblastoma multiforme by inducing CCL3 levels resulting in CCL21 upregulation and increased lymph node migration of the DC vaccine. The increased DC vaccine migration was associated with improved overall survival. However, the immunogenicity may partly be attributed to the viral antigen targeted by the DC vaccine. Therefore, generating a local inflammatory response at the site of the DC vaccine injection may serve to improve DC migration. Further studies are required to comprehend how these strategies might be incorporated into DC vaccines in order to enhance the arrival of DCs into the lymph nodes. Other unknown factors are whether these DCs directly elicit T cell responses as suggested in animal studies192 or if a component of the maturing DCs is in turn cross-presented by resident DCs either through uptake of dying cells, trogocytosis or through a process called “cross-dressing”193. In animal tumor models, it is clear that the CD103+ DCs acquire antigen from tumor cells and migrate to the draining lymph nodes to activate CD8+ T cells50. Intravenous injection of DCs has the advantage that injected cells can rapidly access secondary lymphoid tissue. Mouse studies of melanoma have shown that intra-tumorally injected mature DCs can modulate the TME, reversing an immune suppressive TME into one that supports T cell attraction and ultimately tumor control194. In this case a deficiency of CCL4 due to Wnt signaling prevented the recruitment of DCs into the tumor bed, which could be bypassed by restoring DC numbers in the tumor itself. The administration of Flt3L under such circumstances together with supplying maturation stimuli may alleviate these barriers in vivo195.
In vivo approaches to DC vaccination
Direct targeting of antigens to in vivo DCs to induce tumor-specific immune responses196,197 bypasses the expensive, labor-intensive, and often difficult to standardize and scale up ex vivo DC generation process. In vivo targeting allows for vaccines to be produced on a larger scale and perhaps, more importantly, allows for direct stimulation of the activation of natural DC subsets at multiple sites in vivo. Early approaches of targeting in vivo DCs involved engineering irradiated tumor cells to secrete GM-CSF198,199 to stimulate the recruitment and enhance the function of APCs. Delivered in a neoadjuvant setting, GVAX comprising an allogeneic pancreatic cell line secreting GM-CSF resulted in the recruitment of T cells into pancreatic tumors200. In a phase II trial of GVAX combined with and without recombinant live attenuated L. monocytogenes engineered to secrete mesothelin (CRS-207) and with low dose cyclophosphamide, CD8+ T cell responses induced were associated with longer overall survival in patients with advanced pancreatic cancer201. However, a phase 2b study failed to show improved overall survival in patients treated with the combination or CRS-207 alone compared with chemotherapy. Prolonged GM-CSF production in the TME has been associated with disease progression in some experimental models and in a phase III trial in patients with hormone therapy-refractory prostate cancer199, possibly due to recruitment of myeloid suppressor cells or differentiation of myeloid precursors into immature tolerogenic DCs202,203. The effect of GM-CSF is therefore context-dependent. Systemically elevated GM-CSF is not effective whereas when locally delivered, as in the form of GVAX, can recruit DCs.
Newer approaches involve the in vivo targeting of DCs via ligation of CD40, and CLRs. Agonist CD40 antibodies have been used to mimic the activation of DCs, which is required for the release of pro-inflammatory cytokines and enhancement of T cell activation. CD40 antibody was given after chemotherapy in mouse models and was shown to have a synergistic effect on inducing CD8+ T cell responses that eliminated the tumor204. A phase I study of the agonist CD40 antibody in combination with gemcitabine in patients with advanced pancreatic cancer demonstrated some clinical activity with associated cytokine release syndrome that is manageable in an outpatient setting205. Interestingly, anti-CD40 may modulate the TME by activating monocytes206. In a mouse model, CD40 agonistic antibody could overcome resistance to checkpoint blockade and improve survival207. The production of anti-CD40 antibodies that preferentially target DCs and convert M2 macrophages into M1 macrophages, and have reduced toxicity in vivo, remains challenging.
CLRs are also attractive targets as DC subsets are known to express different CLRs, which are involved in the recognition and capture of many glycosylated antigens208. Studies in animals targeting antigens to CLRs (DEC205, Langerin, and Clec9A) resulted in effective generation of T cell responses209. Antigen targeting using Clec9a in animal models showed the induction of cellular and humoral immune responses, which is essential for effective vaccination210,211,212. Antigen-specific T cell responses were reported in humanized mouse models of Clec9a-mediated antigen targeting indicating the potential of clinical application of this strategy213.
Recent studies using DEC205-targeting antibodies have successfully induced immune responses to both cancer antigens214 and HIV antigens215,209. Although the majority of these studies were done in mice, human studies using antigens targeted to DC-SIGN216, MR217, and DEC205218 have demonstrated successful induction of tumor-specific T cell responses in subsets of patients, although in some the responses were weak. The correlation with clinical responses remains unclear and larger studies will be needed to further evaluate the efficacy. Clinical trials of anti-DEC205-NY-ESO-1 are currently ongoing in ovarian cancer (NCT02166905), acute myeloid leukemia (NCT01834248), various solid cancers (NCT01522820, NCT02661100), and melanoma (NCT02129075). Preliminary analysis of the clinical trial in melanoma showed that Flt3L with anti-DEC205-NY-ESO-1 treatment considerably increased the numbers of innate immune cells and induced NY-ESO-1-specific immune responses (N Bhardwaj, personal communication). Another potential approach is via targeting of antigen with the chemokine for XCR1, a chemokine receptor exclusively expressed on CD141+ DCs219,220,221.
Newer approaches of targeting in vivo DCs may involve delivery of tumor antigens via systemic administration of RNA lipoplexes. The lipoplexes protect the RNA from degradation, whereas the RNA triggers activation of pDCs and the subsequent release of type I IFNs222. A phase I dose escalation study evaluating the safety of intravenous administration of the RNA-LPX vaccine, which encodes four TAAs in melanoma patients (NCT02410733) is ongoing. Preliminary immune monitoring data from the study shows induction of IFNα and strong antigen-specific T cell responses in the first three patients evaluated222.
Finally, given their safety profile, DC vaccines are being tested in immune prevention. Immune prevention is an intervention designed to halt cancer recurrence. It is currently being evaluated in patients with Lynch syndrome or colorectal cancer with microsatellite instability. These patients have germline mutations in DNA repair genes. In an ongoing clinical trial (NCT 01885702) mutations in various genes, e.g., genes encoding caspase, TGFβ receptor, and CEA, have been identified in these patients. Some of these patients have T cells that recognize these mutated antigens in the circulation suggesting the development of spontaneous immunity and justifying an immunization approach in these high-risk cohorts223. Other studies have delivered DC vaccine into ductal carcinoma in situ lesions targeting HER-2/neu. A decline and/or eradication of HER-2/neu expression was demonstrated224. A long-term goal, therefore, in high risk of recurrence settings is to immunize patients prophylactically.
