
Abstract
Na+,K+-ATPase (NKA) is required to generate the resting membrane potential in neurons. Nociceptive afferent neurons express not only the α and β subunits of NKA but also the γ subunit FXYD2. However, the neural function of FXYD2 is unknown. The present study shows that FXYD2 in nociceptive neurons is necessary for maintaining the mechanical allodynia induced by peripheral inflammation. FXYD2 interacted with α1NKA and negatively regulated the NKA activity, depolarizing the membrane potential of nociceptive neurons. Mechanical allodynia initiated in FXYD2-deficient mice was abolished 4 days after inflammation, whereas it persisted for at least 3 weeks in wild-type mice. Importantly, the FXYD2/α1NKA interaction gradually increased after inflammation and peaked on day 4 post inflammation, resulting in reduction of NKA activity, depolarization of neuron membrane and facilitation of excitatory afferent neurotransmission. Thus, the increased FXYD2 activity may be a fundamental mechanism underlying the persistent hypersensitivity to pain induced by inflammation.
Similar content being viewed by others

Introduction
The Na+,K+-ATPase (NKA, Na+/K+ pump) expels three Na+ ions in exchange for two K+ ions and derives energy from the hydrolysis of an ATP molecule. This maintenance of the Na+ and K+ gradients across the plasma membrane is required to generate the resting membrane potential in neurons. The minimum functional unit of NKA is a heterodimer of α and β subunits. The catalytic, transport and pharmacological properties of NKA reside in the α subunit, while β subunit is required for the efficient expression of α subunit and assembly of functional NKA1,2. Four isoforms of the α subunit (α1-α4) and three isoforms of the β subunit (β1-β3) are expressed in a tissue- and cell-dependent manner in mammals2,3. NKA activity is regulated by direct modulators ATP, Na+ and K+ and indirect modulators (catecholamines, insulin, angiotensin II and morphine) through receptor-mediated mechanisms4. NKA can be inhibited by plant cardiac glycosides such as ouabain and digoxin5 and digitalis-like steroid hormones released from the adrenal gland6. Recent studies show that the α1 subunit-containing NKA (α1NKA) can be activated directly by an endogenous protein, follistatin-like 1 (FSTL1)7,8.
FXYD proteins are single transmembrane proteins and contain an FXYD motif near the N-terminus9,10. FXYD proteins bind to the α subunit and modify transport properties of NKA in a tissue- and isoform-specific way10,11. Seven FXYD proteins (FXYD1-7) have been found in mammals10. FXYD2 was originally characterized as a small acidic proteolipid that co-purified with renal NKA12. FXYD2 was then found to be expressed in the distal convoluted tubule of kidney13 and was shown to reduce the affinity of NKA for both Na+ and K+ in renal cells13,14,15. Therefore, we investigated whether FXYD2 can regulate neuronal activity by modulating NKA activity.
Small-diameter neurons of the dorsal root ganglion (DRG) give rise to unmyelinated (C-fibers) and thinly myelinated axons (Aδ-fibers) that terminate in laminae I-II of the spinal cord. These axons convey the somatic sensory signals generated by nociceptors, thermoreceptors and sensitive mechanoreceptors16,17,18. The α1, α3 and β1 subunits of NKA are expressed in the DRG19,20. The α1 subunit is present in both small and large DRG neurons, whereas the α3 subunit is mainly distributed in large neurons21. The membrane current produced by NKA activity in DRG neurons is primarily mediated by the α1NKA22. Recent studies show that an endogenous activator, FSTL1, can be released from nociceptive afferent terminals and activate presynaptic α1NKA and suppress excitatory synaptic transmission, resulting in the negative regulation of nociceptive responses. Therefore, the regulation of NKA activity in nociceptive afferent neurons may be an important mechanism for modulating pain sensation. Our previous microarray analysis revealed FXYD2 as one of the genes expressed at a high level in the DRG7. A recent study reports that FXYD2 is expressed in the isolectin B4 (IB4)-positive (non-peptidergic) subset of small DRG neurons23. However, the functions of FXYD2 in somatosensory neurons remain unknown.
The present study shows that FXYD2 negatively regulates the activity of NKA, depolarizing the membrane potential of small DRG neurons. In the persistent inflammatory pain model induced by intraplantar injection of complete Freund's adjuvant (CFA), the FXYD2/α1NKA interaction increased, thereby maintaining the inflammation-induced membrane depolarization of small DRG neurons, as well as facilitating afferent synaptic transmission and long-lasting mechanical allodynia. Therefore, FXYD2 contributes to the inflammation-induced persistent pain hypersensitivity.
Results
FXYD2 is expressed in subsets of small DRG neurons
Microarray analysis showed a high level of FXYD2 mRNA in the DRG7. Next, the expression levels of FXYD2 in the mouse tissues were studied with real-time PCR. The highest level of FXYD2 mRNA was found in the kidney (data not shown), consistent with the microarray analysis and previous reports10. In the nervous system, the highest level of FXYD2 mRNA was found in the DRG (Figure 1A).

