
Abstract
Harnessing the immune system to eradicate malignant cells is becoming a most powerful new approach to cancer therapy. FDA approval of the immunotherapy-based drugs, sipuleucel-T (Provenge), ipilimumab (Yervoy, anti-CTLA-4), and more recently, the programmed cell death (PD)-1 antibody (pembrolizumab, Keytruda), for the treatment of multiple types of cancer has greatly advanced research and clinical studies in the field of cancer immunotherapy. Furthermore, recent clinical trials, using NY-ESO-1-specific T cell receptor (TCR) or CD19-chimeric antigen receptor (CAR), have shown promising clinical results for patients with metastatic cancer. Current success of cancer immunotherapy is built upon the work of cancer antigens and co-inhibitory signaling molecules identified 20 years ago. Among the large numbers of target antigens, CD19 is the best target for CAR T cell therapy for blood cancer, but CAR-engineered T cell immunotherapy does not yet work in solid cancer. NY-ESO-1 is one of the best targets for TCR-based immunotherapy in solid cancer. Despite the great success of checkpoint blockade therapy, more than 50% of cancer patients fail to respond to blockade therapy. The advent of new technologies such as next-generation sequencing has enhanced our ability to search for new immune targets in onco-immunology and accelerated the development of immunotherapy with potentially broader coverage of cancer patients. In this review, we will discuss the recent progresses of cancer immunotherapy and novel strategies in the identification of new immune targets and mutation-derived antigens (neoantigens) for cancer immunotherapy and immunoprecision medicine.
Similar content being viewed by others
Introduction
Cancer immunotherapy is designed to boost or engineer immune cells (in particular T cells) to fight against cancer cells while leaving normal cells untouched. The host immune system consisting of arms of innate and adaptive immunity plays an essential role in immunosurveillance, recognition and destruction of cancer cells1,2. Among immune cells, tumor-reactive T-cells have been demonstrated to play a direct role in tumor rejection in murine tumor models and human cancers 30 years ago by adoptive transfer experiments3,4,5. More recently, T cell-based immunotherapy has been successfully used to treat many human cancers, such as melanoma, renal cell carcinoma, and lymphoma with varying degrees of tumor regression5,6,7,8. Although CD4+ and CD8+ T cells are the major components of T cell-mediated antitumor immunity, natural killer (NK) and NKT cells may also play a role in immunosurveillance against cancer2,9,10,11,12.
To understand and develop tumor-specific immunity for cancer therapy, great efforts have been made towards identification of cancer antigens recognized by immune cells or antibodies. Using tumor-reactive T cells, many immunogenic tumor antigens have been identified13,14,15,16,17,18,19. Clinical studies using molecularly defined tumor antigens show that cancer vaccines can induce antigen-specific immune responses in patients, but overall immune responses are weak and transient20. More than 100 cancer vaccine clinical trials using proteins, peptides, RNA, antigen-expressing viruses and whole tumor cells have been conducted in the United States21,22,23. In 2010, APCs loaded with a tumor antigen was approved by US Food and Drug Administration (FDA) for the treatment of metastatic prostate cancer. However, because of the expensive cost and limited extension of patient survival of 4.1 months, this FDA-approved vaccine did not have a broad application in the treatment of prostate cancer patients. Thus, therapeutic cancer vaccines with broad application and improved clinical efficacy are urgently needed.
Among the many factors that contribute to the low clinical response rates of cancer vaccines, immune suppression plays a major role. These include checkpoints or roadblocks (cytotoxic T lymphocyte antigen-4 (CTLA-4), programmed cell death-1 (PD-1) and its ligand PD-L1) for T cell activation and function24,25,26, regulatory T cells, other immunosuppressive cells and inhibitory cytokines27,28,29,30,31,32,33. To enhance antitumor immunity, it is necessary to remove these roadblocks so that T cells can be fully activated and functional for the eradication of cancer cells. Importantly, anti-CTLA-4 antibody therapy has shown therapeutic effects on several types of cancer34,35,36,37. The first checkpoint inhibitor, ipilimumab (Yervoy, anti-CTLA-4), was approved by US FDA for the treatment of metastatic melanoma in 2011. More recently, pembrolizumab (Keytruda, anti-PD-1) and Nivolumab were approved for the treatment of metastatic melanoma in 2014. These checkpoint inhibitors have now been approved for the treatment of many types of cancer, including melanoma, lung cancer, bladder cancer and renal cell carcinoma36,38,39. It should be noted that these checkpoint blockade therapies rely on the presence of tumor-specific T cells at tumor sites, which is highly correlated with PD-L1 expression on cancer cells38. Furthermore, recent clinical trials, using antigen-specific T cell receptor (TCR) for the treatment of patients with synovial sarcoma, metastatic melanoma or myeoloma, and CD19-chimeric antigen receptor (CAR) for leukemia and lymphoma, have shown promising clinical responses40,41,42,43,44,45,46,47,48.
Despite these significant progresses of cancer immunotherapy, more than 50% of cancer patients fail to respond to checkpoint blockade therapy. There are only limited numbers of immune targets (checkpoint targets: CTLA-4 and PD-1/PD-L1, and TCR/CAR T cell therapy targets: NY-ESO-1 and CD19) that have demonstrated clinical success, although many immune targets and cancer antigens have been identified. Because of their expression patterns, many of these cancer antigens are not suitable for TCR-based immunotherapy due to potential toxicities associated with self-antigens or cross-reactivity with unanticipated antigens in essential tissues. CD19-CAR T-cell immunotherapy works well in blood cancer, but this CAR-T technology does not yet work well in solid cancer so far. In this article, we intend to discuss recent progresses in the rapid-moving field of cancer immunotherapy, with an emphasis on how to identify cancer-specific antigens, in particular mutation-derived neoantigens, as potential targets for vaccines and TCR-engineered T cell immunotherapy approaches, and how to develop novel strategies for enhancing antitumor immune responses by overcoming immune suppression in the tumor microenvironment.
The principles and unique features of T cell activation and immunotherapy
The goals of cancer immunotherapy are to activate and expand cancer-specific T cells, which will eliminate cancer cells by recognizing antigen targets expressed on them. In the past 25 years, accumulating evidence indicates that the development of cancer-specific immunity requires three sequential signaling events (Figure 1). First, antigen-presenting cells (APCs) such as dendritic cells (DCs) capture and process cancer antigens from tumor cells, and then present antigenic peptides through the complex with major histocompatibility complex (MHC) molecules for recognition by TCR of a T cell (signal 1). In general, antigenic peptides for MHC class I (MHC-I) presentation for CD8+ T cell recognition are approximately 9-amino acid long and presented through human leukocyte antigen (HLA) molecules with closed pocket ends. Antigenic peptides for CD4+ T cell recognition are of various lengths and are presented through MHC class II (MHC-II) molecules with open pocket ends. Second, T cell activation requires further stimulation of the co-stimulatory surface molecules B7 on APCs and CD28 on T cells (signal 2). Thus, both signals 1 and 2 are required to achieve full T cell activation. Conversely, antigenic peptide stimulation (signal 1) in the absence of co-stimulation (signal 2) cannot induce full T cell activation, thus resulting in T cell tolerance. In addition to co-stimulatory molecules, there are also inhibitory molecules, such as CTLA-4 and PD-1, which induce negative signals to prevent T cell activation36,49. Finally, optimal T cell activation is also instructed by innate immune signaling-induced cytokines (so called signal 3). Innate immune response is the first defense system against invading pathogens and cancer cells by sensing pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) through germline-encoded pattern-recognition receptors (PRRs), including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), Nod-like receptors (NLRs), AIM2-like receptors (ALRs), C-type lectin receptors (CLRs) and other DNA sensors50,51,52,53. Antigenic peptide stimulation initiates T cell activation, but the degree of T cell activation is further determined by the balance between co-stimulation and co-inhibitory signaling and proinflammatory cytokines induced by innate immune signaling. Besides T-cell-intrinsic regulation, T cell activation is also controlled by external factors (extrinsic regulation), such as CD4+ regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs), which inhibit the development and function of effector T cells27,30,54. These immunosuppressive cells are usually present in high abundance in the tumor microenvironment and inhibit antitumor immunity. Therefore, blockade of Treg cell-mediated suppression as another checkpoint of immune activation might be critically important for the development of successful immunotherapy. Based on immune targets and checkpoints of T cell activation and regulatory signaling, we propose three types of cancer immunotherapy drugs: 1) cancer vaccines targeting T cell activation signal 1; 2) blockade of co-inhibitory signaling pathways; and 3) blockade of Treg cells- or MDSCs-mediated immunosuppression in the tumor microenvironment.

Signaling and regulatory mechanisms of T cell activation and inhibition. T cell activation requires MHC/peptide-TCR stimulation (as signal 1) and co-stimulatory signaling between B7 and CD28 (as signal 2). Full activation of T cells is instructed by innate-immune signaling-induced cytokines (as signal 3). Meanwhile, T cell activation is regulated and suppressed by co-inhibitory signaling such as B7-CTLA-4 and PD-L1-PD-1 signaling, as well as by regulatory T cells.
The features of cancer immunotherapy are 1) specificity or precise recognition of tumor antigens on cancer cells; 2) memory function that allows generation of long-lasting antitumor immunity; and 3) that major targets of immunotherapy are on immune cells regulating their activation and function, but not directly on tumor cells for destruction. Thus, the goal of cancer immunotherapy is to educate and boost tumor-specific immune cells, which, in turn, recognize and kill tumor cells to achieve therapeutic efficacy of cancer treatment.
Adoptive T cell therapy with tumor-infiltrating lymphocytes (TILs)
Adoptive cell therapy (ACT) has been pioneered by Rosenberg and his co-workers at National Cancer Institute (NCI), NIH, and has been one of the most effective treatments for patients with metastatic melanoma55,56,57. The clinical efficacy of ACT has been dramatically improved by pre-conditioning of patients with a non-myeloablative immunodepleting regimen consisting of cyclophosphamide and fludarabine before T cell infusion58. Objective clinical response rate of 50% has been achieved for patients with metastatic melanoma, who are refractory to all other treatments5,6. This pre-conditioning regimen is critically important because it depletes circulating lymphocytes as well as those at tumor sites for a short period of time before host hematopoietic cell recovery, thus likely removing suppressive Treg cells and allowing antitumor T cells to survive and expand after adoptive T cell transfer. Further modifications such as body irradiation result in the highest objective clinical responses (72%) in all patients receiving the treatment59. However, for many cancer patients ACT with autologous TILs may not be suitable because of variable availability of tumor tissues and difficulties in the successful generation and expansion of TILs from other types of cancer. Nonetheless, tumor-specific TILs have been generated from epithelial cancers, and used for the treatment of cancer patients60. Overall, the success rate of generating TILs from other types of cancer is much lower than that seen in melanoma. Thus, despite the impressive clinical response rate of ACT, it has not been approved as an anticancer immunotherapy drug. Alternative approaches are to identify immune targets recognized by tumor-reactive T cells or to clone tumor-specific TCRs for use in the engineered T cell immunotherapy.