Future approaches
The safety of DC vaccines has been well established in clinical trials conducted thus far. Observed side effects are relatively mild and transient; they usually include fever, injection site reactions, adenopathy, and fatigue. The immunogenicity of DC vaccines has also been established in most clinical trials with evidence of induction of tumor-specific T cell responses in many patients. A comprehensive meta-analysis of DC vaccination for prostate cancer and renal cancer has shown that DC vaccines have induced tumor-specific immune responses in 77% and 61% of the vaccinated patients, respectively225. However, in our opinion, the clinical efficacy of DC vaccines remains unsubstantiated. Many reasons account for this but the most likely contributing factor is the quality of the DC vaccine. We still lack a consensus on the optimal method of DC preparation for majority of the DC vaccines described thus far. The DC vaccines may be phenotypically mature as evidenced by upregulation of costimulatory molecules113. They may also produce IL-12, a key factor for inducing vaccine efficacy113. But it is unknown how much IL-12 is actually sufficient and whether the DC vaccine will still be producing the IL-12 upon injection or encounter with T cells. Given the correlation between IL-12 production and vaccine efficacy, it will be important to know that IL-12 is indeed still in production after injection113. Furthermore, the majority of the DC vaccine may not migrate to the lymph nodes to stimulate the T cells, and the DC vaccine itself may not directly function to prime immunity, which requires cross-presentation by lymph node resident DCs instead4. Those that do reach the lymph nodes, i.e., after exposure to PGE2, may induce regulatory T cells and Th2 responses. Additional factors that contribute to the limited success of the DC vaccines are intrinsic to the tumors. The TME employs mechanisms that include loss of tumor antigen expression, alteration of MHC molecules, lack of costimulation, expression of inhibitory ligands, induction of regulatory T cells, expression of IDO, and/or production of immunosuppressive cytokines58,226,227,228,229; these may inhibit the capacity of DC vaccines to induce immune responses against the tumor cells. Therefore, studies focused on optimizing the DC preparation in combination with approaches that overcome immune evasive mechanisms by the TME will likely improve efficacies of DC vaccines in vivo. Newer strategies addressing these issues are in development.
In combination
Combination strategies may be the key to the success of DC vaccines. The FDA approvals of checkpoint blockades made the most significant impact on immunotherapy but not just as a monotherapy option and also as part of combination therapy. A recent study showed that DCs electroporated with mRNAs encoding CD40 ligand, CD70, constitutively activated TLR4 and one of four melanoma-associated antigens (gp100, tyrosinase, MAGE-A3, or MAGE-C2) fused to an HLA class II-targeting signal (DC-LAMP) (also called TriMixDC-MEL) in combination with ipilimumab in patients with pretreated advanced melanoma achieved a very encouraging overall response rate of 38%230. A number of DC vaccine combination studies with other checkpoint blockade agents are currently ongoing (NCT02677155, NCT01067287, and NCT01441765). A systematic approach to testing the combination is needed to determine the optimal timing of vaccination or whether using one or two checkpoint inhibitors would be more effective.
Reducing tumor-elicited suppression first may be the key to the success of DC vaccination. By first modulating the TME to make it more receptive, DC vaccination may have a better chance to induce a more effective immune response that can infiltrate the tumors. Talimogene laherparepvec (T-VEC or OncoVEX), derived from human herpes simplex virus 1 and encoding GM-CSF, became the first oncolytic virus that was FDA-approved for the treatment of metastatic melanoma231. Oncolytic viruses have had an impact on melanoma presumably through their effects not only directly on tumor cells but also by activating innate immunity and inducing tumor-specific immunity thus effectively reducing immune suppression in the TME232. Alternatively, intratumoral approaches that mimic viral infection such as intratumoral injections of poly-ICLC233 or CpG234, but without its associated side effects of inducing dominant antiviral immunity would be preferable235. Another possible method to modulate the TME is through direct activation of the STING pathway. Intratumoral administration of STING agonists stimulated IFNβ production leading to regression of established and distant tumors in mouse models236. Finally as alluded to earlier, intratumoral administration of Flt3L may have multipronged effect of increasing DC numbers, mobilization of DCs into the TME, and providing a maturation stimulus, which altogether can substantially improve anti-tumor T cell priming.
New DCs
Another tactic being explored is the differentiation of DCs from human pluripotent stem cells (hPSCs) including induced pluripotent stem cells and embryonic stem cells. There are currently two methods for differentiating DCs from hPSCs: one involves the use of embryoid bodies, an aggregate-like structure mimicking embryonic development, and the other depends on co-culture with stromal cell lines. Both involve a multi-step process utilizing various growth factors such as BMP-4, VEGF, GM-CSF, SCF, Flt3L, and IL-4 at key intervals to induce the differentiation237. Most importantly, this novel source of DCs has potential for large-scale production using bioreactors238, unlike the currently used methods, which are highly dependent on operational quality standards. Most recently, a lentiviral vector was used to transduce hPSCs to express the tumor antigen MART-1. The transduced hPSCs were differentiated into DCs that expressed typical DC markers and matured in response to TNF stimulation. Furthermore, the DCs were able to prime MART-specific CD8+ T cell responses239. Therefore, this new technology has the potential to improve DC vaccination by ensuring an unlimited source of DCs with the added feature of being able to transduce the antigen directly, ensure presentation on MHC class I and stimulate CD8+ T cell responses. The development of stem cell-derived DCs for clinical purposes will also enable the isolation of DC subsets that are profoundly more immunogenic, e.g., the CD141+ DC subset.
The clustered regulatory interspaced short palindromic repeat-associated 9 (Cas9) system is a powerful and versatile genetic engineering technology that gives the most exceptional control over genome editing. Briefly, the guide RNA binds to the target sequence and subsequently, Cas9, an endonuclease, targets the specific DNA sequence that pairs with the guide RNA240. This technology can be used to manipulate DCs to prevent expression of inhibitory molecules (i.e., PD-L1) and cytokines (i.e., IL-10) to improve their effectiveness in vivo, or to preferentially drive CD8+ T cell differentiation. Our laboratory has successfully applied this approach to delete target genes in DCs.
Conclusion
The plethora of different methods used in clinical trials and lack of controlled comparative studies make it virtually impossible to determine the most effective method to use DC vaccines. However, these earlier studies have provided valuable information and contributed to expansion of our knowledge of DCs and tumor biology. Combinatorial approaches and development of new technologies should continue to improve efficacy of DC vaccines in the clinic. Phase III trials testing the efficacy of MDDCs-based vaccines pulsed with autologous tumor preparations in renal cancer (NCT01582672) and GBM (NCT00045968) are ongoing and should provide valuable information.