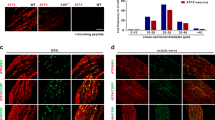
FXYD2 is expressed in subsets of small DRG neurons. (A) Real-time PCR showed that the highest level of FXYD2 mRNA (Fxyd2) in all tested tissues was observed in the DRG, and very low levels were observed in the spinal cord and brain of adult mice. In situ hybridization showed that FXYD2 mRNA was mainly present in small DRG neurons. Scale bar, 50 μm. (B) Quantitative analysis showed that almost all of FXYD2 mRNA-containing neurons were small DRG neurons with cross-neuronal area less than 600 μm2. (C) Co-localization of FXYD2 with neuronal markers by in situ hybridization combined with immunostaining or double fluorescent in situ hybridization. Most of FXYD2 mRNA-positive DRG neurons were peripherin-positive neurons and only a few FXYD2-expressing neurons were NF200-positive large neurons. About half of FXYD2 mRNA-positive neurons bound to IB4 and were non-peptidergic neurons. Some FXYD2-expressing neurons contained mRNA encoding Mrgprd or Mrgprb4. FXYD2 mRNA was also present in TH mRNA-positive neurons. Some CGRP-positive peptidergic neurons also contained FXYD2 mRNA. Scale bar, 50 μm. (D) Diagram showing the profile of FXYD2-expressing neurons in subsets of DRG neurons.
In situ hybridization showed that 47.4% of DRG neurons in lumbar (L) 4 and L5 DRGs contained FXYD2 mRNA (Figure 1A). These FXYD2 mRNA-containing neurons were small neurons (cross-sectional area of neuron profiles < 600 μm2; Figure 1B). FXYD2 mRNA was found in 58.1% of small DRG neurons (n = 2 685). Using in situ hybridization combined with immunostaining, we further found that 80.6% of FXYD2 mRNA-containing neurons (n = 2 123) were peripherin-immunoreactive small DRG neurons, and 77.1% of peripherin-positive small DRG neurons (n = 2 218) expressed FXYD2 (Figure 1C). Only 3.0% of FXYD2 mRNA-containing DRG neurons (n = 2 415) were neurofilament 200 (NF200)-positive, and 3.4% of NF200-positive neurons (n = 2 133) expressed FXYD2 (Figure 1C).
Small DRG neurons are often classified into peptidergic and IB4-positive (non-peptidergic) subsets. We observed that more than half of FXYD2 mRNA-containing neurons (55.2%, n = 2 804) were IB4-positive, and 81.6% of IB4-positive neurons (n = 1 434) expressed FXYD2 (Figure 1C and 1D). Subpopulations of IB4-positive small DRG neurons expressed either MAS-related G-protein-coupled receptor member D (MRGPRD) or member B4 (MRGPRB4)24. FXYD2 mRNA was present in the subpopulations containing MRGPRD mRNA or MRGPRB4 mRNA (Figure 1C). We also found that 22.4% of FXYD2 mRNA-positive neurons (n = 1 691) contained neuropeptide calcitonin gene-related peptide (CGRP), and 24.9% of CGRP-immunoreactive small DRG neurons (n = 1 086) expressed FXYD2 (Figure 1C and 1D). Furthermore, FXYD2 mRNA was also found in the tyrosine hydroxylase (TH) mRNA-containing subset of small DRG neurons (Figure 1C and 1D), which are also IB4-negative25. Therefore, FXYD2 is expressed in both IB4-positive and TH-containing subsets of small DRG neurons and in some peptidergic small DRG neurons under normal circumstances.
Immunoblot analysis showed that FXYD2 (∼7 kDa) was present in the dorsal horn of spinal cord (Figure 2A), although FXYD2 mRNA was absent in the dorsal spinal cord (Figure 1A). To confirm the transport of FXYD2 from the neuron soma in the DRG to the dorsal spinal cord, the dorsal roots were transected. Three days after surgery, the proteins in the spinal dorsal horn were examined with immunoblotting. The FXYD2 level in the ipsilateral dorsal horn of spinal cord was lower than that in the contralateral dorsal horn (Figure 2B), indicating that FXYD2 synthesized in small DRG neurons is transported to the dorsal horn of spinal cord.

FXYD2 is transported to the dorsal spinal cord and interacts with α1NKA. (A) Immunoblotting (IB) showed that FXYD2 was present in both the DRG and the dorsal horn of spinal cord. This image represents five experiments with similar results. (B) Immunoblotting showed that the level of FXYD2 in the ipsilateral dorsal horn of spinal cord was decreased after dorsal root transection. Four experiments showed similar results. (C) Double fluorescent in situ hybridization showed that Fxyd2 mRNA and α1 subunit mRNA (Atp1a1) were co-localized in small DRG neurons. Scale bar, 50 μm. (D) Co-immunoprecipitation (IP) showed FXYD2 among proteins precipitated with the α1 subunit antibody from mouse DRGs. Similar results were observed in three experiments. (E) In COS7 cells, overexpressed FXYD2 was co-immunoprecipitated with the α1 subunit of NKA. This image is representative of three experiments.
FXYD2 interacts with the α1 subunit and negatively regulates NKA activity
The cellular relationship between FXYD2 and the α1 subunit of NKA in the DRG was analyzed with in situdouble hybridization. We found that almost all small DRG neurons containing FXYD2 mRNA also expressed the α1 subunit mRNA (Figure 2C). Co-immunoprecipitation (IP) experiment showed that FXYD2 interacted with the α1 subunit of NKA in the lysate of mouse DRGs (Figure 2D). Thus, there is a cellular basis for the FXYD2/α1NKA interaction in nociceptive afferent neurons.