The first wave of tumor antigen discovery using tumor-reactive T cells
Tumor antigens recognized by CD8+ T cells
In parallel to ACT, great efforts have been made in the last 25 years in cancer antigen discovery with goals to develop cancer vaccines. The first human tumor antigen was identified by screening a cDNA expression library with tumor-reactive HLA-restricted CD8+ T cells in 199161. Since then, many tumor antigens have been identified by using T cells with an antitumor activity14,16,62,63 (Table 1). Meanwhile, antibody-reactive tumor antigens have also been identified by serological analysis of expression cDNA libraries (SEREX)64. These tumor antigens can be classified into several types: 1) tissue-specific tumor antigens with higher expression in cancer cells compared with normal cells, which include tyrosinase, MART-1, gp100, TRP-1/gp75 and TRP-2 proteins. 2) Tumor-specific and shared antigens that are expressed in cancer and testis, but not in other normal tissues, which include MAGE-A1 and NY-ESO-1. These antigens are also called cancer-testis (CT) antigens based on their expression patterns (Table 1). However, many CT antigens have been found in other normal tissues with low expression. 3) Tumor-specific and unique antigens that are mutated antigens, including CDK4, catenin and caspase-8 antigens. 4) Overexpressed tumor antigens that are overexpressed in cancer cells compared with in normal cells. Due to the cytotoxic property of CD8+ T cells and with an emphasis on broad and shared antigens for vaccine development, the majority of tumor antigens identified in the last two decades are shared nonmutated antigens (Table 1), with limited numbers of antigens being mutated or unique antigens.
Different mechanisms responsible for T-cell epitope generation
Screening cDNA expression library for tumor antigen discovery with tumor-reactive T cells derived from cancer patients has revealed several distinct mechanisms by which MHC-I-restricted peptides are generated for T cell recognition.
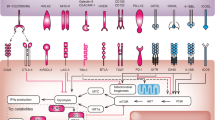
T cell epitopes translated from alternative open reading frames (ORFs) In general, one gene is transcribed into one mRNA (also spliced isoforms for many genes), which then is translated into one polypeptide from the normal ORF. However, in some cases, an mRNA can also be translated into more than one gene products or proteins (Figure 2). Using T cells with in vivo antitumor activity, we found that two gene products were translated from the TRP-1/gp75 cDNA using the normal and alternative ORFs. Importantly, tumor-reactive T cells recognized an antigenic epitope derived from alternative ORF product65,66. This represents the first example of a human cancer antigen that is generated from an alternative ORF and recognized by T cells from cancer patients. A second example is NY-ESO-1, an immunogenic CT antigen independently identified by antibody and T cell screening, respectively67,68. We found that some CTL clones from TIL586 recognized antigenic peptides derived from the primary ORF (ORF1) of CTAG1B (NY-ESO-1), which encodes a protein of 180 amino acids, while other T cell clones failed to recognize peptides from the NY-ESO-1 protein, but were still capable of recognizing NY-ESO-1 cDNA-transfected cells and tumor cells68. Further experiments revealed that these T cells recognized a peptide derived from a 58-amino acid polypeptide translated from NY-ESO-1 ORF2. Thus, NY-ESO-1 mRNA can be translated into two gene products (180 amino acids and 58 amino acids). In addition, LAGE-1, a homolog of NY-ESO-1 with 87% amino acid identity69, was also found to be translated into two gene products. The alternative ORF of LAGE-1, designated as CAMEL, produces T cell epitopes recognized by HLA-A2- and HLA-DR-restricted T cells70,71. T cell epitope from the primary ORF of LAGE-1 is recognized by HLA-DR13-restricted T cells72. Both NY-ESO-1 and LAGE epitopes from primary and alternative ORFs can be efficiently presented for T cell recognition68,72,73,74. Other examples of alternative ORF products as immune targets include M-CSF and intestinal carboxyl esterase75,76. These studies suggest that many proteins may be translated from alternative ORFs in vivo, but the biological significance of these gene products requires further investigation besides their roles as immune targets. It appears that the host immune system not only provides surveillances for gene products from primary ORFs, but also monitors proteins or peptides from alternative ORFs.

T cell epitopes are generated from the primary and alternative open reading frames (ORFs) of mRNAs encoding tyrosinase-related protein 1 (TRP-1), NY-ESO-1, LAGE-1 and LAGE-1b.
T cell epitopes derived from somatic mutations (neoantigens) The first human mutated antigen (neoantigen) was identified from the mutated CDK4 protein in a cDNA expression library screening77. Subsequently, several neoantigens have been reported from mutated β-catenin and CASP 8 proteins78,79 (Table 1). Using an improved sensitive screening assay and multi-specific mixed lymphocyte-tumor cell cultures (MLTCs), five antigens derived from mutations of distinct genes and two nonmutated antigens (tyrosinase and gp100) were identified80. In MLTCs T cell reactivity against neoantigens prevailed over response to nonmutated tumor antigens. TILs with antitumor activity in vivo also recognized two neoantigens derived from mutated growth arrest-specific gene 7 (GAS7) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH)81. It should be noted that identification of neoantigens using cDNA expression screening is not efficient, and the utility of this approach in clinical settings is unclear.
T cell epitopes from intronic sequences or untranslated regions Aberrant RNA splicing could generate different mRNAs or mRNA containing introns, which can be translated into protein products that can be detected by the immune system. Genome-wide analyses of alternative splicing show that 40%-60% of human genes undergo alternative splicing, contributing to functional complexity of the human genome82. T cell epitopes could be generated from an aberrant mRNA or the intron of an incompletely spliced mRNA. For example, a cryptic promoter present in one of the introns of the N-acetylglucosaminyltransferase V (GnT-V) gene is responsible for the generation of an aberrant transcript. A T cell epitope of the gene product of 74 amino acids predicted from the intronic transcript of GnT-V was identified from a cDNA library using HLA-A2-restricted T cells83. Similarly, T-cell epitopes were identified from an intron of an incompletely spliced form of the gp100 RNA (gp100-intron 4) and a partially spliced form of TRP-2 (TRP-2-INT2)84,85. Thus, alternative promoter usage or aberrant splicing events in cancer cells can result in the translation of aberrant mRNAs or intron-containing mRNAs into proteins or peptides that are then presented by MHC class I molecules to T cells. A recent study shows that peptides can be produced for the MHC class I pathway by a translation event occurring before mRNA splicing86. We recently identified a T cell epitope from the untranslated region of an mRNA (unpublished data). These results suggest that the immune system monitors antigen expression and processing beyond the coding regions.
Antigenic peptides derived from protein splicing Several antigenic epitopes have been identified to result from protein splicing148,149,150. In these cases, T cell epitopes are generated from the fusion of the N-terminal and C-terminal peptides, while the central portion of the original peptide is not required for T cell epitope formation. We recently also identified several antigenic peptides that are generated from protein splicing for T cell recognition (unpublished data). Even though proteasome-generated spliced epitopes have been reported, such protein-splicing derived epitopes are generally rare events. Interestingly, a recent study shows that the proteasome-generated spliced peptide pool could account for one-third of the entire HLA class I immunopeptidome in terms of diversity and one-fourth in terms of abundance151. However, it is not clear how many or what percentages of tumor antigens are generated through this mechanism. Nonetheless, proteasome-generated MHC class I-restricted spliced cancer peptides may represent a previously untapped source of epitopes for use in vaccines and cancer immunotherapy151. MHC class II-restricted spliced T cell epitopes from cancer cells have not been reported so far.
Tumor antigens recognized by CD4+ T cells
Given the importance of CD4+ T cells in antitumor immunity, many MHC class II-restricted tumor antigens capable of stimulating CD4+ T helper (Th) cells have been identified9,16. As shown in Figure 3, CD4+ T cells recognize peptides bound to MHC class II molecules on the cell surface of APCs or tumor cells through a multi-step process, which is distinct from MHC class I endogenous antigen presentation and favors presentation of antigens derived from exogenous proteins152. MHC-II α and β molecules form a dimer in endoplasmic reticulum (ER) followed by association with an invariant chain (Ii). Ii chain is a nonpolymorphic type II transmembrane glycoprotein and exists as several isoforms due to alternative splicing and alternative usage of start codons for translation153. Association of Ii with MHC-II αβ molecules prevents antigenic peptide binding in the ER. A targeting sequence in the cytoplasmic tail of Ii is responsible for the transport of nonameric (αβIi)3 complexes from the ER to acidic endosomal/lysosomal-like structures called MHC class II compartments (MIIC)152. Some endogenous antigens can be directly targeted to MIIC or late endosome through lysosomal targeting sequence, while most MHC-II antigens are delivered to MIIC/late endosome through multiple pathways, including phagocytosis, macropinocytosis, endocytosis and autophagy pathways152. Therefore, most proteins expressed from regular cDNA libraries will not reach MIIC/late endosome for antigen processing. MHC class II antigen processing and presentation require products of at least five genes (DRα, DRβ, Ii, DMα, and DMβ) in the specialized MIIC (Figure 3). Ii chain is cleaved in acidic endosome or MIIC, but a 20-amino acid peptide of Ii still remains associated with MHC-II molecules, called CLIP (class II-associated invariant chain peptide). DMα and DMβ promote CLIP release from MHC-II, thus making MHC-II molecules ready for antigenic peptide loading. Furthermore, DM molecules can remove low-affinity peptide from MHC-II molecules, and thus only MHC-II/high-affinity antigenic peptide complexes will be presented on the surface of a cell for T cell recognition (Figure 3). Autologous B cells or DCs derived from the same patient can be used as APCs for transfection with minigene RNAs or loading with purified proteins, but conventional expression systems for endogenous protein expression in cells are not suitable for MHC class II antigen processing and presentation. Due to poor transfection efficiency of primary cells, we produced 293ECIIDR cells expressing DRα, DRβ, DMα, DMβ, and Ii as “professional” APCs. This system has been successfully used to identify many MHC class II-restricted antigens recognized by tumor-reactive T cells (Figure 4 and Table 2).

Processing and presentation of MHC class I and II restricted antigens for CD8+ and CD4+ T cell recognition, respectively. (A) Endogenously expressed proteins are degraded through proteasomal degradation pathways to generate protein fragments, which are further trimmed and processed in ER to generate short (8-10 amino acids) peptides for loading onto MHC class I molecules. MHC-I/peptide complexes are transported to the cell surface for CD8+ T cell recognition. Exogenous proteins or peptides can also be processed and cross-presented for T cell recognition. (B) MHC class II α and β chains form a complex with the invariant chain (Ii) with a targeting sequence to MHC class II compartments (MIIC). The α/β/Ii complexes are transported to the MIIC, where Ii is processed and replaced with an antigenic peptide. Antigenic peptides can be generated from some endogenously expressed proteins with targeting sequence to the MIIC, or proteins targeted to autophagosomes through an autophagy-mediated pathway for protein degradation. Exogenous proteins are generally captured into early endosomes via endocytosis, and then further processed in the MIIC for loading onto MHC class II molecules. MHC-II/peptide complexes are finally transported to the cell surface and presented for recognition by CD4+ T cells.