References
Steinman RM . Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 2012; 30:1–22.
Liu K, Nussenzweig MC . Origin and development of dendritic cells. Immunol Rev 2010; 234:45–54.
Lee J, Breton G, Oliveira TY, et al. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J Exp Med 2015; 212:385–399.
Breton G, Lee J, Zhou YJ, et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J Exp Med 2015; 212:401–413.
Schlitzer A, Sivakamasundari V, Chen J, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol 2015; 16:718–728.
Breton G, Lee J, Liu K, Nussenzweig MC . Defining human dendritic cell progenitors by multiparametric flow cytometry. Nat Protoc 2015; 10:1407–1422.
Merad M, Sathe P, Helft J, Miller J, Mortha A . The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 2013; 31:563–604.
Collin M, McGovern N, Haniffa M . Human dendritic cell subsets. Immunology 2013; 140:22–30.
Haniffa M, Shin A, Bigley V, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 2012; 37:60–73.
Murphy TL, Grajales-Reyes GE, Wu X, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol 2016; 34:93–119.
Meixlsperger S, Leung CS, Ramer PC, et al. CD141+ dendritic cells produce prominent amounts of IFN-α after dsRNA recognition and can be targeted via DEC-205 in humanized mice. Blood 2013; 121:5034–5044.
Zhang S, Kodys K, Li K, Szabo G . Human type 2 myeloid dendritic cells produce interferon-λ and amplify interferon-α in response to hepatitis C virus infection. Gastroenterology 2013; 144:414–425.e7.
Huysamen C, Willment JA, Dennehy KM, Brown GD . CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J Biol Chem 2008; 283:16693–16701.
Sancho D, Joffre OP, Keller AM, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009; 458:899–903.
Cohn L, Chatterjee B, Esselborn F, et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J Exp Med 2013; 210:1049–1063.
McKenna K, Beignon AS, Bhardwaj N . Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol 2005; 79:17–27.
Tel J, Schreibelt G, Sittig SP, et al. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 2013; 121:459–467.
Lui G, Manches O, Angel J, Molens JP, Chaperot L, Plumas J . Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS One 2009; 4:e7111.
O'Brien M, Manches O, Wilen C, et al. CD4 receptor is a key determinant of divergent HIV-1 sensing by plasmacytoid dendritic cells. PLoS Pathog 2016; 12:e1005553.
Salio M, Cella M, Vermi W, et al. Plasmacytoid dendritic cells prime IFN-gamma-secreting melanoma-specific CD8 lymphocytes and are found in primary melanoma lesions. Eur J Immunol 2003; 33:1052–1062.
Hartmann E, Wollenberg B, Rothenfusser S, et al. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res 2003; 63:6478–6487.
Gerlini G, Urso C, Mariotti G, et al. Plasmacytoid dendritic cells represent a major dendritic cell subset in sentinel lymph nodes of melanoma patients and accumulate in metastatic nodes. Clin Immunol 2007; 125:184–193.
Battaglia A, Buzzonetti A, Baranello C, et al. Metastatic tumour cells favour the generation of a tolerogenic milieu in tumour draining lymph node in patients with early cervical cancer. Cancer Immunol Immunother 2009; 58:1363–1373.
Sharma MD, Baban B, Chandler P, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest 2007; 117:2570–2582.
Munn DH, Sharma MD, Hou D, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest 2004; 114:280–290.
Segura E, Amigorena S . Inflammatory dendritic cells in mice and humans. Trends Immunol 2013; 34:440–445.
McGovern N, Schlitzer A, Gunawan M, et al. Human dermal CD14(+) cells are a transient population of monocyte-derived macrophages. Immunity 2014; 41:465–477.
Jaensson E, Uronen-Hansson H, Pabst O, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med 2008; 205:2139–2149.
Agace WW, Persson EK . How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol 2012; 33:42–48.
Reis e Sousa C . Dendritic cells in a mature age. Nat Rev Immunol 2006; 6:476–483.
Jego G, Pascual V, Palucka AK, Banchereau J . Dendritic cells control B cell growth and differentiation. Curr Dir Autoimmun 2005; 8:124–139.
Munz C, Dao T, Ferlazzo G, de Cos MA, Goodman K, Young JW . Mature myeloid dendritic cell subsets have distinct roles for activation and viability of circulating human natural killer cells. Blood 2005; 105:266–273.
Fujii S, Shimizu K, Kronenberg M, Steinman RM . Prolonged IFN-gamma-producing NKT response induced with α-galactosylceramide-loaded DCs. Nat Immunol 2002; 3:867–874.
Akira S, Takeda K . Toll-like receptor signalling. Nat Rev Immunol 2004; 4:499–511.
Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006; 441:101–105.
Pedra JH, Cassel SL, Sutterwala FS . Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol 2009; 21:10–16.
Skoberne M, Beignon AS, Bhardwaj N . Danger signals: a time and space continuum. Trends Mol Med 2004; 10:251–257.
Gallo PM, Gallucci S . The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol 2013; 4:138.
Blum JS, Wearsch PA, Cresswell P . Pathways of antigen processing. Annu Rev Immunol 2013; 31:443–473.
Segura E, Amigorena S . Cross-presentation in mouse and human dendritic cells. Adv Immunol 2015; 127:1–31.
Nair-Gupta P, Baccarini A, Tung N, et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 2014; 158:506–521.
Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol 2000; 18:767–811.
Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S . Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol 2002; 20:621–667.
Joffre OP, Segura E, Savina A, Amigorena S . Cross-presentation by dendritic cells. Nat Rev Immunol 2012; 12:557–569.
Brentville VA, Metheringham RL, Gunn B, et al. Citrullinated vimentin presented on MHC-II in tumor cells is a target for CD4+ T-cell-mediated antitumor immunity. Cancer Res 2016; 76:548–560.
Cobbold M, De La Pena H, Norris A, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med 2013; 5:203ra125.
Geijtenbeek TB, Gringhuis SI . Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 2009; 9:465–479.
Benvenuti F . The dendritic cell synapse: a life dedicated to T cell activation. Front Immunol 2016; 7:70.
Penna G, Vulcano M, Sozzani S, Adorini L . Differential migration behavior and chemokine production by myeloid and plasmacytoid dendritic cells. Hum Immunol 2002; 63:1164–1171.
Roberts EW, Broz ML, Binnewies M, et al. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 2016; 30:324–336.