We then used a cell line to investigate whether FXYD2 regulated the NKA activity. RT-PCR showed that FXYD2 mRNA was not detected in COS7 cells (data not shown), which contained low levels of mRNAs encoding the α1 and β1 subunits of NKA but no detectable levels of mRNAs for the α2, α3 and α4 subunits7. COS7 cells were transfected with a plasmid expressing Myc-tagged FXYD2 at C-terminus (FXYD2-Myc) together with a plasmid expressing Flag-tagged α1 subunit at C-terminus (α1 subunit-Flag). Co-IP showed that FXYD2 interacted with the α1 subunit in the lysate of the transfected COS7 cells (Figure 2E). Then, COS7 cells were transfected with the plasmids expressing the α1 and β1 subunits together with the plasmid expressing FXYD2. The NKA activity in the microsome samples prepared from these transfected COS7 cells was measured. A reduction of ∼16% (15.6% ± 1.2%, n = 5) in NKA activity was observed in the COS7 cells overexpressing FXYD2 compared with the vector (Figure 3A). These results suggest that NKA activity is negatively regulated by FXYD2.

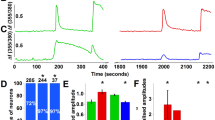
FXYD2 negatively regulates NKA activity and depolarizes the membrane potential. (A) Co-expression of FXYD2 with α1 and β1 subunits of NKA in COS7 cells reduced NKA activity. Treatment with FSTL1 increased NKA activity in COS7 cells transfected with the plasmids expressing α1 and β1 subunits, and partially reversed the reduction of NKA activity in COS7 cells expressing FXYD2 together with the α1 and β1 subunits. n = 5, **P < 0.01 vs vector with vehicle and ##P < 0.01 vs indicated. (B) Immunoblotting and immunostaining showed that FXYD2 protein was absent in the DRG of Fxyd2−/− mice, whereas the expression of the α1 subunit of NKA was not apparently changed. Scale bar, 50 μm. (C) NKA activity in the DRG of Fxyd2−/− mice was higher than that of Fxyd2+/+ mice. FSTL1 activated NKA in the DRG of Fxyd2+/+ mice and also increased NKA activity in Fxyd2−/− DRGs. n = 3, *P < 0.05 vs Fxyd2+/+with vehicle and #P < 0.05 vs Fxyd2−/− with vehicle. (D) The traces recorded from IB4-positive DRG neurons by current-clamp were averaged and compared between Fxyd2+/+ (n = 36) and Fxyd2−/− mice (n = 50). (E) Quantitative analysis showed that the IB4-positive small DRG neurons of Fxyd2−/− mice were hyperpolarized (n = 63 for Fxyd2+/+ and n = 54 for Fxyd2−/− mice, ***P < 0.001). (F) A long duration (200 ms) current was injected to induce sustained firing of small DRG neurons. The number of IB4-positive small DRG neurons with a low firing frequency of AP (0-3 APs per stimulation) was increased slightly in Fxyd2−/− mice, whereas the number of IB4-positive small DRG neurons with high firing frequency (4-7 APs per stimulation) was reduced.
To further confirm the effect of FXYD2 in neurons, we measured the NKA activity in the DRGs of FXYD2-knockout (Fxyd2−/−) mice. Immunoblotting and immunostaining showed that FXYD2 was absent in the DRG of Fxyd2−/− mice (Figure 3B). The expression of the α1 subunit of NKA in DRGs of Fxyd2−/− mice was not obviously different from that of wild-type (Fxyd2+/+) mice (Figure 3B). The NKA activity was ∼19% higher (18.9% ± 4.1%, n = 3) in the microsome prepared from the DRGs of Fxyd2−/− mice than that from the Fxyd2+/+littermates (Figure 3C). These results indicate that NKA activity can be negatively regulated by FXYD2 in small DRG neurons.
Additional expression of FXYD2 did not markedly affect the FSTL1-induced facilitation of NKA activity in the COS7 cells expressing the α1 and β1 subunits of NKA (Figure 3A). Treatment with FSTL1 (60 nM) induced an increase of the NKA activity in the microsome collected from the DRGs of both Fxyd2+/+ and Fxyd2−/− mice (14.8% ± 3.6% for Fxyd2+/+ and 22.5% ± 5.8% for Fxyd2−/−, n = 3; Figure 3C). These data suggest that FXYD2 is not directly involved in the FSTL1-induced activation of α1 subunit of NKA. Therefore, α1NKA activity can be regulated independently by FXYD2 and FSTL1.