Three types of mutations or alterations trigger CD4+ T cell responses. (A, B) Mutations in fibronectin (FN) and triosephosphate isomerase (TPI) directly constitute neoepitopes (neoantigens) for T cell recognition. (C) Mutations in CDC27 do not directly give rise to neoepitopes, but rather allow a nonmutated epitope to be processed and presented for T cell recognition. (D) Genetic alteration (chromosomal inversion) generates a fusion protein (LDLR-FUT), which gives rise to a neoepitope from the polypeptide translated from the FUT gene but in an antisense direction.
Mutated antigens recognized by patient-derived CD4+ T cells
New epitopes derived from fusion proteins We have developed the genetic targeting expression (GTE) system for identification of antigens recognized by CD4+ T cells14,154,155. One of these antigens is the fusion gene product LDFP recognized by HLA-DR1-restricted CD4+T cells (Table 2)154. DNA and RNA analyses show that LDFP was generated by fusing a portion of low-density-lipid receptor (LDLR) gene at the 5′ end to a portion of GDP-L-fucose:β-D-galactoside 2-α-L-fucosyltransferase (FUT) gene in an antisense orientation at the 3′ end (Figure 4D). The fusion gene encodes the first five ligand-binding repeats of LDLR in the N-terminus followed by a new polypeptide translated in frame with LDLR from the FUT gene in an antisense direction154. Recently, it has been reported that pathogenic CD4+ T cells in type 1 diabetes recognize epitopes formed by covalent cross-linking of proinsulin peptides to other peptides present in beta cell secretory granules156. These results suggest that fusion proteins play a critical role in elicting immune responses in cancer and other diseases.
Neoantigens derived from somatic mutations One example of mutation-derived neoantigens is a mutated form of triosephosphate isomerase (TPI) identified by both biochemical and GTE approaches (Figure 4B and Table 2)155,157. The somatic point-mutation gives rise to an HLA-DR1-restricted peptide recognized by CD4+ T cells. TPI plays an important role in glycolysis and is essential for efficient energy production, suggesting that the mutated TPI may be involved in cancer cell metabolism. A mutated fibronectin (FN) is another example that mutation-derived epitope is recognized by tumor-reactive CD4+ T cells158. The mutation resulting in the substitution of a Glu residue with Lys creates a neoepitope for T cell recognition (Figure 4A). Interestingly, the mutated FN alters the extracellular matrix and promotes tumor metastasis158.
Nonmutated epitopes derived from mutated antigens The third type of mutated antigens recognized by CD4+ T cells is represented by mutated human CDC27155, an important component of the anaphase-promoting complex involved in cell cycle regulation (Table 2). Sequencing analysis revealed 5 mutations within the CDC27 coding region, resulting in amino acid changes. In this case, the mutations do not constitute a T-cell epitope, but alter protein trafficking and cellular distribution, thus allowing a nonmutated peptide within CDC27 to be processed and presented to T cells (Figure 4C). Therefore, the location or trafficking of a protein can determine whether it is processed and presented for T-cell recognition.
T epitopes derived from mutated mitochondrial proteins Most tumor antigens identified to date are coded by genomic DNA. However, our previous study shows that a mutated mitochondrial protein (cytochrome b), which is coded by mitochondrial DNA, can serve as an antigen recognized by CD4+ T cells159. Notably, this antigen is a translational product of an alternative ORF of cytochrome b mRNA. Since cytochrome b functions as part of the electron transport chain and is the main subunit of transmembrane cytochrome bc1 and b6f complexes, mutations in cytochrome b suggest that energy metabolism may be altered in these cancer cells. Although many mutations in the mitochondrial genome have been identified160, little is known as to whether these mutations trigger immune responses.
Nonmutated epitopes derived from tumor-specific antigens or noncoding RNA Besides mutation-derived neoantigens, cDNA expression library screening allowed us to identify LAGE-1 as a tumor-specific antigen and ARTC1 as an antigenic peptide translated from a noncoding RNA (Table 2)31,161. Several MHC class II-restricted epitopes have been identified by using HLA-DR transgenic (Tg) mice in combination with peptide stimulation in vitro162,163. NY-ESO-1 is one of the most immunogenic cancer antigens recognized by both antibodies and T cells67,68,130. Notably, 10%-13% of patients with advanced cancer developed high titer antibody response against the NY-ESO-1 protein163,164. Importantly, we identified a T-cell epitope in NY-ESO-1 presented by HLA-DP4, a predominant allele expressed in 40%-70% of the population165. Due to the sequence similarity between NY-ESO-1 and LAGE-1, some of the T cell epitopes are shared between these two proteins. Nevertheless, we also identified a unique HLA-DR13-restricted peptide from LAGE-131. Both LAGE-1 and NY-ESO-1 can be recognized by Th1 and Treg cells31,166, suggesting that these cancer antigens can activate Th1 and Treg cells, depending on particular epitope affinity and cytokine milieu in the tumor microenvironment.
Clinical development of cancer antigen-based vaccines and engineered T cell immunotherapy
Cancer vaccine development
Professional APCs such as DCs can potently induce antigen-specific T cell responses. DCs loaded with cancer antigens or tumor lysates have been used to vaccinate cancer patients with limited success. Although the US FDA approved the first therapeutic cancer vaccine drug (Provenge) for the treatment of metastatic castration-resistant prostate cancer in 2010 based on an overall survival benefit of 4.1 months, the sipuleucel-T vaccine comprising blood monocytes loaded with a fusion protein of the prostate antigen prostatic acid phosphatase linked to an immunostimulatory factor granulocyte-macrophage colony-stimulating factor (GM-CSF) failed to show strong clinical therapeutic evidence, such as tumor regression and PSA reduction. Interestingly, some evidence for a therapeutic effect as indicated by tumor growth inhibition and regression was observed in patients who received peptide vaccines182,183. However, objective complete clinical responses were sporadic, even though CTL reactivity was clearly evident after one round of stimulation in vitro of PBMCs from the majority of vaccinated patients20. Analysis of the infiltrating lymphocytes in skin and tumor biopsies using specific peptide-MHC tetramers showed the generation of antigen-specific CD8+ T cells184. A multi-center phase II clinical trial using gp100 peptide with or without IL-2 shows that the gp100 peptide vaccine plus IL-2 group had a significant improvement in centrally verified overall clinical response as compared with the IL-2-only group (16% vs 6%), as well as longer progression-free survival185. MAGE-A3 peptide/protein vaccines were also tested in phases II and III clinical trials186,187,188, but a MAGE-A3 phase III clinical trial failed to meet the projected requirement in patients with resected non-small cell lung cancer. Consistent with these clinical development of cancer vaccines, phase I trials using NY-ESO-1 recombinant protein or synthetic peptides have been conducted in melanoma and ovarian cancer patients189,190,191,192, showing that NY-ESO-1 vaccines can induce antigen-specific immune responses, but are insufficient to eradicate cancer cells. We recently completed a phase I clinical trial to evaluate the safety and feasibility of combined use of MHC class I and class II NY-ESO-1 peptides in metastatic prostate cancer patients. Nine patients were enrolled in this study. We found that peptide vaccines were well-tolerated193. We observed prolonged median PSA doubling time compared with baseline in 6 patients and a decrease in PSA level in 2 patients. Strong NY-ESO-1-specific T cell responses were observed in 6 of the 9 patients193. These encouraging clinical results provide great impetus to investigate whether antitumor immunity induced by NY-ESO-1 peptide vaccines can be further enhanced by a combination therapy with anti-PD-1 or by blocking immune suppression of Treg cells.
CAR-engineered T cell immunotherapy
In light of the sporadic and limited success of cancer vaccines, recent success in the CAR-engineered T cell immunotherapy targeting the B cell lineage differentiation antigen CD19 in B cell malignancies has provided new opportunities to treat cancer. Since the first report of CD19-CAR T cell therapy48, this immunotherapy strategy has been successfully extended to treating patients with many types of chemotherapy-refractory B cell malignancies, including acute lymphoblastic B cell leukemias (ALLs), chronic lymphocytic leukemias (CLLs), aggressive B cell lymphoma, and marginal zone lymphoma41,43,194,195. A major toxicity associated with CAR T cell therapy is cytokine release syndrome (CRS) resulting from an exuberant release of cytokines such as IFN-γ and IL-6 from infused T cells when they encounter CD19-positive tumor cells46,47. Clinical symptoms of CRS include high fevers, kidney failure, electrolyte abnormalities, hypotension, cardiac dysfunction, and seizures. Clinical toxicity associated with CD19-CAR therapy could be managed with anti-IL-6 antibody or immune suppression agents. Alternatively, Lim and his coworkers have designed “ON-switch” CARs that enable small-molecule control over T cell therapeutic functions while still retaining antigen specificity196. In these split constructs, antigen-binding and intracellular signaling components are separated without a dimerization agent, and then assembled together only in the presence of a heterodimerization-promoting small molecule, thus allowing one to precisely control the timing, location, and dosage of T cell activity for controlling potential toxicity196. Furthermore, several new designs of the CAR technology based on a synthetic Notch receptor system have been reported to enable T cell activation through recognition of combinational antigens, and to allow engineering of T cells with customized therapeutic response programs197,198,199. These studies illustrate the potential of combining cellular engineering with synthetic biology tools to produce safer therapeutic CAR-T cells for cancer immunotherapy. CAR T immunotherapy is extensively reviewed by a separate article in this issue, and thus is only briefly discussed here. Although CAR T cell immunotherapy works well in many types of blood cancer, there are two major issues that remain to be resolved. First, approximately 40%-50% of patients who received CD19-CAR T cell therapy will have recurrence within one year, and the underlying mechanisms remain unclear. The second issue is that CAR T cell therapy does not yet work well in solid cancers, even though many CAR T cell therapies target surface molecules expressed by solid cancers46. Thus, immune suppression in the tumor microenvironment is a major obstacle for T-cell-based immunotherapy.
TCR-engineered T cell immunotherapy
As mentioned above, NY-ESO-1 is one of the best cancer antigens for cancer immunotherapy; clinical trials using NY-ESO-1 peptides, proteins and viruses encoding NY-ESO-1 show that NY-ESO-1-specific T cell responses can be readily induced in cancer patients and are safe, but not sufficient to eliminate cancer. To improve clinical response, recent clinical trials using HLA-A2-restricted NY-ESO-1 TCR-transduced T cells showed clinical response rates of 55%-60% in metastatic synovial sarcoma, melanoma, and myeloma without toxicity40,44,200, suggesting that adoptive transfer of a large number of NY-ESO-1-specific TCR-transduced T cells can produce therapeutic immunity and mediate tumor regression. A previous study shows that adoptive transfer of HLA-DP4-restricted NY-ESO-1-specific CD4+ T cells results in tumor regression in a metastatic melanoma patient201. Indeed, a recent study shows that adoptive transfer of mutation-specific CD4+ T cells results in the regression of metastatic cholangiocarcinoma (an epithelial cancer) in a cancer patient202, suggesting that tumor-specific CD4+ T cells are capable of mediating tumor regression in epithelial cancers.