Gerner MY, Torabi-Parizi P, Germain RN . Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity 2015; 42:172–185.
Colino J, Shen Y, Snapper CM . Dendritic cells pulsed with intact Streptococcus pneumoniae elicit both protein- and polysaccharide-specific immunoglobulin isotype responses in vivo through distinct mechanisms. J Exp Med 2002; 195:1–13.
Yamazaki C, Miyamoto R, Hoshino K, et al. Conservation of a chemokine system, XCR1 and its ligand, XCL1, between human and mice. Biochem Biophys Res Commun 2010; 397:756–761.
Fox JC, Nakayama T, Tyler RC, Sander TL, Yoshie O, Volkman BF . Structural and agonist properties of XCL2, the other member of the C-chemokine subfamily. Cytokine 2015; 71:302–311.
Skoberne M, Beignon AS, Larsson M, Bhardwaj N . Apoptotic cells at the crossroads of tolerance and immunity. Curr Top Microbiol Immunol 2005; 289:259–292.
Cools N, Ponsaerts P, Van Tendeloo VF, Berneman ZN . Balancing between immunity and tolerance: an interplay between dendritic cells, regulatory T cells, and effector T cells. J Leukoc Biol 2007; 82:1365–1374.
Mellor AL, Munn DH . IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 2004; 4:762–774.
Munn DH, Mellor AL . IDO and tolerance to tumors. Trends Mol Med 2004; 10:15–18.
Levings MK, Gregori S, Tresoldi E, Cazzaniga S, Bonini C, Roncarolo MG . Differentiation of Tr1 cells by immature dendritic cells requires IL-10 but not CD25+CD4+ Tr cells. Blood 2005; 105:1162–1169.
Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH . Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med 2000; 192:1213–1222.
Woo EY, Chu CS, Goletz TJ, et al. Regulatory CD4+CD25+ T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res 2001; 61:4766–4772.
Liyanage UK, Moore TT, Joo HG, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol 2002; 169:2756–2761.
Tang Q, Bluestone JA . The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol 2008; 9:239–244.
Idoyaga J, Fiorese C, Zbytnuik L, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest 2013; 123:844–854.
Skoberne M, Somersan S, Almodovar W, et al. The apoptotic-cell receptor CR3, but not αvβ5, is a regulator of human dendritic-cell immunostimulatory function. Blood 2006; 108:947–955.
Frleta D, Ochoa CE, Kramer HB, et al. HIV-1 infection-induced apoptotic microparticles inhibit human DCs via CD44. J Clin Invest 2012; 122:4685–4697.
Kalialis LV, Drzewiecki KT, Klyver H . Spontaneous regression of metastases from melanoma: review of the literature. Melanoma Res 2009; 19:275–282.
Wang RF, Rosenberg SA . Human tumor antigens for cancer vaccine development. Immunol Rev 1999; 170:85–100.
Kim R, Emi M, Tanabe K . Cancer immunoediting from immune surveillance to immune escape. Immunology 2007; 121:1–14.
Dunn GP, Old LJ, Schreiber RD . The three Es of cancer immunoediting. Annu Rev Immunol 2004; 22:329–360.
Dhodapkar MV, Steinman RM, Sapp M, et al. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J Clin Invest 1999; 104:173–180.
Constantino J, Gomes C, Falcao A, Cruz MT, Neves BM . Antitumor dendritic cell-based vaccines: lessons from 20 years of clinical trials and future perspectives. Transl Res 2016; 168:74–95.
Lundberg K, Albrekt AS, Nelissen I, et al. Transcriptional profiling of human dendritic cell populations and models--unique profiles of in vitro dendritic cells and implications on functionality and applicability. PLoS One 2013; 8:e52875.
Carpentier S, Vu Manh TP, Chelbi R, et al. Comparative genomics analysis of mononuclear phagocyte subsets confirms homology between lymphoid tissue-resident and dermal XCR1+ DCs in mouse and human and distinguishes them from Langerhans cells. J Immunol Methods 2016; 432:35–49.
Gandhi RT, O'Neill D, Bosch RJ, et al. A randomized therapeutic vaccine trial of canarypox-HIV-pulsed dendritic cells vs. canarypox-HIV alone in HIV-1-infected patients on antiretroviral therapy. Vaccine 2009; 27:6088–6094.
Palucka AK, Ueno H, Connolly J, et al. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J Immunother 2006; 29:545–557.
Redman BG, Chang AE, Whitfield J, et al. Phase Ib trial assessing autologous, tumor-pulsed dendritic cells as a vaccine administered with or without IL-2 in patients with metastatic melanoma. J Immunother 2008; 31:591–598.
O'Neill DW, Bhardwaj N . Differentiation of peripheral blood monocytes into dendritic cells. Curr Protoc Immunol 2005; Chapter 22:Unit 22F.24.
O'Neill D, Bhardwaj N . Generation of autologous peptide- and protein-pulsed dendritic cells for patient-specific immunotherapy. Methods Mol Med 2005; 109:97–112.
Florcken A, Kopp J, van Lessen A, et al. Allogeneic partially HLA-matched dendritic cells pulsed with autologous tumor cell lysate as a vaccine in metastatic renal cell cancer: a clinical phase I/II study. Hum Vaccin Immunother 2013; 9:1217–1227.
Kumar J, Kale V, Limaye L . Umbilical cord blood-derived CD11c(+) dendritic cells could serve as an alternative allogeneic source of dendritic cells for cancer immunotherapy. Stem Cell Res Ther 2015; 6:184.
Pinzon-Charry A, Maxwell T, Lopez JA . Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol Cell Biol 2005; 83:451–461.
Wells JW, Cowled CJ, Darling D, et al. Semi-allogeneic dendritic cells can induce antigen-specific T-cell activation, which is not enhanced by concurrent alloreactivity. Cancer Immunol Immunother 2007; 56:1861–1873.
Fabre JW . The allogeneic response and tumor immunity. Nat Med 2001; 7:649–652.
Ratzinger G, Baggers J, de Cos MA, et al. Mature human Langerhans cells derived from CD34+ hematopoietic progenitors stimulate greater cytolytic T lymphocyte activity in the absence of bioactive IL-12p70, by either single peptide presentation or cross-priming, than do dermal-interstitial or monocyte-derived dendritic cells. J Immunol 2004; 173:2780–2791.
Romano E, Rossi M, Ratzinger G, et al. Peptide-loaded Langerhans cells, despite increased IL15 secretion and T-cell activation in vitro, elicit antitumor T-cell responses comparable to peptide-loaded monocyte-derived dendritic cells in vivo. Clin Cancer Res 2011; 17:1984–1997.