Loss of FXYD2 hyperpolarizes the membrane potential in small DRG neurons
Given that FXYD2 negatively regulated the activity of NKA, we further examined whether FXYD2 regulated the electrical properties of the neuronal plasma membrane, by using whole-cell patch clamp recording. To induce a single action potential (AP), a brief current (15 ms, 100 pA) was injected into dissociated IB4-positive small DRG neurons from Fxyd2+/+ or Fxyd2−/− mice. Then, we analyzed the parameters such as membrane potential, AP duration and afterhyperpolarization (AHP) duration. All recorded traces from current-clamp recording were averaged, and the traces for Fxyd2+/+ and Fxyd2−/− mice were compared (Figure 3D). We found that the mean membrane potential changed from −44 mV (−43.9 ± 0.8, n = 63) in IB4-positive small DRG neurons cultured from Fxyd2+/+ mice to −48 mV (−48.0 ± 0.7, n = 54) in that from Fxyd2−/− mice (Figure 3D and 3E), whereas in IB4-negetive, including capsaicin-sensitive, small DRG neurons, the mean membrane potential did not differ significantly between Fxyd2+/+ and Fxyd2−/− mice (Supplementary information, Figure S1A and S1B). AHP duration was higher in the neurons from Fxyd2−/− mice than those from Fxyd2+/+ mice, whereas other analyzed parameters were not significantly different (Supplementary information, Figure S2A-S2D). Thus, loss of FXYD2 in small DRG neurons can result in membrane hyperpolarization.
To examine changes in the firing frequency in the absence of FXYD2, we injected a current with extended duration and increased intensity (200 ms, 70 pA) into IB4-positive small DRG neurons, to induce repetitive firing of AP. The number of neurons with a low firing frequency of AP (0-3 APs per stimulation) was only slightly higher in Fxyd2−/− mice than in Fxyd2+/+ mice, and the number of neurons with higher firing frequency (4-7 APs per stimulation) was slightly lower in Fxyd2−/− mice than in Fxyd2+/+ mice (Figure 3F). Taken together, these results suggest that FXYD2 is required for regulating the membrane potential and the excitability of certain populations of small DRG neurons. However, the effect of FXYD2 on the neuronal excitability may be limited under normal circumstances.
FXYD2 is required for maintaining inflammation-induced mechanical allodynia
We used Fxyd2−/− mice to test whether FXYD2 is involved in the modulation of nociception. The accelerated rotarod test showed no apparent motor defect in Fxyd2−/− mice (Figure 4A). The radiant heat test and hotplate test showed that the latency to the noxious heat stimulus in Fxyd2−/− mice was similar to that in Fxyd2+/+ littermates (Figure 4B and 4C), suggesting that FXYD2 is not involved in the regulation of basal thermal nociceptive responses. The von Frey test showed that the mean mechanical threshold was slightly higher in Fxyd2−/− mice (Figure 4D), suggesting that FXYD2 has a limited effect on the basal mechanical nociceptive response.

FXYD2 is required for maintaining inflammatory nociceptive responses. (A) The drop latency of Fxyd2−/− mice was similar to that of Fxyd2+/+ mice in rotarod test (n = 14 for Fxyd2+/+ and n = 10 for Fxyd2−/− mice). (B) The Hargreaves test showed that the thermal latency of Fxyd2−/− mice was not altered compared to Fxyd2+/+ mice (n = 10 for both Fxyd2+/+ and Fxyd2−/− mice). (C) The hotplate test showed that thermal latency at 50, 52, and 55 °C was unchanged between Fxyd2+/+ and Fxyd2−/− mice (n = 11 for Fxyd2+/+ and n = 12 for Fxyd2−/− mice). (D) The mechanical threshold of Fxyd2−/− mice was slightly increased compared to that of Fxyd2+/+ mice (n = 14 for Fxyd2+/+ and n = 16 for Fxyd2−/− mice). (E) The Fxyd2−/− mice did not show any apparent changes in the number of flinches in either the first or the second phase of the formalin test (n = 11 for both Fxyd2+/+ and Fxyd2−/− mice). (F) The radiant heat test showed that recovery from the thermal hyperalgesia was facilitated in Fxyd2−/− mice 2 days after CFA injection (n = 10 for both Fxyd2+/+ and Fxyd2−/− mice). (G) The von Frey test showed that the mechanical allodynia was maintained only in Fxyd2+/+ mice and not in Fxyd2−/− mice 2 days after CFA injection (n = 9 for both Fxyd2+/+ and Fxyd2−/− mice, ***P < 0.001).
Next, we examined the FXYD2 function using the formalin test and the CFA-induced persistent inflammation model. We first investigated the effect of FXYD2 loss in acute pain model induced by intraplantar injection with formalin, which causes a stereotypic two-phase pattern of nociceptive response. The Fxyd2−/− mice did not show any apparent changes in the number of flinches in either the first or second phases of the formalin test (Figure 4E).
We then analyzed the nociceptive behavior of Fxyd2−/− mice 2 days after intraplantar injection of CFA at the hindpaw, and we found that Fxyd2+/+ and Fxyd2−/− mice developed thermal hyperalgesia and mechanical allodynia to a similar degree (Figure 4F and 4G), suggesting that FXYD2 is not required for initiating the inflammatory nociceptive response. On day 4 and day 7 after CFA injection, Fxyd2−/− mice showed no significant difference in the thermal hyperalgesia from Fxyd2+/+ mice (Figure 4F). Unexpectedly, the inflammation-induced mechanical allodynia was absent in Fxyd2−/− mice 4 days after CFA treatment, whereas the mechanical allodynia in Fxyd2+/+ mice lasted for at least 21 days, the longest time interval used in the experiment (Figure 4G). Thus, the Fxyd2−/− mice recovered much more quickly from inflammatory pain than Fxyd2+/+ mice, suggesting that FXYD2 expressed in small DRG neurons contributes to the maintenance of mechanical allodynia following inflammation.