Despite robust antitumor immunity using TCRs targeting diverse cancer antigens, there are severe unanticipated adverse effects resulting from T cell recognition of tissue-specific antigens gp100 and MART-1 expressed by both normal melanocytes and melanoma203, or from the cross-reactivity of higher affinity MAGE-A3 TCR-transduced T cells against epitopes in unintended targets in normal and essential tissues204,205. Vaccination of cancer patients with gp100 and MART-1 resulted in objective clinical response without serious toxicity to melanocytes182,185. However, clinical studies using high-affinity MART-1 TCR-engineered T cells showed severe toxicity, including the destruction of normal melanocytes in the skin, eye, and ear, and sometimes requiring local steroid administration to treat uveitis and hearing loss203, compared with patient-derived TCR-engineered T cell therapy206. Similarly, serious toxicity was not observed with endogenous MAGE-A3-reactive T cells from cancer patients or patients vaccinated with a MAGE-A3 protein188,207. MAGE-A3 is highly expressed in cancer cells, but not in normal cells. Notably, other members of MAGE family proteins such as MAGE-A12 show low levels of expression in normal tissues including the brain. HLA-A2-restricted affinity-enhanced MAGE-A3 TCR-engineered T cell therapy showed unanticipated toxicities, including mental status changes, comas and subsequent death; further experiments reveal that the engineered T cells have cross-reactivity with MAGE-A12 and MAGE-A9204, which are weakly expressed in the brain tissue. In another clinical trial using HLA-A1-restricted high-affinity MAGE-A3 TCR, two patients died of cardiovascular-related issues due to cross-reactivity with a peptide derived from the muscle-specific protein TITIN205. Thus, while cancer vaccines using tissue-specific antigens or CT antigens such as MAGE-A3 are safe, but not potent enough to eradicate cancer, clinical studies using affinity-enhanced TCRs show the increased ability to recognize target tumor cells, but they also increase the risks of self-destruction of normal tissues or cross-reactivity against unanticipated targets expressed in essential normal tissues. For these reasons, it is better to include an inducible safety cassette or a regulatory switch in these TCR constructs, as demonstrated in CAR-based constructs196,197,208.
Based on its expression profile and clinical studies, NY-ESO-1 is considered one of the best targets identified for solid cancer immunotherapy with NY-ESO-1 TCR-engineered T cells. Although NY-ESO-1 is widely expressed in many types of solid cancer, the expression frequency in melanoma, prostate cancer, triple-negative breast cancer, liver cancer and lung cancer is in the range of 20%-40%. It is not frequently expressed in gastric, colon and pancreatic cancers. Therefore, it is an urgent need to develop therapeutic vaccines that can target many tumor antigens and augment antitumor immunity to eliminate cancer cells by acting alone or in combination with checkpoint blockade. Alternatively, we need to identify more cancer-specific neoantigens for cancer immunotherapy, as described below.
The second wave of tumor antigen discovery: mutation-derived neoantigens
In the last 25 years, tumor immunologists have paid more attention to nonmutated and shared cancer antigens than mutation-derived neoantigens for the development of broad-coverage cancer vaccines. Several mutation-derived neoantigens such as mutant CDK4, β-catenin and caspase-8 serving as targets for CD8+ T cell recognition of tumor cells have been documented long time ago. Similarly, our group has identified an array of mutated antigens from human cancer as targets recognized by CD4+ T cells through the screening of a tumor-derived cDNA expression library with T cells. The majority of tumor-reactive CD4+ T cells recognize mutated antigens. However, how to rapidly identify neoantigens and how to use them as immunotherapy targets remain an important challenge because of their private and patient-specific nature.
Recent large-scale sequencing projects have identified many mutations and genetic alterations in major cancer types and provided a comprehensive view of somatic mutational landscapes in cancer and potential therapeutic targets for cancer treatment209,210,211,212,213,214,215. It appears that the frequencies of somatic mutations vary widely among tumor types ranging from very few mutations in blood cancer in children to very high mutation levels in solid cancer, such as lung cancer, colon cancer, and melanoma209,210. Analysis of exome sequencing data from breast and colon cancers predicted that many mutations could be presented as neoantigens for T cell recognition216,217. Notably, not all mutations can be processed for T cell recognition. Proteins containing somatic mutations must be processed to short peptides (generally 9 amino acids for MHC-I and variable length (average 13-15 amino acids) for MHC-II) to be presented by MHC molecules on the cell surface for T cell recognition (Figure 3). Thus, whether mutated peptides can be recognized by T cells depends on several factors: 1) whether mutated peptides can be naturally processed to a short peptide with a specific MHC-binding motif; 2) the binding affinity of mutated peptides to MHC molecules (peptide/MHC density on the cell surface); and 3) the affinity of TCR binding to mutated peptide/MHC complexes.
Using exome sequencing and computer-assisted epitope prediction, several neoantigens have been identified from B16 melanoma, methylcholanthrene-induced sarcoma and a transgenic tumor model218,219,220. These studies demonstrate that neoantigens can be rapidly identified within a few weeks and can serve as tumor-specific targets for T cell-mediated recognition and destruction of tumor cells. Similarly, several neoantigens have been identified from human cancer recognized by CD8+ and CD4+ T cells through exome sequencing and screening for T cell responses221,222,223. More recently, it has been further demonstrated that neoantigen-specific T cells can be isolated from patient PBMCs using FACS analysis and purification, although their frequencies are much lower than neoantigen-specific T cells in the corresponding TIL populations45,224. These studies raise the possibility to obtain neoantigen-specific T cells from patient's blood for personalized immunotherapy and precision cancer medicine (Figure 5).

A schematic presentation of development of neoantigen-specific immunotherapy. Tumor samples and normal control tissues are obtained from a cancer patient for genomic DNA and RNA isolation. Whole-exome sequencing and bioinformatics analysis allows identification of non-synonymous mutations present in the tumor cells, but not in normal cells. RNA-seq will determine the expression profile of the mutated genes in cancer cells. Protein sequences with amino acid substitutions will be used to generate neoantigen-containing long peptides or tandem minigenes (TMGs), which are used to pulse autologous antigen presenting cells (APCs) in the form of peptides or transfected into autologous APCs or HLA-matched APCs. T cells obtained from the same patient will be used to screen against peptides-pulsed or TMGs-transfected APCs based on cytokine release from T cells or upregulation of activation makers (PD-1, 4-1BB and OX-40) on T cells after co-culture of T cells with APCs. Meanwhile, a single T cell can be isolated and tested for its reactivity against newly identified neoantigens. Neoantigen-specific T cells can be expanded for adoptive T cell therapy, as demonstrated for ERBB2IP (E805G) and KRAS (G12D)60,202,240, or used to identify neoantigen-specific T cell receptors (TCRs). These neoantigen-specific TCRs are cloned into expression vectors for further engineering of T cells in TCR-based immunotherapy. In addition, neoantigens can be used in synthetic (peptide, DNA and RNA) vaccines to elicit therapeutic immunity against cancer.
Neoantigen-specific T cells may play a critical role in tumor regression observed in checkpoint blockade therapy, which relies on the presence of tumor-infiltrating T cells at tumor sites. Using next-generation sequencing, peptide motif prediction and mass spectrometry validation, one study reports the identification of three neoantigens from MC38 tumor cells and demonstrates that vaccines comprising these neoantigens plus polyinosinic:polycytidylic acid (poly(I:C)) and anti-CD40 induce therapeutic immunity225. In another study, two neoantigens were identified from MCA sarcoma cells. Vaccines containing these two neoantigens and poly(I:C) induced protective immunity against subsequent MCA sarcoma challenge, and generated therapeutic immunity in combination with anti-PD-1 and anti-CTLA-4 treatment226. Similarly, MHC class II-restricted neoantigens have been used to generate therapeutic immunity against neoantigen-expressing cancers using peptide- or RNA-based vaccine strategy227,228,229. Mutation-derived neoantigens may either alter the binding affinity to MHC molecules or the binding affinity of mutated peptide/MHC complexes to TCR. It is not clear what kind of mutations could generate therapeutic immunity. However, a recent study suggests that neoantigens with improved binding affinity to MHC alleles likely can generate protective immunity230. To gain better understanding of therapeutic immunity in human cancer patients, a recent study identified several neoantigens from melanoma tissues collected at different time points. After vaccination with DCs loaded with neoepitope peptides, several neoantigen-specific T cell populations could be detected and expanded from the PBMCs of melanoma patients231. However, clinical responses in these melanoma patients were not presented. Although there is no direct evidence that neoantigen-specific T cells mediate tumor regression in the vaccine study or checkpoint blockade therapy, several studies show that mutational landscapes of cancer patients correlate with clinical response rates in checkpoint blockade therapies232,233, suggesting that cancer cells with more mutations may generate more targets for T cell induction and recognition, thus becoming more immunogenic. Furthermore, T cells recognizing clonal neoantigens can be detected in patients with durable clinical benefit of checkpoint blockade234. These studies indicate that mutation-derived neoantigens may play an important role in inducing tumor-specific immune responses in checkpoint blockade immunotherapy.
Identifying neoantigens recognized by CD8+ T cells
Whole-exome sequencing of cancer cells and normal cells is the first step to identify somatic mutations. In general, surgically removed tumor tissues and normal cells such as PBMCs from the same patient are used for constructing whole-exome library and subjected to deep sequencing, which permits the identification of thousands of mutations and human variations against the 1000 human genome databases. Further direct comparison of tumor and normal cells will eliminate potential human variations presented in both cancer and normal cells. After further filtering analysis, true somatic mutations that are present in cancer cells, but not in normal cells will be identified (Figure 5). RNA sequencing of tumor cells will determine whether the mutated genes are expressed in cancer cells. For a mutation to be presented for T cell response, a mutated protein must be expressed with amino acid changes due to non-synonymous mutations, and processed into a short peptide with high binding affinity to one of the patient's MHC molecules. Once somatic mutations are identified, these mutation-containing proteins will be analyzed by computer-assisted epitope prediction program to identify high-affinity binding mutated peptides as potential candidate neoantigens (Figure 5). In subsequent validation steps, tumor-reactive T cells are the key to screening and identification of T cell-recognized neoantigens (Table 3). TILs can be isolated from freshly obtained tumor tissues and used for screening candidate neoantigens221. Alternatively, neoantigen-specific T cells may be isolated from the patient's PBMCs based on upregulation of activation markers such as 4-1BB, OX-40 and PD-1 on T cells224,235. For screening of neoantigens, long peptides that contain a mutated amino acid can be pulsed onto autologous APCs, which are then co-cultured with T cells to determine their reactivity based on cytokine release or upregulation of co-stimulatory molecules. Alternatively, minigenes encoding the mutated peptides are transfected into autologous APCs, and tested for T cell recognition60,202. However, neoantigens could not be defined if the patient's T cells fail to recognize candidate neoantigens or if tumor-reactive T cells are not available. To overcome the limitation of poor T cell activity or unavailability of tumor-reactive T cells, we previously used in vitro peptide stimulation of cells from healthy donors to define immunogenic peptides with strong ability to stimulate CD4+ or CD8+ T cell responses236,237,238. More recently, this approach has been used to identify immunogenic neoantigens after in vitro stimulation of HLA class I-matched donor T cells with predicted neoepitopes239. Hence, if TILs or tumor-reactive T cells cannot be obtained from cancer patients, one can use the HLA-matched donor T cells to identify immunogenic neoepitopes after stimulation of T cells with predicted neoepitopes. An alternative approach is to elute neoantigen peptides from tumor cells obtained from the same patient, followed by reversed-phase HPLC fractionation and mass spectrometry (MS) analysis. If candidate neoantigens identified by exome sequencing and HLA-binding motif prediction are present in the pool of MS-identified peptides, it is likely that these candidate neoantigens are immunogenic225. However, definitive evidence for such neoantigens still requires T cell recognition (Figure 5). Some of the neoantigens recognized by tumor-reactive T cells from TILs or PBMCs are summarized in Table 3.