Yuan J, Latouche JB, Reagan JL, et al. Langerhans cells derived from genetically modified human CD34+ hemopoietic progenitors are more potent than peptide-pulsed Langerhans cells for inducing antigen-specific CD8+ cytolytic T lymphocyte responses. J Immunol 2005; 174:758–766.
Lee J, Breton G, Aljoufi A, et al. Clonal analysis of human dendritic cell progenitor using a stromal cell culture. J Immunol Methods 2015; 425:21–26.
Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010; 363:411–422.
Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol 2006; 24:3089–3094.
Small EJ, Fratesi P, Reese DM, et al. Immunotherapy of hormone-refractory prostate cancer with antigen-loaded dendritic cells. J Clin Oncol 2000; 18:3894–3903.
Sheikh NA, Jones LA . CD54 is a surrogate marker of antigen presenting cell activation. Cancer Immunol Immunother 2008; 57:1381–1390.
GuhaThakurta D, Sheikh NA, Fan LQ, et al. Humoral immune response against nontargeted tumor antigens after treatment with sipuleucel-T and its association with improved clinical outcome. Clin Cancer Res 2015; 21:3619–3630.
Sheikh NA, Petrylak D, Kantoff PW, et al. Sipuleucel-T immune parameters correlate with survival: an analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol Immunother 2013; 62:137–147.
Fong L, Carroll P, Weinberg V, et al. Activated lymphocyte recruitment into the tumor microenvironment following preoperative sipuleucel-T for localized prostate cancer. J Natl Cancer Inst 2014; 106.
Marroquin CE, Westwood JA, Lapointe R, et al. Mobilization of dendritic cell precursors in patients with cancer by flt3 ligand allows the generation of higher yields of cultured dendritic cells. J Immunother 2002; 25:278–288.
Anandasabapathy N, Breton G, Hurley A, et al. Efficacy and safety of CDX-301, recombinant human Flt3L, at expanding dendritic cells and hematopoietic stem cells in healthy human volunteers. Bone Marrow Transplant 2015; 50:924–930.
Chen W, Antonenko S, Sederstrom JM, et al. Thrombopoietin cooperates with FLT3-ligand in the generation of plasmacytoid dendritic cell precursors from human hematopoietic progenitors. Blood 2004; 103:2547–2553.
Tel J, Aarntzen EH, Baba T, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res 2013; 73:1063–1075.
Schreibelt G, Bol KF, Westdorp H, et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin Cancer Res 2016; 22:2155–2166.
Carreno BM, Magrini V, Becker-Hapak M, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; 348:803–808.
De Vries IJ, Krooshoop DJ, Scharenborg NM, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res 2003; 63:12–17.
Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N . Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med 2001; 193:233–238.
Dhodapkar MV, Steinman RM . Antigen-bearing immature dendritic cells induce peptide-specific CD8(+) regulatory T cells in vivo in humans. Blood 2002; 100:174–177.
de Vries IJ, Lesterhuis WJ, Scharenborg NM, et al. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin Cancer Res 2003; 9:5091–5100.
Lee AW, Truong T, Bickham K, et al. A clinical grade cocktail of cytokines and PGE2 results in uniform maturation of human monocyte-derived dendritic cells: implications for immunotherapy. Vaccine 2002; 20 Suppl 4:A8–A22.
Jongmans W, Tiemessen DM, van Vlodrop IJ, Mulders PF, Oosterwijk E . Th1-polarizing capacity of clinical-grade dendritic cells is triggered by Ribomunyl but is compromised by PGE2: the importance of maturation cocktails. J Immunother 2005; 28:480–487.
Krause P, Singer E, Darley PI, Klebensberger J, Groettrup M, Legler DF . Prostaglandin E2 is a key factor for monocyte-derived dendritic cell maturation: enhanced T cell stimulatory capacity despite IDO. J Leukoc Biol 2007; 82:1106–1114.
Morelli AE, Thomson AW . Dendritic cells under the spell of prostaglandins. Trends Immunol 2003; 24:108–111.
Scandella E, Men Y, Gillessen S, Forster R, Groettrup M . Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood 2002; 100:1354–1361.
Krause P, Bruckner M, Uermosi C, Singer E, Groettrup M, Legler DF . Prostaglandin E(2) enhances T-cell proliferation by inducing the costimulatory molecules OX40L, CD70, and 4-1BBL on dendritic cells. Blood 2009; 113:2451–2460.
Ma DY, Clark EA . The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol 2009; 21:265–272.
Carreno BM, Becker-Hapak M, Huang A, et al. IL-12p70-producing patient DC vaccine elicits Tc1-polarized immunity. J Clin Invest 2013; 123:3383–3394.
Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R . Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2001; 2:947–950.
Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A . selected toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol 2005; 6:769–776.
Boullart AC, Aarntzen EH, Verdijk P, et al. Maturation of monocyte-derived dendritic cells with toll-like receptor 3 and 7/8 ligands combined with prostaglandin E2 results in high interleukin-12 production and cell migration. Cancer Immunol Immunother 2008; 57:1589–1597.
Mailliard RB, Wankowicz-Kalinska A, Cai Q, et al. α-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res 2004; 64:5934–5937.
Lee JJ, Foon KA, Mailliard RB, Muthuswamy R, Kalinski P . Type 1-polarized dendritic cells loaded with autologous tumor are a potent immunogen against chronic lymphocytic leukemia. J Leukoc Biol 2008; 84:319–325.
Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol 2011; 29:330–336.
Chiang CL, Kandalaft LE, Tanyi J, et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: from bench to bedside. Clin Cancer Res 2013; 19:4801–4815.
Kolanowski ST, Sritharan L, Lissenberg-Thunnissen SN, Van Schijndel GM, Van Ham SM, ten Brinke A . Comparison of media and serum supplementation for generation of monophosphoryl lipid A/interferon-gamma-matured type I dendritic cells for immunotherapy. Cytotherapy 2014; 16:826–834.
Pantel A, Cheong C, Dandamudi D, et al. A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T-cell immunity in vivo. Eur J Immunol 2012; 42:101–109.
Schreibelt G, Benitez-Ribas D, Schuurhuis D, et al. Commonly used prophylactic vaccines as an alternative for synthetically produced TLR ligands to mature monocyte-derived dendritic cells. Blood 2010; 116:564–574.
Bol KF, Aarntzen EH, Pots JM, et al. Prophylactic vaccines are potent activators of monocyte-derived dendritic cells and drive effective anti-tumor responses in melanoma patients at the cost of toxicity. Cancer Immunol Immunother 2016; 65:327–339.
Langenkamp A, Messi M, Lanzavecchia A, Sallusto F . Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol 2000; 1:311–316.