Increase in the FXYD2/α1NKA interaction after peripheral inflammation
Given that FXYD2 could regulate the inflammation-induced mechanical allodynia, we investigated the mechanism underlying this function. We were particularly interested in the mechanism for the differential action of FXYD2 on day 2 and day 4 post inflammation, because FXYD2 seemed to be mainly involved in the development of mechanical allodynia 4 days after inflammation. First, we tested whether inflammation alters the expression of FXYD2, FSTL1, NKA subunits, and other related molecules in DRG neurons. After intraplantar CFA injection at the hindpaw, the expression levels of the α1, α3 and β1 subunits of NKA, FSTL1 and FXYD2 did not change in L4 and L5 DRGs (Supplementary information, Figure S3A-S3C). The numbers of DRG neurons that expressed FXYD2 and α1NKA appeared unchanged after peripheral inflammation (Supplementary information, Figure S3B). Furthermore, immunoblotting showed no apparent change in the FXYD2 level in the DRG after peripheral inflammation (Figure 5A). Thus, it is unlikely that the action of FXYD2 in inflammatory pain is due to changes in the expression of FXYD2 in DRG neurons.

FXYD2 is required for maintaining the inflammation-induced membrane depolarization and excitability. (A) Co-IP showed that the interaction between FXYD2 and the α1 subunit of NKA increased gradually after peripheral inflammation and peaked on day 4 after injection. (B) Quantitation showed that the interaction between FXYD2 and the α1 subunit of NKA increased gradually and peaked on day 4 (n = 4, ***P < 0.001). The increased interaction lasted for 14 days, although the average interaction level on day 14 was lower than that on day 4 (n = 3, *P < 0.05). (C) The NKA activity gradually decreased in the DRGs of Fxyd2+/+ mice treated with CFA for 2 and 4 days (n = 3 for CFA day 2 and n = 6 for CFA day 4, ***P < 0.001 vs Fxyd2+/+mice without CFA treatment). However, 4 days after CFA treatment, the NKA activity in the DRGs of Fxyd2−/− mice was not significantly different from that in the DRGs of Fxyd2+/+ mice without CFA treatment. The reduction of NKA activity in the DRGs of Fxyd2+/+ mice was reversed by deletion of Fxyd2 (n = 5, ***P < 0.001 vs Fxyd2+/+mice without CFA treatment and ###P < 0.001 vs indicated). (D) Whole-cell patch clamp recording at current-clamp mode showed that the membrane potential of small DRG neurons cultured from Fxyd2+/+mice was depolarized after CFA injection (n = 52 for control, n = 55 for CFA 2 days and n = 65 for CFA 4 days, *P < 0.05 and ***P < 0.001 vs Fxyd2+/+mice without CFA treatment). The membrane potential of small DRG neurons cultured from Fxyd2−/− mice was also depolarized 2 days after CFA injection (n = 54 for control and n = 49 for CFA 2 days, ###P < 0.001 vs indicated). However, 4 days after CFA injection, the depolarized membrane potential was reversed in small DRG neurons of Fxyd2−/− mice (n = 67 for CFA 4 days; N.S., non-significant difference). The data were shown as the change of membrane potential and statistic analysis was performed for the membrane potential between the indicated groups. (E) Bath-applied ouabain (1 mM) depolarized the membrane potential of small DRG neurons of both Fxyd2+/+ and Fxyd2−/− mice 4 days after CFA injection (n = 13 for Fxyd2+/+ mice and n = 14 for Fxyd2−/− mice, ***P < 0.001 vs Fxyd2+/+ mice 4 days after CFA injection, before ouabain treatment and ###P < 0.001 vs indicated). The data were shown as the change of membrane potential and statistic analysis was performed for the membrane potential between the indicated groups. (F) The current (70 pA, 200 ms) was injected to induce sustained firing of IB4-positive small DRG neurons. Representative traces showed that the firing frequency of AP was increased in IB4-positive small DRG neurons from Fxyd2+/+ mice 2 and 4 days after CFA injection, whereas the firing rate of AP was decreased in the neurons from Fxyd2−/− mice 4 days after CFA injection. The number of IB4-positive small DRG neurons with a low firing frequency of AP (0-3 APs per stimulation) was decreased gradually in Fxyd2+/+ mice after CFA injection, but was increased in Fxyd2−/− mice 4 days after CFA injection (n = 42 for control, n = 39 for CFA 2 days and n = 50 for CFA 4 days in Fxyd2+/+ mice; n = 59 for control, n = 46 for CFA 2 days and n = 57 for CFA 4 days in Fxyd2−/− mice).