Identifying neoantigens recognized by CD4+ T cells
In a recent study, neoantigens recognized by CD4+ T cells were identified by exome sequencing and screening with peptide-loaded B cells223; however, the use of TILs from three melanoma patients identified only a few (0-3) neoantigens, even though 125-312 mutations were identified in their tumor cells223. In a second study, two neoantigens were identified by screening tandem minigenes with CD4+ T cells, using minigene RNA-transduced B cells or DCs202. Furthermore, several neoantigens have been identified from human gastrointestinal cancers60. Based on these studies, it appears that the prediction program for MHC class II-restricted peptides is not accurate. Therefore, relatively fewer neoantigens recognized by CD4+ T cells have been identified so far. To overcome this issue, we recently developed a strategy to identify many neoantigens recognized by CD4+ T cells: we constructed a minigene library based on somatic mutations identified by whole-exome sequencing analysis, and screened the library with tumor-reactive TILs or T cell clones in an engineered 293-based APC system expressing mutation-containing minigenes (unpublished data).
Advantages and limitations of using neoantigens for cancer immunotherapy
Immunotherapy drugs including anti-CTLA-4 (ipilimumab/Yervoy®) and anti-PD-1 (pembrolizumab/Keytruda®, Nivolumab/Opdivo®) have been approved for the treatment of metastatic melanoma, lung cancer, and other malignancies. However, more than 50%-80% cancer patients fail to respond to checkpoint inhibitor therapy. Furthermore, TCR-based immunotherapy is currently limited to those expressing NY-ESO-1 and HLA-A2 molecules. For many types of solid cancer, alternative immunotherapy is not available for those who do not respond to checkpoint inhibitor therapy or those whose tumors do not present suitable targets. Given the cost-effective nature of next-generation sequencing, identification of genetic mutations is becoming a routine practice, thus providing an exceptional opportunity to develop personalized immunotherapy for cancer treatment. Uncovering cancer-specific neoantigens not only provides therapeutic targets for immunotherapy, but also can reveal the molecular mechanisms by which checkpoint blockade therapy activates neoantigen-specific T cells for clinical benefit. Cancer-specific mutated antigens are present in every tumor type regardless of the mutation frequency of the tumor, some of which can be identified as neoantigens recognized by the patient's own T cells. Therefore, rapid identification of neoantigens would provide excellent targets for personalized immunotherapy. In this regard, a wave of companies are developing personalized vaccines based on tumor-specific neoantigens, thus providing a timely boost for an important but failed area of cancer immunotherapy241. One key challenge is how to rapidly identify neoantigens recognized by T cells and how to select candidates from the many identified neoantigens for vaccines or T cell immunotherapy. There are many limitations associated with neoantigen-based immunotherapy. First of all, the whole-exome sequencing-based approach will miss many immunogenic antigens, including cancer-specific and nonmutated shared antigens such as NY-ESO-1 and LAGE-1, as well as antigenic peptides derived from alternative ORFs and untranslated regions of mRNAs, incompletely spliced transcripts and introns, noncoding RNAs and mitochondrial proteins. Therefore, whole-genome sequencing plus RNA sequencing, and other approaches may be used to identify cancer-specific shared antigens for the broad coverage of cancer patients by immunotherapy. Secondly, because of the private or unique nature of neoantigens, it is necessary to identify neoantigens for each patient individually. Thus, high throughput and robust methods such as more reliable and accurate prediction programs are needed. Thirdly, once neoantigens have been identified for a specific patient, we need to develop neoantigen-specific vaccines using RNA, DNA or peptides, or generate neoantigen-specific TCR-based immunotherapy. Development of such personalized immunotherapies is very expensive, and requires new regulatory guidelines from the FDA. It is ideal to target multiple neoantigens with vaccines or TCR-based approach for clinical benefits, even though clinical studies show that therapy targeting a single target such as NY-ESO-1 TCR- or CD19-CAR-engineered T cell therapy can induce durable clinical responses. We recently observed that a single TCR can recognize multiple neoantigens, but not their wild-type counterparts, raising the possibility that a single or few TCRs can cover several neoantigens and recognize heterogenetic populations of cancer cells (unpublished data). Thus, cancer immunotherapy should consider both cancer-specific shared antigens and patient-specific unique neoantigens. Identification of neoantigens from large numbers of patients may lead to the discovery of shared neoantigens among certain groups of cancer patients. Establishment of neoantigen-specific TCR banks may allow one to cover the majority of cancer patients by “off the shelf” immunotherapy. Although further studies are needed to address these issues in the near future, it is certain that identification of both cancer-specific shared antigens and unique neoantigens will be the key to the development of immunotherapy for many types of cancers.
Concluding remarks
In the last 25 years, significant progress has been made in the field of cancer immunotherapy. In particular, several immunotherapy drugs have been approved for the treatment of several types of cancer with impressive and durable clinical responses. With encouraging results from CD19-CAR- and NY-ESO-1 TCR-engineered T cell immunotherapy, major university cancer centers and companies have joined the force to develop more effective immunotherapies for many types of cancer. It is projected that immunotherapy will become the mainstream of cancer treatment in the near future. The major challenges are to broaden cancer populations responding to checkpoint blockade therapy, and to target shared cancer antigens and unique neoantigens for the development of cancer vaccines or CAR and TCR immunotherapy. The key issues will be how to direct the trafficking of tumor-reactive T cells to tumor sites and to enable their long-term persistence for tumor destruction. To achieve these goals, 1) we need to develop methods for in vivo targeted delivery of peptides and RNAs into APCs such as DCs for inducing tumor-specific T cell responses; 2) it is critically important to overcome immune suppression mediated by Treg cells and other immune cells, besides the negative signaling of PD-1 and PD-L1; 3) importantly, shared antigen- or neoantigen-specific vaccines should combine with strategies that aim to overcome negative regulators or immune suppression to achieve maximal antitumor immunity and clinical responses; 4) finally, it is important to prolong T cell persistence through epigenetic and metabolic reprograming of T cells for cancer immunotherapy. Therefore, it is reasonable to believe that the combined use of immunotherapy with innate immune stimulation, chemotherapy or radiation therapy may enhance immune responses and clinical outcomes. The recent advances and rapid progress in the field represent an unprecedented opportunity for the development of cancer immunotherapy by offering the possibility to target novel immune targets and neoantigens for every cancer patient in the next few years.
References
Schreiber RD, Old LJ, Smyth MJ . Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565–1570.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ . Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011; 29:235–271.
Rosenberg SA . Adoptive immunotherapy for cancer. Scientific American (May) 1990; 262:62–69.
Greenberg PD . Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol 1991; 49:281–355.
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME . Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 2008; 8:299–308.
Rosenberg SA . Cell transfer immunotherapy for metastatic solid cancer — what clinicians need to know. Nat Rev Clin Oncol 2011; 8:577–585.
Lesterhuis WJ, Haanen JB, Punt CJ . Cancer immunotherapy — revisited. Nat Rev Drug Discov 2011; 10:591–600.
Tey SK, Bollard CM, Heslop HE . Adoptive T-cell transfer in cancer immunotherapy. Immunol Cell Biol 2006; 84:281–289.
Wang RF . The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends in Immunology 2001; 22:269–276.
Diefenbach A, Raulet DH . Strategies for target cell recognition by natural killer cells. Immunol Rev 2001; 181:170–184.
Childs RW, Carlsten M . Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov 2015; 14:487–498.
Heczey A, Liu D, Tian G, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014; 124:2824–2833.
Boon T, Cerottini JC, Van Den Eynde B, Van der Bruggen P, Van Pel A . Tumor antigens recognized by T lymphocytes. Annu Rew Immunol 1994; 12:337–365.
Wang RF, Rosenberg SA . Human tumor antigens for cancer vaccine development. Immunol Rev 1999; 170:85–100.
Finn OJ . Vaccines for cancer prevention: a practical and feasible approach to the cancer epidemic. Cancer Immunol Res 2014; 2:708–713.
Wang HY, Wang RF . Enhancing cancer immunotherapy by intracellular delivery of cell-penetrating peptides and stimulation of pattern-recognition receptor signaling. Adv Immunol 2012; 114:151–176.
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T . Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014; 14:135–146.
Hinrichs CS, Restifo NP . Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol 2013; 31:999–1008.
Novellino L, Castelli C, Parmiani G . A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother 2005; 54:187–207.
Rosenberg SA, Yang JC, Restifo NP . Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10:909–915.
Goldman B, DeFrancesco L . The cancer vaccine roller coaster. Nat Biotechnol 2009; 27:129–139.
Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH . Therapeutic cancer vaccines. J Clin Invest 2015; 125:3401–3412.
Melero I, Gaudernack G, Gerritsen W, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 2014; 11:509–524.
Callahan MK, Wolchok JD, Allison JP . Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin Oncol 2010; 37:473–484.
Chambers CA, Kuhns MS, Egen JG, Allison JP . CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 2001; 19:565–594.
Zhu Y, Yao S, Chen L . Cell surface signaling molecules in the control of immune responses: a tide model. Immunity 2011; 34:466–478.
Wang HY, Wang RF . Regulatory T cells and cancer. Curr Opin Immunol 2007; 19:217–223.
Joyce JA, Fearon DT . T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015; 348:74–80.
Ugel S, De Sanctis F, Mandruzzato S, Bronte V . Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 2015; 125:3365–3376.
Marvel D, Gabrilovich DI . Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 2015; 125:3356–3364.
Wang HY, Lee DA, Peng G, et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implication for immunotherapy. Immunity 2004; 20:107–118.
Peng G, Wang HY, Peng W, Kiniwa Y, Seo K, Wang RF . Tumor-infiltrating gamma-delta T cells suppress T and dendritic cell function via mechanisms controlled by a unique Toll-like receptor signaling pathway. Immunity 2007; 27:334–348.
Sakaguchi S . Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 2004; 22:531–562.
Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010; 33:828–833.
Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723.
Sharma P, Allison JP . The future of immune checkpoint therapy. Science 2015; 348:56–61.
Buchbinder E, Hodi FS . Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest 2015; 125:3377–3383.
Chen L, Han X . Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015; 125:3384–3391.
Topalian SL, Drake CG, Pardoll DM . Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015; 27:450–461.
Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011; 29:917–924.
Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra225.
Rosenberg SA, Restifo NP . Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015; 348:62–68.
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507–1517.
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015; 21:914–921.
Klebanoff CA, Rosenberg SA, Restifo NP . Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 2016; 22:26–36.
Jackson HJ, Rafiq S, Brentjens RJ . Driving CAR T-cells forward. Nat Rev Clin Oncol 2016; 13:370–383.
Sadelain M . CAR therapy: the CD19 paradigm. J Clin Invest 2015; 125:3392–3400.
Porter DL, Levine BL, Kalos M, Bagg A, June CH . Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725–733.
Chen L, Flies DB . Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13:227–242.
Goubau D, Deddouche S, Reis e Sousa C . Cytosolic sensing of viruses. Immunity 2013; 38:855–869.
Takeuchi O, Akira S . Pattern recognition receptors and inflammation. Cell 2010; 140:805–820.
Cui J, Chen Y, Wang HY, Wang RF . Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother 2014; 10:3270–3285.
Paludan SR, Bowie AG . Immune sensing of DNA. Immunity 2013; 38:870–880.
Wang M, Yin B, Wang HY, Wang RF . Current advances in T-cell-based cancer immunotherapy. Immunotherapy 2014; 6:1265–1278.
Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst 1994; 86:1159–1166.
Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. Preliminary report. N Engl J Med 1988; 319:1676–1680.
Rosenberg SA . Interleukin-2 and the development of immunotherapy for the treatment of patients with cancer. Cancer J Sci Am 2000; 6 Suppl 1:S2–S7.
Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer Regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298:850–854.
Wrzesinski C, Paulos CM, Kaiser A, et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother 2010; 33:1–7.
Tran E, Ahmadzadeh M, Lu YC, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015; 350:1387–1390.
Van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytiv T lymphocytes on a hunan melanoma. Science 1991; 254:1643–1647.
Renkvist N, Castelli C, Robbins PF, Parmiani G . A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother 2001; 50:3–15.
Vigneron N, Stroobant V, Van den Eynde BJ, van der Bruggen P . Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun 2013; 13:15.
Sahin U, Tureci O, Schmitt H, et al. Human neoplasms elicit multiple immune responses in the autologous host. Proc Natl Acad Sci USA 1995; 92:11810–11813.
Wang RF, Robbins PF, Kawakami Y, Kang XQ, Rosenberg SA . Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J Exp Med 1995; 181:799–804.
Wang RF, Parkhurst MR, Kawakami Y, Robbins PF, Rosenberg SA . Utilization of an alternative open reading frame of a normal gene in generating a novel human cancer antigen. J Exp Med 1996; 183:1131–1140.
Chen YT, Scanlan MJ, Sahin U, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA 1997; 94:1914–1918.
Wang RF, Johnston SL, Zeng G, Schwartzentruber DJ, Rosenberg SA . A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol 1998; 161:3596–3606.
Lethe B, Lucas S, Michaux L, et al. LAGE-1, a new gene with tumor specificity. Int J Cancer 1998; 76:903–908.
Aarnoudse CA, van den Doel PB, Heemskerk B, Schrier PI . Interleukin-2-induced, melanoma-specific T cells recognize CAMEL, an unexpected translation product of LAGE-1. Int J Cancer 1999; 82:442–448.
Slager EH, Borghi M, van der Minne CE, et al. CD4+ Th2 cell recognition of HLA-DR-restricted epitopes derived from CAMEL: a tumor antigen translated in an alternative open reading frame. J Immunol 2003; 170:1490–1497.
Wang HY, Deen A. Lee, Guangyong Peng, et al. Tumor-specific human CD4+ regulatory T cells and their ligands: implication for immunotherapy. Immunity 2004; 20:107–118.
Rimoldi D, Rubio-Godoy V, Dutoit V, et al. Efficient simultaneous presentation of NY-ESO-1/LAGE-1 primary and nonprimary open reading frame-derived CTL epitopes in melanoma. J Immunol 2000; 165:7253–7261.
Valmori D, Dutoit V, Lienard D, et al. Naturally occurring human lymphocyte antigen-A2 restricted CD8+ T-cell response to the cancer testis antigen NY-ESO-1 in melanoma patients. Cancer Res 2000; 60:4499–4506.
Probst-Kepper M, Stroobant V, Kridel R, et al. An alternative Open Reading Frame of the Human Macrophage Colony- stimulating Factor Gene Is Independently Translated and Codes for an Antigenic Peptide of 14 Amino Acids Recognized by Tumor-infiltrating CD8 T Lymphocytes. J Exp Med 2001; 193:1189–1198.
Ronsin C, Chung-Scott V, Poullion I, Aknouche N, Gaudin C, Triebel F . A non-AUG-defined alternative open reading frame of the intestinal carboxyl esterase mRNA generates an epitope recognized by renal cell carcinoma-reactive tumor-infiltrating lymphocytes in situ. J Immunol 1999; 163:483–490.
Wolfel T, Hauer M, Schneider J, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995; 269:1281–1284.
Robbins PF, El-Gamil M, Li YF, et al. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med 1996; 183:1185–1192.
Mandruzzato S, Brasseur F, Andry G, Boon T, van der Bruggen P . A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J Exp Med 1997; 186:785–793.
Lennerz V, Fatho M, Gentilini C, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA 2005; 102:16013–16018.
Zhou J, Dudley ME, Rosenberg SA, Robbins PF . Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J Immunother 2005; 28:53–62.
Modrek B, Lee C . A genomic view of alternative splicing. Nat Genet 2002; 30:13–19.
Guilloux Y, Lucas L, Brichard VG, et al. A peptide recognized by human cytolytic T lymphocytes on HLA-A2 melanomas is encoded by an intron sequence of the N-acetylglucosaminyltransferase V gene. J Exp Med 1996; 183:1173–1183.
Robbins P, El-Gamil M, Li YF, Fitzgerald E, Kawakami Y, Rosenberg SA . The intronic region of an incompletely spliced gp100 gene transcript encodes an epitope recognized by melanoma-reactive tumor-infiltrating lymphocytes. J Immunol 1997; 159:303–308.
Lupetti R, Pisarra P, Verrecchia A, et al. Translation of a retained intron in tyrosinase-related protein (TRP) 2 mRNA generates a new cytotoxic T lymphocyte (CTL)-defined and shared human melanoma antigen not expressed in normal cells of the melanocytic lineage. J Exp Med 1998; 188:1005–1016.
Apcher S, Millot G, Daskalogianni C, Scherl A, Manoury B, Fahraeus R . Translation of pre-spliced RNAs in the nuclear compartment generates peptides for the MHC class I pathway. Proc Natl Acad Sci USA 2013; 110:17951–17956.
Bakker ABH, Schreurs MWJ, de Boer AJ, et al. Melanocyte lineage-specific antigen gp100 is recognized by melanocyte-derived tumor-infiltrating lymphocytes. J Exp Med 1994; 179:1005–1009.
Tsai V, Southwood S, Sidney J, et al. Identification of subdominant CTL epitopes of the GP100 melanoma-associated tumor antigen by primary in vitro immunization with peptide-pulsed dendritic cells. J Immunol 1997; 158:1796–1802.
Kawakami Y, Eliyahu S, Jennings C, et al. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol 1995; 154:3961–3968.
Kawakami Y, Eliyahu S, Delgaldo CH, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA 1994; 91:3515–3519.
Cox AL, Skipper J, Cehn Y, et al. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science 1994; 264:716–719.
Kawakami Y, Robbins PF, Wang X, et al. Identification of new melanoma epitopes on melanosomal proteins recognized by tumor infiltrating T lymphocytes restricted by HLA-A1, - A2, and -A3 alleles. J Immunol 1998; 161:6985–6992.
Kawashima I, Tsai V, Southwood S, Takesako K, Celis E, Sette A . Identification of gp100-derived, melanoma-specific cytotoxic T- lymphocyte epitopes restricted by HLA-A3 supertype molecules by primary in vitro immunization with peptide-pulsed dendritic cells. Int J Cancer 1998; 78:518–524.
Skipper JC, Kittlesen DJ, Hendrickson RC, et al. Shared epitopes for HLA-A3-restricted melanoma-reactive human CTL include a naturally processed epitope from Pmel-17/gp100. J Immunol 1996; 157:5027–5033.
Sensi M, Pellegatta S, Vegetti C, Nicolini G, Parmiani G, Anichini A . Identification of a novel gp100/pMel17 peptide presented by HLA-A*6801 and recognized on human melanoma by cytolytic T cell clones. Tissue Antigens 2002; 59:273–279.
Benlalam H, Linard B, Guilloux Y, et al. Identification of five new HLA-B*3501-restricted epitopes derived from common melanoma-associated antigens, spontaneously recognized by tumor-infiltrating lymphocytes. J Immunol 2003; 171:6283–6289.
Castelli C, Tarsini P, Mazzocchi A, et al. Novel HLA-Cw8-restricted T cell epitopes derived from tyrosinase-related protein-2 and gp100 melanoma antigens. J Immunol 1999; 162:1739–1748.
Coulie PG, Brichard V, Van Pel A, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med 1994; 180:35–42.
Kawakami Y, Eliyahu S, Sakaguchi K, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2 restricted tumor infiltrating lymphocytes. J Exp Med 1994; 180:347–352.
Castelli C, Storkus WJ, Maeurer MJ, et al. Mass spectrometric identification of a naturally processed melanoma peptide recognized by CD8+ cytotoxic T lymphocytes. J Exp Med 1995; 181:363–368.
Schneider J, Brichard V, Boon T, Meyer zum Buschenfelde KH, Wolfel T . Overlapping peptides of melanocyte differentiation antigen Melan-A/MART- 1 recognized by autologous cytolytic T lymphocytes in association with HLA-B45.1 and HLA-A2.1. Int J Cancer 1998; 75:451–458.
Parkhurst MR, Fitzgerald EB, Southwood S, Sette A, Rosenberg SA, Kawakami Y . Identification of a shared HLA-A*0201-restricted T-cell epitope from the melanoma antigen tyrosinase-related protein 2 (TRP2). Cancer Res 1998; 58:4895–4901.
Noppen C, Levy F, Burri L, et al. Naturally processed and concealed HLA-A2.1-restricted epitopes from tumor-associated antigen tyrosinase-related protein-2. Int J Cancer 2000; 87:241–246.
Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA . Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med 1996; 184:2207–2216.
Wang RF, Johnston S, Southwood S, Sette A, Rosenberg SA . Recognition of an antigenic peptide derived from TRP-2 by cytotoxic T lymphocytes in the context of HLA-A31 and -A33. J Immunol 1998; 160:890–897.