Dohnal AM, Graffi S, Witt V, et al. Comparative evaluation of techniques for the manufacturing of dendritic cell-based cancer vaccines. J Cell Mol Med 2009; 13:125–135.
Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, van der Burg SH, Offringa R . Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur J Immunol 2008; 38:1033–1042.
Linnemann C, van Buuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 2015; 21:81–85.
Kenter GG, Welters MJ, Valentijn AR, et al. Phase I immunotherapeutic trial with long peptides spanning the E6 and E7 sequences of high-risk human papillomavirus 16 in end-stage cervical cancer patients shows low toxicity and robust immunogenicity. Clin Cancer Res 2008; 14:169–177.
Welters MJ, Kenter GG, Piersma SJ, et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res 2008; 14:178–187.
Kenter GG, Welters MJ, Valentijn AR, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 2009; 361:1838–1847.
de Vos van Steenwijk PJ, Ramwadhdoebe TH, Lowik MJ, et al. A placebo-controlled randomized HPV16 synthetic long-peptide vaccination study in women with high-grade cervical squamous intraepithelial lesions. Cancer Immunol Immunother 2012; 61:1485–1492.
Leffers N, Lambeck AJ, Gooden MJ, et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer 2009; 125:2104–2113.
Sabbatini P, Tsuji T, Ferran L, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res 2012; 18:6497–6508.
Zeestraten EC, Speetjens FM, Welters MJ, et al. Addition of interferon-α to the p53-SLP(R) vaccine results in increased production of interferon-gamma in vaccinated colorectal cancer patients: a phase I/II clinical trial. Int J Cancer 2013; 132:1581–1591.
Speetjens FM, Kuppen PJ, Welters MJ, et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res 2009; 15:1086–1095.
Rosario M, Borthwick N, Stewart-Jones GB, et al. Prime-boost regimens with adjuvanted synthetic long peptides elicit T cells and antibodies to conserved regions of HIV-1 in macaques. AIDS 2012; 26:275–284.
Barrou B, Benoit G, Ouldkaci M, et al. Vaccination of prostatectomized prostate cancer patients in biochemical relapse, with autologous dendritic cells pulsed with recombinant human PSA. Cancer Immunol Immunother 2004; 53:453–460.
Salcedo M, Bercovici N, Taylor R, et al. Vaccination of melanoma patients using dendritic cells loaded with an allogeneic tumor cell lysate. Cancer Immunol Immunother 2006; 55:819–829.
Mahdian R, Kokhaei P, Najar HM, Derkow K, Choudhury A, Mellstedt H . Dendritic cells, pulsed with lysate of allogeneic tumor cells, are capable of stimulating MHC-restricted antigen-specific antitumor T cells. Med Oncol 2006; 23:273–282.
Schnurr M, Galambos P, Scholz C, et al. Tumor cell lysate-pulsed human dendritic cells induce a T-cell response against pancreatic carcinoma cells: an in vitro model for the assessment of tumor vaccines. Cancer Res 2001; 61:6445–6450.
Thumann P, Moc I, Humrich J, et al. Antigen loading of dendritic cells with whole tumor cell preparations. J Immunol Methods 2003; 277:1–16.
Schnurr M, Chen Q, Shin A, et al. Tumor antigen processing and presentation depend critically on dendritic cell type and the mode of antigen delivery. Blood 2005; 105:2465–2472.
Vandenberk L, Belmans J, Van Woensel M, Riva M, Van Gool SW . Exploiting the immunogenic potential of cancer cells for improved dendritic cell vaccines. Front Immunol 2015; 6:663.
Somersan S, Larsson M, Fonteneau JF, Basu S, Srivastava P, Bhardwaj N . Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J Immunol 2001; 167:4844–4852.
Hatfield P, Merrick AE, West E, et al. Optimization of dendritic cell loading with tumor cell lysates for cancer immunotherapy. J Immunother 2008; 31:620–632.
Garg AD, Vandenberk L, Koks C, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 2016; 8:328ra327.
Rosenblatt J, Vasir B, Uhl L, et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood 2011; 117:393–402.
Jenne L, Schuler G, Steinkasserer A . Viral vectors for dendritic cell-based immunotherapy. Trends Immunol 2001; 22:102–107.
Brockstedt DG, Dubensky TW . Promises and challenges for the development of Listeria monocytogenes-based immunotherapies. Expert Rev Vaccines 2008; 7:1069–1084.
Bellone S, El-Sahwi K, Cocco E, et al. Human papillomavirus type 16 (HPV-16) virus-like particle L1-specific CD8+ cytotoxic T lymphocytes (CTLs) are equally effective as E7-specific CD8+ CTLs in killing autologous HPV-16-positive tumor cells in cervical cancer patients: implications for L1 dendritic cell-based therapeutic vaccines. J Virol 2009; 83:6779–6789.
Carrasco J, Van Pel A, Neyns B, et al. Vaccination of a melanoma patient with mature dendritic cells pulsed with MAGE-3 peptides triggers the activity of nonvaccine anti-tumor cells. J Immunol 2008; 180:3585–3593.
Butterfield LH, Comin-Anduix B, Vujanovic L, et al. Adenovirus MART-1-engineered autologous dendritic cell vaccine for metastatic melanoma. J Immunother 2008; 31:294–309.
Veron P, Allo V, Riviere C, Bernard J, Douar AM, Masurier C . Major subsets of human dendritic cells are efficiently transduced by self-complementary adeno-associated virus vectors 1 and 2. J Virol 2007; 81:5385–5394.
Skoberne M, Yewdall A, Bahjat KS, et al. KBMA Listeria monocytogenes is an effective vector for DC-mediated induction of antitumor immunity. J Clin Invest 2008; 118:3990–4001.
Oldham RA, Berinstein EM, Medin JA . Lentiviral vectors in cancer immunotherapy. Immunotherapy 2015; 7:271–284.
Breckpot K, Heirman C, De Greef C, van der Bruggen P, Thielemans K . Identification of new antigenic peptide presented by HLA-Cw7 and encoded by several MAGE genes using dendritic cells transduced with lentiviruses. J Immunol 2004; 172:2232–2237.
He Y, Zhang J, Mi Z, Robbins P, Falo LD Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol 2005; 174:3808–3817.
Dullaers M, Van Meirvenne S, Heirman C, et al. Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther 2006; 13:630–640.
Schroers R, Sinha I, Segall H, et al. Transduction of human PBMC-derived dendritic cells and macrophages by an HIV-1-based lentiviral vector system. Mol Ther 2000; 1:171–179.