However, it was interesting that the interaction between FXYD2 and the α1 subunit of NKA gradually increased in the DRGs after peripheral inflammation (Figure 5A). This increase in the FXYD2/α1NKA interaction peaked (∼6.3 times of the control level) 4 days after CFA injection (Figure 5B). On day 14 post inflammation, the FXYD2/1NKA interaction still exhibited an ∼4.7-fold increase, although its average level was reduced compared with that on day 4 post CFA injection (Figure 5B). Consistent with this finding, we found that the NKA activity gradually decreased in the DRG neurons of Fxyd2+/+ mice after peripheral CFA treatment. Especially on day 4 post CFA treatment, the NKA activity decreased 14% (13.9% ± 1.5%, n = 6) compared to that in the DRG neurons of Fxyd2+/+ mice without CFA treatment (Figure 5C). Four days after CFA treatment, the NKA activity in the Fxyd2−/− DRGs was 12% (12.1% ± 0.1%, n = 5) higher than that in the DRG neurons of Fxyd2+/+ mice without CFA treatment (Figure 5C). These results indicate a role of FXYD2 in the inflammation-induced reduction of NKA activity. Taken together, the FXYD2/α1NKA interaction in the DRG neurons was increased after peripheral inflammation and peaked 4 days after inflammation, resulting in a decrease in NKA activity.
FXYD2 is required for maintaining the inflammation-induced membrane depolarization and excitability
To examine whether there were changes in the membrane potential of small DRG neurons that was correlated with the increase in the FXYD2/α1NKA interaction, we recorded the membrane potential of small DRG neurons prepared from CFA-treated mice. The mean membrane potential of IB4-positive small DRG neurons dissociated from Fxyd2+/+ mice gradually changed from −44 mV (44.2 ± 0.9 mV, n = 52) to −40 mV (−40.4 ± 2.2 mV, n = 55) 2 days after CFA treatment and to −39 mV (−39.1 ± 1.2 mV, n = 65) 4 days after CFA treatment (Figure 5D), suggesting that peripheral inflammation can induce membrane depolarization in IB4-positive small DRG neurons.
In IB4-positive small DRG neurons cultured from Fxyd2−/− mice treated with CFA for 2 days, the membrane potential was depolarized from −48 mV (−48.0 ± 0.8 mV, n = 54) to −40 mV (−40.2 ± 1.1 mV, n = 49; Figure 5D). However, 4 days after CFA treatment, the mean membrane potential of cultured IB4-positive small DRG neurons from Fxyd2−/− mice was more negative (−47.6 ± 0.9 mV, n = 67) than that of IB4-positive small DRG neurons dissociated from Fxyd2+/+ mice without CFA treatment (Figure 5D), leading to membrane hyperpolarization. The time curve of changes in the averaged membrane potential of IB4-positive small DRG neurons dissociated from Fxyd2−/− mice was correlated with the change in NKA activity (Figure 5C and 5D). Therefore, the peak level of FXYD2/1NKA interaction on day 4 post CFA treatment contributes to the membrane depolarization of nociceptive afferent neurons, and loss of the FXYD2-induced negative regulation increases the NKA activity, leading to the membrane hyperpolarization.
To further test whether FXYD2 induced the membrane depolarization through negative regulation of α1NKA activity in small DRG neurons, we used a high dose of NKA inhibitor ouabain (1 mM) to block the α1NKA activity22. Bath-applied ouabain induced a depolarization of ∼6 mV (6.1 ± 0.3, n = 4) in the membrane potential of IB4-positive small DRG neurons. Four days after inflammation, the membrane potential of IB4-positive small DRG neurons from Fxyd2+/+ mice was able to be further depolarized to a small range (3.3 ± 0.5 mV, from −39.1 ± 1.2 to −35.8 ± 1.0 mV, n = 13) by the same dose of ouabain, and the depolarization effect of ouabain was also observed on DRG neurons from Fxyd2−/− mice (6.7 ± 0.7 mV, from −48.9 ± 1.2 to −42.2 ± 1.1 mV, n = 21; Figure 5E). Thus, the membrane depolarization observed 4 days after inflammation is attributed to the effect of FXYD2 on α1NKA activity.
Next, we examined the effect of FXYD2 on the excitability of small DRG neurons after peripheral inflammation. The number of IB4-positive small DRG neurons with a low frequency of AP firing (0-3 APs per stimulation) decreased 2 days after inflammation, whereas the number of neurons with higher frequency of AP firing (4-10 APs per stimulation) increased (Figure 5F). Such changes became more pronounced 4 days after inflammation (Figure 5F). However, loss of FXYD2 reversed these changes on day 4 post inflammation and resulted in more neurons with low frequency of AP firing (0-3 APs per stimulation) compared with control mice (Figure 5F). Thus, changes in the membrane potential and excitability of small DRG neurons are highly correlated with the increase in FXYD2/α1NKA interaction and the regulatory effect of FXYD2 on mechanical allodynia after peripheral inflammation.