Kittlesen DJ, Thompson LW, Gulden PH, et al. Human melanoma patients recognize an HLA-A1-restricted CTL epitope from tyrosinase containing two cysteine residues: implications for tumor vaccine development. J Immunol 1998; 160:2099–2106.
Wolfel T, Van Pel A, Brichard V, et al. Two tyrosinase nonapeptides recognized on HLA-A2 melanomas by autologous cytolytic T lymphocytes. Eur J Immunol 1994; 24:759–764.
Kang X-Q, Kawakami Y, Sakaguchi K, et al. Identification of a tyrosinase epitope reocognized by HLA-A24 restricted tumor-infiltrating lymphocytes. J Immunol 1995; 155:1343–1348.
Brichard VG, Herman J, Van Pel A, et al. A tyrosinase nonpeptide presented by HLA-B44 is recognized on a human melanoma by autologous cytolytic T lymphocytes. Eur J Immunol 1996; 26:224–230.
Boel P, Wildmann C, Sensi ML, et al. BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity 1995; 2:167–175.
Traversari C, Van der Bruggen P, Luescher IF, et al. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J Exp Med 1992; 176:1453–1457.
Chaux P, Luiten R, Demotte N, et al. Identification of five MAGE-A1 epitopes recognized by cytolytic T lymphocytes obtained by in vitro stimulation with dendritic cells transduced with MAGE-A1. J Immunol 1999; 163:2928–2936.
Fujie T, Tahara K, Tanaka F, Mori M, Takesako K, Akiyoshi T . A MAGE-1-encoded HLA-A24-binding synthetic peptide induces specific anti-tumor cytotoxic T lymphocytes. Int J Cancer 1999; 80:169–172.
Tanzarella S, Russo V, Lionello I, et al. Identification of a promiscuous T-cell epitope encoded by multiple members of the MAGE family. Cancer Res 1999; 59:2668–2674.
Van der Bruggen P, Szikora JP, Boel P, et al. Autologous cytolytic T lymphocytes recognize a MAGE-1 nonapeptide on melanomas expressing Cw1601. Eur J Immunol 1994; 24:2134–2140.
Visseren MJ, van der Burg SH, van der Voort EI, et al. Identification of HLA-A*0201-restricted CTL epitopes encoded by the tumor-specific MAGE-2 gene product. Int J Cancer 1997; 73:125–130.
Tahara K, Takesako K, Sette A, Celis E, Kitano S, Akiyoshi T . Identification of a MAGE-2-encoded human leukocyte antigen-A24-binding synthetic peptide that induces specific antitumor cytotoxic T lymphocytes. Clin Cancer Res 1999; 5:2236–2241.
Gaugler B, Van Den Eynde B, Van der Bruggen P, et al. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med 1994; 179:921–930.
Van der Bruggen P, Bastin J, Gajewski T, et al. A peptide encoded by human gene MAGE-3 and presented by HLA-A2 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE-3. Eur J Immunol 1994; 24:3038–3043.
Oiso M, Eura M, Katsura F, et al. A newly identified MAGE-3-derived epitope recognized by HLA-A24-restricted cytotoxic T lymphocytes. Int J Cancer 1999; 81:387–394.
Tanaka F, Fujie T, Tahara K, et al. Induction of antitumor cytotoxic T lymphocytes with a MAGE-3-encoded synthetic peptide presented by human leukocytes antigen-A24. Cancer Res 1997; 57:4465–4468.
Herman J, van der Bruggen P, Luescher IF, et al. A peptide encoded by the human MAGE3 gene and presented by HLA-B44 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE3. Immunogenetics 1996; 43:377–383.
Fleischhauer K, Avila D, Vilbois F, Traversari C, Bordignon C, Wallny HJ . Characterization of natural peptide ligands for HLA-B*4402 and -B*4403: implications for peptide involvement in allorecognition of a single amino acid change in the HLA-B44 heavy chain. Tissue Antigens 1995; 44:311–317.
Russo V, Tanzarella S, Dalerba P, et al. Dendritic cells acquire the MAGE-3 human tumor antigen from apoptotic cells and induce a class I-restricted T cell response. Proc Natl Acad Sci USA 2000; 97:2185–2190.
Duffour MT, Chaux P, Lurquin C, Cornelis G, Boon T, van der Bruggen P . A MAGE-A4 peptide presented by HLA-A2 is recognized by cytolytic T lymphocytes. Eur J Immunol 1999; 29:3329–3337.
Zorn E, Hercend T . A MAGE-6-encoded peptide is recognized by expanded lymphocytes infiltrating a spontaneously regressing human primary melanoma lesion. Eur J Immunol 1999; 29:602–607.
Huang LQ, Brasseur F, Serrano A, et al. Cytolytic T lymphocytes recognize an antigen encoded by MAGE-A10 on a human melanoma. J Immunol 1999; 162:6849–6854.
Panelli MC, Bettinotti MP, Lally K, et al. A tumor-infiltrating lymphocyte from a melanoma metastasis with decreased expression of melanoma differentiation antigens recognizes MAGE-12. J Immunol 2000; 164:4382–4392.
Heidecker L, Brasseur F, Probst-Kepper M, Gueguen M, Boon T, Van den Eynde BJ . Cytolytic T lymphocytes raised against a human bladder carcinoma recognize an antigen encoded by gene MAGE-A12. J Immunol 2000; 164:6041–6045.
Jager E, Chen YT, Drijfhout JW, et al. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med 1998; 187:265–270.
Ayyoub M, Stevanovic S, Sahin U, et al. Proteasome-assisted identification of a SSX-2-derived epitope recognized by tumor-reactive CTL infiltrating metastatic melanoma. J Immunol 2002; 168:1717–1722.
Coulie PG, Lehmann F, Lethe B, et al. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc Natl Acad Sci USA 1995; 92:7976–7980.
Chiari R, Foury F, De Plaen E, Baurain JF, Thonnard J, Coulie PG . Two antigens recognized by autologous cytolytic T lymphocytes on a melanoma result from a single point mutation in an essential housekeeping gene. Cancer Res 1999; 59:5785–5792.
Baurain JF, Colau D, van Baren N, et al. High frequency of autologous anti-melanoma CTL directed against an antigen generated by a point mutation in a new helicase gene. J Immunol 2000; 164:6057–6066.
Kawakami Y, Wang X, Shofuda T, et al. Isolation of a new melanoma antigen, MART-2, containing a mutated epitope recognized by autologous tumor-infiltrating T lymphocytes. J Immunol 2001; 166:2871–2877.
Vigneron N, Ooms A, Morel S, Degiovanni G, Van Den Eynde BJ . Identification of a new peptide recognized by autologous cytolytic T lymphocytes on a human melanoma. Cancer Immun 2002; 2:9.
Huang J, El-Gamil M, Dudley ME, Li YF, Rosenberg SA, Robbins PF . T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J Immunol 2004; 172:6057–6064.
Sensi M, Nicolini G, Zanon M, et al. Immunogenicity without immunoselection: a mutant but functional antioxidant enzyme retained in a human metastatic melanoma and targeted by CD8(+) T cells with a memory phenotype. Cancer Res 2005; 65:632–640.
Corbiere V, Chapiro J, Stroobant V, et al. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res 2011; 71:1253–1262.
Lu YC, Yao X, Li YF, et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol 2013; 190:6034–6042.
Hogan KT, Eisinger DP, Cupp SB 3rd, et al. The peptide recognized by HLA-A68.2-restricted, squamous cell carcinoma of the lung-specific cytotoxic T lymphocytes is derived from a mutated elongation factor 2 gene. Cancer Res 1998; 58:5144–5150.
Echchakir H, Mami-Chouaib F, Vergnon I, et al. A point mutation in the alpha-actinin-4 gene generates an antigenic peptide recognized by autologous cytolytic T lymphocytes on a human lung carcinoma. Cancer Res 2001; 61:4078–4083.
Karanikas V, Colau D, Baurain JF, et al. High frequency of cytolytic T lymphocytes directed against a tumor-specific mutated antigen detectable with HLA tetramers in the blood of a lung carcinoma patient with long survival. Cancer Res 2001; 61:3718–3724.
Takenoyama M, Baurain JF, Yasuda M, et al. A point mutation in the NFYC gene generates an antigenic peptide recognized by autologous cytolytic T lymphocytes on a human squamous cell lung carcinoma. Int J Cancer 2006; 118:1992–1997.
Brandle D, Brasseur F, Weynants P, Boon T, Van den Eynde B . A mutated HLA-A2 molecule recognized by autologous cytotoxic T lymphocytes on a human renal cell carcinoma. J Exp Med 1996; 183:2501–2508.
Gaudin C, Kremer F, Angevin E, Scott V, Triebel F . A hsp70-2 mutation recognized by CTL on a human renal cell carcinoma. J Immunol 1999; 162:1730–1738.
Tatsumi T, Kierstead LS, Ranieri E, et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4(+) T cell responses against MAGE-6 in HLA-DRB10401(+) patients with renal cell carcinoma or melanoma. J Exp Med 2002; 196:619–628.
Vigneron N, Stroobant V, Chapiro J, et al. An antigenic peptide produced by peptide splicing in the proteasome. Science 2004; 304:587–590.
Hanada K, Yewdell JW, Yang JC . Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 2004; 427:252–256.
Warren EH, Vigneron NJ, Gavin MA, et al. An antigen produced by splicing of noncontiguous peptides in the reverse order. Science 2006; 313:1444–1447.
Liepe J, Marino F, Sidney J, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016; 354:354–358.
Neefjes J, Jongsma ML, Paul P, Bakke O . Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 2011; 11:823–836.
Germain RN . MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 1994; 76:287–299.
Wang RF, Wang X, Rosenberg SA . Identification of a novel MHC class II-restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4+ T cells. J Exp Med 1999; 189:1659–1667.
Wang RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA . Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science 1999; 284:1351–1354.
Delong T, Wiles TA, Baker RL, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016; 351:711–714.
Pieper R, Christian RE, Gonzales MI, et al. Biochemical identification of a mutated human melanoma antigen recognized by CD4(+) T cells. J Exp Med 1999; 189:757–766.
Wang HY, Zhou J, Zhu K, Riker AI, Marincola FM, Wang RF . Identification of a mutated fibronectin as a tumor antigen recognized by CD4+ T cells: its role in extracellular matrix formation and tumor metastasis. J Exp Med 2002; 195:1397–1406.
Voo KS, Zeng G, Mu JB, Zhou J, Su XZ, Wang RF . CD4+ T-cell response to mitochondrial cytochrome B in human melanoma. Cancer Res 2006; 66:5919–5926.
Lightowlers RN, Taylor RW, Turnbull DM . Mutations causing mitochondrial disease: What is new and what challenges remain? Science 2015; 349:1494–1499.
Wang HY, Peng G, Guo Z, Shevach EM, Wang RF . Recognition of a new ARTC1 peptide ligand uniquely expressed in tumor cells by antigen-specific CD4+ gegulatory T cells. J Immunol 2005; 174:2661–2670.