Dyall J, Latouche JB, Schnell S, Sadelain M . Lentivirus-transduced human monocyte-derived dendritic cells efficiently stimulate antigen-specific cytotoxic T lymphocytes. Blood 2001; 97:114–121.
Lizee G, Gonzales MI, Topalian SL . Lentivirus vector-mediated expression of tumor-associated epitopes by human antigen presenting cells. Hum Gene Ther 2004; 15:393–404.
Bobadilla S, Sunseri N, Landau NR . Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther 2013; 20:514–520.
Varela-Rohena A, Carpenito C, Perez EE, et al. Genetic engineering of T cells for adoptive immunotherapy. Immunol Res 2008; 42:166–181.
Sundarasetty BS, Chan L, Darling D, et al. Lentivirus-induced 'Smart' dendritic cells: pharmacodynamics and GMP-compliant production for immunotherapy against TRP2-positive melanoma. Gene Ther 2015; 22:707–720.
Woller N, Knocke S, Mundt B, et al. Virus-induced tumor inflammation facilitates effective DC cancer immunotherapy in a Treg-dependent manner in mice. J Clin Invest 2011; 121:2570–2582.
Nair SK, Morse M, Boczkowski D, et al. Induction of tumor-specific cytotoxic T lymphocytes in cancer patients by autologous tumor RNA-transfected dendritic cells. Ann Surg 2002; 235:540–549.
Muller MR, Tsakou G, Grunebach F, Schmidt SM, Brossart P . Induction of chronic lymphocytic leukemia (CLL)-specific CD4- and CD8-mediated T-cell responses using RNA-transfected dendritic cells. Blood 2004; 103:1763–1769.
Nencioni A, Muller MR, Grunebach F, et al. Dendritic cells transfected with tumor RNA for the induction of antitumor CTL in colorectal cancer. Cancer Gene Ther 2003; 10:209–214.
Milazzo C, Reichardt VL, Muller MR, Grunebach F, Brossart P . Induction of myeloma-specific cytotoxic T cells using dendritic cells transfected with tumor-derived RNA. Blood 2003; 101:977–982.
Gilboa E, Vieweg J . Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev 2004; 199:251–263.
Heiser A, Maurice MA, Yancey DR, Coleman DM, Dahm P, Vieweg J . Human dendritic cells transfected with renal tumor RNA stimulate polyclonal T-cell responses against antigens expressed by primary and metastatic tumors. Cancer Res 2001; 61:3388–3393.
Strobel I, Berchtold S, Gotze A, Schulze U, Schuler G, Steinkasserer A . Human dendritic cells transfected with either RNA or DNA encoding influenza matrix protein M1 differ in their ability to stimulate cytotoxic T lymphocytes. Gene Ther 2000; 7:2028–2035.
Koido S, Kashiwaba M, Chen D, Gendler S, Kufe D, Gong J . Induction of antitumor immunity by vaccination of dendritic cells transfected with MUC1 RNA. J Immunol 2000; 165:5713–5719.
Heiser A, Coleman D, Dannull J, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest 2002; 109:409–417.
Routy JP, Boulassel MR, Yassine-Diab B, et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin Immunol 2010; 134:140–147.
Amin A, Dudek AZ, Logan TF, et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): phase 2 study results. J Immunother Cancer 2015; 3:14.
Obeid J, Hu Y, Slingluff CL Jr. Vaccines, adjuvants, and dendritic cell activators--current status and future challenges. Semin Oncol 2015; 42:549–561.
Schumacher TN, Schreiber RD . Neoantigens in cancer immunotherapy. Science 2015; 348:69–74.
Brown SD, Warren RL, Gibb EA, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014; 24:743–750.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N . Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015; 160:48–61.
Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124–128.
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371:2189–2199.
Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015; 350:207–211.
Hugo W, Zaretsky JM, Sun L, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016; 165:35–44.
Carreno BM, Magrini V, Becker-Hapak M, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; 348:803–808.
Verdijk P, Aarntzen EH, Lesterhuis WJ, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res 2009; 15:2531–2540.
Fujiwara S, Wada H, Miyata H, et al. Clinical trial of the intratumoral administration of labeled DC combined with systemic chemotherapy for esophageal cancer. J Immunother 2012; 35:513–521.
Lesterhuis WJ, de Vries IJ, Schreibelt G, et al. Route of administration modulates the induction of dendritic cell vaccine-induced antigen-specific T cells in advanced melanoma patients. Clin Cancer Res 2011; 17:5725–5735.
Aarntzen EH, Srinivas M, Bonetto F, et al. Targeting of 111in-labeled dendritic cell human vaccines improved by reducing number of cells. Clin Cancer Res 2013; 19:1525–1533.
Mitchell DA, Batich KA, Gunn MD, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015; 519:366–369.
Headley MB, Bins A, Nip A, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 2016; 531:513–517.
Yewdall AW, Drutman SB, Jinwala F, Bahjat KS, Bhardwaj N . CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One 2010; 5:e11144.
Spranger S, Bao R, Gajewski TF . Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015; 523:231–235.
Salmon H, Idoyaga J, Rahman A, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 2016; 44:924–938.
Shortman K, Lahoud MH, Caminschi I . Improving vaccines by targeting antigens to dendritic cells. Exp Mol Med 2009; 41:61–66.
Tacken PJ, Torensma R, Figdor CG . Targeting antigens to dendritic cells in vivo. Immunobiology 2006; 211:599–608.
Jinushi M, Hodi FS, Dranoff G . Enhancing the clinical activity of granulocyte-macrophage colony-stimulating factor-secreting tumor cell vaccines. Immunol Rev 2008; 222:287–298.
Jinushi M, Tahara H . Cytokine gene-mediated immunotherapy: current status and future perspectives. Cancer Sci 2009; 100:1389–1396.
Lutz ER, Wu AA, Bigelow E, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2014; 2:616–631.
Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol 2015; 33:1325–1333.
Filipazzi P, Valenti R, Huber V, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol 2007; 25:2546–2553.
Sica A, Bronte V . Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 2007; 117:1155–1166.
Nowak AK, Robinson BW, Lake RA . Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res 2003; 63:4490–4496.
Beatty GL, Torigian DA, Chiorean EG, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res 2013; 19:6286–6295.
Bajor DL, Xu X, Torigian DA, et al. Immune activation and a 9-year ongoing complete remission following CD40 antibody therapy and metastasectomy in a patient with metastatic melanoma. Cancer Immunol Res 2014; 2:1051–1058.
Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res 2015; 3:399–411.
van Kooyk Y . C-type lectins on dendritic cells: key modulators for the induction of immune responses. Biochem Soc Trans 2008; 36:1478–1481.
Idoyaga J, Lubkin A, Fiorese C, et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc Natl Acad Sci USA 2011; 108:2384–2389.