FXYD2 is required for facilitating afferent synaptic transmission during inflammation
The lamina II in the dorsal horn of the spinal cord receives projections of nociceptive primary afferent axons, including fine myelinated A (Aδ)- and unmyelinated C-afferent fibers. The sensory afferent terminals in the lamina II then form synapses with the local neurons to convey sensory information to the central nervous system. Therefore, the lamina II of spinal cord plays an important role in the modulation of nociceptive synaptic transmission26,27. We then examined the effect of FXYD2 loss on the synaptic transmission between afferent terminals and local neurons in lamina II, which appeared to be a translucent band in the superficial dorsal horn of lumbar spinal cord under the microscope. The recorded spinal lamina II neurons showed sustained firing of APs induced by current injection under a current-clamp mode. We found that the mean frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in Fxyd2+/+ mice increased 2 and 4 days after CFA treatment, and the mean amplitude of sEPSCs increased 2 days after inflammation (Figure 6A and 6B). The mean frequency of sEPSCs in Fxyd2−/− mice was 58% of the sEPSC frequency in Fxyd2+/+ mice, and 2 days after inflammation the increase in the sEPSC frequency in Fxyd2−/− mice was less pronounced than that in Fxyd2+/+ mice (Figure 6B), suggesting that FXYD2 is involved in the regulation of excitatory afferent synaptic transmission. Furthermore, the increase in sEPSC frequency on day 4 post inflammation was absent in Fxyd2−/− mice (Figure 6A and 6B), consistent with the marked reduction in nociceptive responses at the same timepoint. The facilitation of afferent synaptic transmission was no longer observed in the spinal cord slices prepared from both Fxyd2+/+ and Fxyd2−/− mice 14 days after CFA injection (Figure 6A and 6B). Taken together, changes in the afferent synaptic transmission in Fxyd2−/− mice are highly correlated with the regulatory effect of FXYD2 on the inflammation-induced persistent mechanical allodynia.

FXYD2 is required for facilitating afferent synaptic transmission during inflammation. (A) Whole-cell recording from lamina II neurons in the spinal slice showed that the sEPSC frequency in Fxyd2+/+ mice was increased 2 and 4 days after CFA injection, and it was reduced to the basal level 14 days after CFA injection (n = 21 for control, n = 11 for CFA 2 days, n = 10 for CFA 4 days and n = 15 for CFA 14 days). In Fxyd2−/− mice, the increase of sEPSC frequency appeared 2 days after CFA injection, but was not detected 4 days after CFA injection (n = 11 for control, n = 12 for CFA 2 days, n = 14 for CFA 4 days and n = 20 for CFA 14 days). (B) The mean frequency of sEPSCs in Fxyd2−/− mice was reduced to 58% of that in Fxyd2+/+ mice. Two days after CFA injection, the increase in sEPSC frequency in Fxyd2−/− mice was less pronounced than that in Fxyd2+/++ mice. The increase of sEPSC frequency was absent in Fxyd2−/− mice 4 days after CFA injection. The changes of sEPSC amplitude in Fxyd2−/− mice were similar to those in Fxyd2+/+ mice. The increase in sEPSC frequency was not detected in either Fxyd2+/+ or Fxyd2−/− mice 14 days after inflammation. *P < 0.05, **P < 0.01 and ***P < 0.001 vs Fxyd2+/+ mice, and #P < 0.01 and ##P < 0.01 vs indicated. (C) Proposed model for two regulatory mechanisms of α1NKA at the axonal terminals of nociceptive afferent fibers. First, membrane depolarization triggers Ca2+-dependent exocytosis of synaptic vesicles and FSTL1 vesicles. Secreted FSTL1 activates the presynaptic α1NKA to hyperpolarize the membrane (Vm) and reduce Ca2+ influx, enabling an inhibitory regulation of the excitatory synaptic transmission. Second, FXYD2 interacts with α1NKA and reduces α1NKA activity. Such an effect of FXYD2 can be facilitated by peripheral inflammation, resulting in membrane depolarization and the facilitation of synaptic transmission.
Discussion
The present study shows that FXYD2 is expressed in most IB4-positive small DRG neurons and some peptidergic small DRG neurons in mice. FXYD2 interacts with the α1 subunit of NKA and negatively regulates the NKA activity, depolarizing the neuronal membrane potential. Four days after peripheral tissue inflammation, the FXYD2/α1NKA interaction peaks, and this level of interaction is required for maintaining the inflammation-induced membrane depolarization and excitability of the neurons and the facilitation of nociceptive afferent transmission. Persistent mechanical allodynia cannot develop in Fxyd2−/− mice 4 days after peripheral inflammation. Thus, FXYD2 plays an important role in the mechanism of chronic inflammatory pain.
FXYD2 mediates a distinct mechanism for regulating the activity of NKA in nociceptive neurons
NKA is regulated indirectly by neurotransmitters through receptor-mediated signaling pathways, and is therefore involved in many physiological and pharmacological functions, including opioid receptor-mediated morphine analgesia28,29. Some neurotransmitters can induce the phosphorylation of the α subunit through stimulating protein kinase A or C, thereby modulating the transport properties of NKA4,30. Moreover, NKA may also act as a signal transducer31. Our recent studies showed that the α1 subunit of NKA is activated directly by FSTL1 released from the sensory afferent terminals of small DRG neurons, enabling presynaptic inhibition of excitatory neurotransmission. In the present study, we attempted to explore the regulatory effect of FXYD2 on NKA in nociceptive DRG neurons. Previous studies showed that the affinity of the α subunit for Na+ and K+ was reduced by binding of FXYD213,14,15. We found that NKA activity was suppressed in both exogenous (COS7 cells) and endogenous (DRG neurons of Fxyd2-knockout mice) systems, suggesting that FXYD2 negatively regulates the NKA activity in IB4-positive subset of small DRG neurons and other FXYD2-expressing small DRG neurons.