Touloukian CE, Leitner WW, Topalian SL, et al. Identification of a MHC class II-restricted human gp100 epitope using DR4-IE transgenic mice. J Immunol 2000; 164:3535–3542.
Zeng G, Touloukian CE, Wang X, Restifo NP, Rosenberg SA, Wang RF . Identification of CD4+ T cell epitopes from NY-ESO-1 presented by HLA-DR molecules. J Immunol 2000; 165:1153–1159.
Stockert E, Jager E, Chen YT, et al. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med 1998; 187:1349–1354.
Zeng G, Wang X, Robbins PF, Rosenberg SA, Wang RF . CD4+ T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA-DP4 allele: association with NY-ESO-1 antibody production. Proc Natl Acad Sci USA 2001; 98:3964–3969.
Vence L, Palucka AK, Fay JW, et al. Circulating tumor antigen-specific regulatory T cells in patients with metastatic melanoma. Proc Natl Acad Sci USA 2007; 104:20884–20889.
Topalian SL, Gonzales MI, Ward Y, Wang X, Wang RF . Revelation of a cryptic major histocompatibility complex class II-restricted tumor epitope in a novel RNA-processing enzyme. Cancer Res 2002; 62:5505–5509.
Novellino L, Renkvist N, Rini F, et al. Identification of a mutated receptor-like protein tyrosine phosphatase kappa as a novel, class II HLA-restricted melanoma antigen. J Immunol 2003; 170:6363–6370.
Saeterdal I, Bjorheim J, Lislerud K, et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci USA 2001; 98:13255–13260.
Topalian SL, Gonzales MI, Parkhurst M, et al. Melanoma-specific CD4+ T cells recognize nonmutated HLA-DR-restricted tyrosinase epitopes. J Exp Med 1996; 183:1965–1971.
Kobayashi H, Kokubo T, Takahashi M, et al. Tyrosinase epitope recognized by an HLA-DR-restricted T-cell line from a Vogt-Koyanagi-Harada disease patient. Immunogenetics 1998; 47:398–403.
Kobayashi H, Lu J, Celis E . Identification of helper T-cell epitopes that encompass or lie proximal to cytotoxic T-cell epitopes in the gp100 melanoma tumor antigen. Cancer Res 2001; 61:7577–7584.
Lapointe R, Royal RE, Reeves ME, Altomare I, Robbins PF, Hwu P . Retrovirally transduced human dendritic cells can generate T cells recognizing multiple MHC class I and class II epitopes from the melanoma antigen glycoprotein 100. J Immunol 2001; 167:4758–4764.
Zarour HM, Kirkwood JM, Kierstead LS, et al. Melan-A/MART-1(51-73) represents an immunogenic HLA-DR4-restricted epitope recognized by melanoma-reactive CD4(+) T cells. Proc Natl Acad Sci USA 2000; 97:400–405.
Manici S, Sturniolo T, Imro MA, et al. Melanoma cells present a MAGE-3 epitope to CD4(+) cytotoxic T cells in association with histocompatibility leukocyte antigen DR11. J Exp Med 1999; 189:871–876.
Chaux P, Vantomme V, Stroobant V, et al. Identification of MAGE-3 epitopes presented by HLA-DR molecules to CD4(+) T lymphocytes. J Exp Med 1999; 189:767–778.
Schultz ES, Lethe B, Cambiaso CL, et al. A MAGE-A3 peptide presented by HLA-DP4 is recognized on tumor cells by CD4+ cytolytic T lymphocytes. Cancer Res 2000; 60:6272–6275.
Kobayashi H, Song Y, Hoon DS, Appella E, Celis E . Tumor-reactive T helper lymphocytes recognize a promiscuous MAGE-A3 epitope presented by various major histocompatibility complex class II alleles. Cancer Res 2001; 61:4773–4778.
Consogno G, Manici S, Facchinetti V, et al. Identification of immunodominant regions among promiscuous HLA-DR-restricted CD4+ T-cell epitopes on the tumor antigen MAGE-3. Blood 2003; 101:1038–1044.
Schroers R, Huang XF, Hammer J, Zhang J, Chen SY . Identification of HLA DR7-restricted epitopes from human telomerase reverse transcriptase recognized by CD4+ T-helper cells. Cancer Res 2002; 62:2600–2605.
Chiari R, Hames G, Stroobant V, et al. Identification of a tumor-specific shared antigen derived from an Eph receptor and presented to CD4 T cells on HLA class II molecules. Cancer Res 2000; 60:4855–4863.
Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Immunologic and therapeutic evaluation of a synthetic tumor-associated peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 1998; 4:321–327.
Marchand M, van Baren N, Weynants P, et al. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA- A1. Int J Cancer 1999; 80:219–230.
Yee C, Thompson JA, Roche P, et al. Melanocyte destruction after antigen-specific immunotherapy of melanoma. Direct evidence of T cell-mediated vitiligo. J Exp Med 2000; 192:1637–1644.
Schwartzentruber DJ, Lawson DH, Richards JM, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med 2011; 364:2119–2127.
Cecco S, Muraro E, Giacomin E, et al. Cancer vaccines in phase II/III clinical trials: state of the art and future perspectives. Curr Cancer Drug Targets 2011; 11:85–102.
Bilusic M, Madan RA . Therapeutic cancer vaccines: the latest advancement in targeted therapy. Am J Ther 2012; 19:e172–e181.
Vansteenkiste J, Zielinski M, Linder A, et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: phase II randomized study results. J Clin Oncol 2013; 31:2396–2403.
Davis ID, Chen W, Jackson H, et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci USA 2004; 101:10697–10702.
Khong HT, Yang JC, Topalian SL, et al. Immunization of HLA-A*0201 and/or HLA-DPbeta1*04 patients with metastatic melanoma using epitopes from the NY-ESO-1 antigen. J Immunother 2004; 27:472–477.
Odunsi K, Qian F, Matsuzaki J, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci USA 2007; 104:12837–12842.
Valmori D, Souleimanian NE, Tosello V, et al. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc Natl Acad Sci USA 2007; 104:8947–8952.
Sonpavde G, Wang M, Peterson LE, et al. HLA-restricted NY-ESO-1 peptide immunotherapy for metastatic castration resistant prostate cancer. Invest New Drugs 2014; 32:235–242.
Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509–1518.
Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012; 119:2709–2720.
Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA . Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015; 350:aab4077.
Roybal KT, Rupp LJ, Morsut L, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016; 164:770–779.
Roybal KT, Williams JZ, Morsut L, et al. Engineering T cells with customized therapeutic response programs using synthetic Notch receptors. Cell 2016; 167:419–432.
Morsut L, Roybal KT, Xiong X, et al. Engineering customized cell sensing and response behaviors using synthetic Notch receptors. Cell 2016; 164:780–791.
Robbins PF, Kassim SH, Tran TL, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 2015; 21:1019–1027.
Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 2008; 358:2698–2703.
Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014; 344:641–645.
Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009; 114:535–546.
Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013; 36:133–151.
Cameron BJ, Gerry AB, Dukes J, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 2013; 5:197ra103.
Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006; 314:126–129.
Germeau C, Ma W, Schiavetti F, et al. High frequency of antitumor T cells in the blood of melanoma patients before and after vaccination with tumor antigens. J Exp Med 2005; 201:241–248.
Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365:1673–1683.
Martincorena I, Campbell PJ . Somatic mutation in cancer and normal cells. Science 2015; 349:1483–1489.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW . Cancer genome landscapes. Science 2013; 339:1546–1558.
Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502:333–339.
George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015; 524:47–53.
Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015; 521:489–494.
Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015; 518:495–501.
Lu YC, Robbins PF . Cancer immunotherapy targeting neoantigens. Semin Immunol 2016; 28:22–27.
Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108–1113.
Segal NH, Parsons DW, Peggs KS, et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008; 68:889–892.
Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; 482:400–404.
Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72:1081–1091.
DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T . Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012; 482:405–409.
Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013; 19:747–752.
van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013; 31:e439–442.
Linnemann C, van Buuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 2015; 21:81–85.
Cohen CJ, Gartner JJ, Horovitz-Fried M, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 2015; 125:3981–3991.
Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014; 515:572–576.
Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014; 515:577–581.
Schumacher T, Bunse L, Pusch S, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014; 512:324–327.
Kreiter S, Vormehr M, van de Roemer N, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520:692–696.
Kranz LM, Diken M, Haas H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016; 534:396–401.
Duan F, Duitama J, Al Seesi S, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 2014; 211:2231–2248.
Carreno BM, Magrini V, Becker-Hapak M, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; 348:803–808.
Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124–128.
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371:2189–2199.
McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016; 351:1463–1469.
Gros A, Parkhurst MR, Tran E, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med 2016; 22:433–438.
Voo KS, Fu T, Heslop HE, Brenner MK, Rooney CM, Wang RF . Identification of HLA-DP3-restricted peptides from EBNA1 recognized by CD4(+) T cells. Cancer Res 2002; 62:7195–7199.
Voo KS, Fu T, Wang HY, et al. Evidence for the presentation of major histocompatibility complex class I-restricted Epstein-Barr virus nuclear antigen 1 peptides to CD8+ T lymphocytes. J Exp Med 2004; 199:459–470.
Voo KS, Peng G, Guo Z, et al. Functional characterization of EBV-encoded nuclear antigen 1-specific CD4+ helper and regulatory T cells elicited by in vitro peptide stimulation. Cancer Res 2005; 65:1577–1586.
Stronen E, Toebes M, Kelderman S, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016; 352:1337–1341.
Tran E, Robbins, PF, Lu YC, et al. T cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med 2016; 375:2255–2262.
Mullard A . The cancer vaccine resurgence. Nat Rev Drug Discov 2016; 15:663–665.
Acknowledgements
This work was supported, in part, by grants from the NCI and NIDA, NIH (R01CA101795, R01DA030338 and U54CA210181), Cancer Prevention and Research Institute of Texas (CPRIT; DP150099, RP150611 and RP170537), Department of Defense (DoD) CDMRP BCRP (BC151081), and Golfer against cancer foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, RF., Wang, H. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res 27, 11–37 (2017). https://doi.org/10.1038/cr.2016.155
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.155
Keywords
This article is cited by
-
Upregulation of PD-1/PD-L1 and downregulation of immune signaling pathways lead to more severe visceral leishmaniasis in undernutrition mice
Parasites & Vectors (2024)
-
The radiomics nomogram predicts the prognosis of pancreatic cancer patients with hepatic metastasis after chemoimmunotherapy
Cancer Immunology, Immunotherapy (2024)
-
Systemic administration of STING agonist promotes myeloid cells maturation and antitumor immunity through regulating hematopoietic stem and progenitor cell fate
Cancer Immunology, Immunotherapy (2023)
-
Innovative drugs promote precision cancer therapy
Clinical Cancer Bulletin (2023)
-
Oncolytic attenuated measles virus encoding NY-ESO-1 induces HLA I and II presentation of this tumor antigen by melanoma and dendritic cells
Cancer Immunology, Immunotherapy (2023)