Kato Y, Zaid A, Davey GM, et al. Targeting antigen to Clec9A primes follicular Th cell memory responses capable of robust recall. J Immunol 2015; 195:1006–1014.
Lahoud MH, Ahmet F, Kitsoulis S, et al. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J Immunol 2011; 187:842–850.
Li J, Ahmet F, Sullivan LC, et al. Antibodies targeting Clec9A promote strong humoral immunity without adjuvant in mice and non-human primates. Eur J Immunol 2015; 45:854–864.
Tullett KM, Leal Rojas IM, Minoda Y, et al. Targeting CLEC9A delivers antigen to human CD141+ DC for CD4+ and CD8+T cell recognition. JCI Insight 2016; 1:e87102.
Wang B, Zaidi N, He LZ, et al. Targeting of the non-mutated tumor antigen HER2/neu to mature dendritic cells induces an integrated immune response that protects against breast cancer in mice. Breast Cancer Res 2012; 14:R39.
Cheong C, Choi JH, Vitale L, et al. Improved cellular and humoral immune responses in vivo following targeting of HIV Gag to dendritic cells within human anti-human DEC205 monoclonal antibody. Blood 2010; 116:3828–3838.
Tacken PJ, de Vries IJ, Gijzen K, et al. Effective induction of naive and recall T-cell responses by targeting antigen to human dendritic cells via a humanized anti-DC-SIGN antibody. Blood 2005; 106:1278–1285.
Ramakrishna V, Treml JF, Vitale L, et al. Mannose receptor targeting of tumor antigen pmel17 to human dendritic cells directs anti-melanoma T cell responses via multiple HLA molecules. J Immunol 2004; 172:2845–2852.
Trumpfheller C, Longhi MP, Caskey M, et al. Dendritic cell-targeted protein vaccines: a novel approach to induce T-cell immunity. J Int Med 2012; 271:183–192.
Hartung E, Becker M, Bachem A, et al. Induction of potent CD8 T cell cytotoxicity by specific targeting of antigen to cross-presenting dendritic cells in vivo via murine or human XCR1. J Immunol 2015; 194:1069–1079.
Terhorst D, Fossum E, Baranska A, et al. Laser-assisted intradermal delivery of adjuvant-free vaccines targeting XCR1+ dendritic cells induces potent antitumoral responses. J Immunol 2015; 194:5895–5902.
Fossum E, Grodeland G, Terhorst D, et al. Vaccine molecules targeting Xcr1 on cross-presenting DCs induce protective CD8+ T-cell responses against influenza virus. Eur J Immunol 2015; 45:624–635.
Kranz LM, Diken M, Haas H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016; 534:396–401.
Saeterdal I, Bjorheim J, Lislerud K, et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci USA 2001; 98:13255–13260.
Sharma A, Koldovsky U, Xu S, et al. HER-2 pulsed dendritic cell vaccine can eliminate HER-2 expression and impact ductal carcinoma in situ. Cancer 2012; 118:4354–4362.
Draube A, Klein-Gonzalez N, Mattheus S, et al. Dendritic cell based tumor vaccination in prostate and renal cell cancer: a systematic review and meta-analysis. PLoS One 2011; 6:e18801.
Bennaceur K, Chapman J, Brikci-Nigassa L, Sanhadji K, Touraine JL, Portoukalian J . Dendritic cells dysfunction in tumour environment. Cancer Lett 2008; 272:186–196.
Aptsiauri N, Cabrera T, Mendez R, Garcia-Lora A, Ruiz-Cabello F, Garrido F . Role of altered expression of HLA class I molecules in cancer progression. Adv Exp Med Biol 2007; 601:123–131.
Bronte V, Mocellin S . Suppressive influences in the immune response to cancer. J Immunother 2009; 32:1–11.
Chang CC, Ogino T, Mullins DW, et al. Defective human leukocyte antigen class I-associated antigen presentation caused by a novel beta2-microglobulin loss-of-function in melanoma cells. J Biol Chem 2006; 281:18763–18773.
Wilgenhof S, Corthals J, Heirman C, et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol 2016; 34:1330–1338.
Pol J, Kroemer G, Galluzzi L . First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016; 5:e1115641.
Lawler SE, Chiocca EA . Oncolytic virus-mediated immunotherapy: a combinatorial approach for cancer treatment. J Clin Oncol 2015; 33:2812–2814.
Salazar AM, Erlich RB, Mark A, Bhardwaj N, Herberman RB . Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: case report, hypothesis, and clinical trial. Cancer Immunol Res 2014; 2:720–724.
Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol 2010; 28:4324–4332.
de Vries CR, Kaufman HL, Lattime EC . Oncolytic viruses: focusing on the tumor microenvironment. Cancer Gene Ther 2015; 22:169–171.
Corrales L, Glickman LH, McWhirter SM, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep 2015; 11:1018–1030.
Senju S, Hirata S, Motomura Y, et al. Pluripotent stem cells as source of dendritic cells for immune therapy. Int J Hematol 2010; 91:392–400.
Li Y, Liu M, Yang ST . Dendritic cells derived from pluripotent stem cells: potential of large scale production. World J Stem Cells 2014; 6:1–10.
Zeng J, Wu C, Wang S . Antigenically modified human pluripotent stem cells generate antigen-presenting dendritic cells. Sci Rep 2015; 5:15262.
White MK, Khalili K . CRISPR/Cas9 and cancer targets: future possibilities and present challenges. Oncotarget 2016; 7:12305–12317.
Acknowledgements
Some of the studies cited in this review were supported by the Cancer Research Institute, the Melanoma Research Alliance, and the NIH (AI044628, CA180913, and AI081848).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sabado, R., Balan, S. & Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res 27, 74–95 (2017). https://doi.org/10.1038/cr.2016.157
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.157
Keywords
This article is cited by
-
Anoikis-related signature predicts prognosis and characterizes immune landscape of ovarian cancer
Cancer Cell International (2024)
-
Pan-cancer analysis reveals PDK family as potential indicators related to prognosis and immune infiltration
Scientific Reports (2024)
-
Immunomodulatory effects of Diospyros peregrina fruit preparation (DFP) in non-small cell lung cancer (NSCLC) by utilizing dendritic cell-mediated antigen presentation and T helper (TH) cell differentiation
Medical Oncology (2024)
-
Combinational delivery of TLR4 and TLR7/8 agonist enhanced the therapeutic efficacy of immune checkpoint inhibitors to colon tumor
Molecular and Cellular Biochemistry (2024)
-
Immune landscape and response to oncolytic virus-based immunotherapy
Frontiers of Medicine (2024)