Notably, the FXYD-mediated regulation appears to be an intrinsic mechanism contributed by a γ subunit component of NKA, which negatively regulates the activity of the α subunit. The activity of the α subunit could simultaneously be positively regulated by endogenous activator FSTL1 in small DRG neurons7. This regulatory mechanism is independent of FXYD: neither FXYD2 overexpression in COS7 cells nor FXYD2 deletion in DRG neurons visibly affected the FSTL1-induced increase in NKA activity. Thus, the α1NKA activity in sensory afferent neurons is regulated directly by at least two distinct mechanisms, FXYD2 for inhibition and FSTL1 for activation (Figure 6C).
FXYD2 regulates the neuronal membrane potential and synaptic transmission
NKA plays an essential role in the maintenance of resting membrane potential. The electrical potential of plasma membrane contributed by NKA is usually less than 10 mV32. One could expect a corresponding change in NKA activity and the membrane potential in neurons. The present study showed that FXYD2 deficiency caused a change of ∼4 mV in the membrane potential in IB4-positive small DRG neurons of mice, leading to membrane hyperpolarization, which would reduce the neuronal excitability (Figure 6C). This function of FXYD2 provides a considerable scope for regulation of the membrane potential by changing the number of this γ subunit in the NKA complex or by altering the biochemical properties or protein conformation of FXYD2. Previous studies have shown that a relatively small change in the membrane potential by modulating the NKA activity can markedly affect the synaptic transmission33. We found that loss of FXYD2 increased the number of small DRG neurons with a low frequency of AP firing, and it reduced the sEPSC frequency in the dorsal spinal cord. Thus, FXYD2 is required for regulating the firing rate of small DRG neurons and maintaining the afferent synaptic transmission within the physiological range.
The above notion is also supported by our findings from the CFA-induced inflammatory pain model. The interaction between FXYD2 and the α1 subunit of NKA was gradually increased after inflammation, and the corresponding reduction in NKA activity led to membrane depolarization in small DRG neurons and the increase of sEPSC frequency in the dorsal spinal cord (Figure 6C). These effects were attenuated in Fxyd2−/− mice, suggesting that FXYD2 is involved in the regulation of nociceptive afferent transmission after peripheral inflammation. Interestingly, we did not find apparent alterations in the expression of α1, α3 and β1 subunits of NKA or FXYD2 and FSTL1 in the DRG after inflammation. Previous studies showed that FXYD2 proteins inhibited NKA activity by reducing its affinity for Na+ in kidney34,35, and FXYD proteins reversed oxidative inhibition of NKA activity via facilitating deglutathionylation of the β1 subunit in cardiac myocytes36. However, in our study the contribution of the increased FXYD2/α1NKA interaction to the inflammation-induced facilitation of sensory afferent neurotransmission provides the first line of evidence that NKA activity can be regulated by changing the strength of interaction with the γ subunit in a state-dependent manner in the nerve system.
FXYD2 is required for maintaining inflammation-induced mechanical allodynia
The predominant expression of FXYD2 in IB4-positive small DRG neurons and its co-localization with MRGPRB4 and MRGPRD suggest that FXYD2 is involved in the regulation of mechanosensation, because MRGPRB4-positive neurons are activated by massage-like stroking of hairy skin and MRGPRD-positive neurons are activated by pinching but not by stroking24. Inflammatory mediators activate protein kinase Cɛ in IB4-positive nociceptors to produce a neuroplastic change that allows pronociceptive inflammatory mediators to produce the enhanced and prolonged mechanical hyperalgesia37,38. Moreover, FXYD2 is found in TH-positive small DRG neurons, which function as C-low-threshold mechanoreceptors25. FXYD2 might also be present in a population of peptidergic small DRG neurons that are involved in both thermal and mechanical hyperalgesia18,39. Therefore, the cellular distribution of FXYD2 suggests a role for FXYD2 in various mechanosensation phenomena and mechanical nociception.
Our study suggests that the FXYD2-induced negative regulation and the FSTL1-triggered activation of α1NKA contribute to the mechanism that homeostatically regulates the NKA activity, membrane potential and afferent synaptic transmission in a state-dependent manner (Figure 6C). Under normal circumstances, FSTL1-triggered activation of α1NKA would be more dominant than the effect of FXYD2 because both thermal and mechanical nociceptive responses were facilitated in FSTL1-deficient mice, but not in Fxyd2−/− mice. Moreover, FXYD2-null mice did not display any apparent defects in the formalin-induced nociceptive responses. Intriguingly, FXYD2 deletion impaired the maintenance, but not the initiation, of inflammation-induced mechanical allodynia, which lasted for at least 3 weeks in Fxyd2+/+ mice40,41. The mechanisms for maintaining the inflammation-induced mechanical allodynia have been largely unclear, though some factors such as activin C released from small DRG neurons can partially alleviate mechanical allodynia40. FSTL1 did not reduce the inflammation-induced nociceptive responses (data not shown), although it reduced the mechanical allodynia induced by nerve injury8. Thus, FXYD2 regulation may contribute to a fundamental mechanism underlying the persistence of mechanical pain following inflammation.
Interestingly, the FXYD2/α1NKA interaction gradually increased to a peak 4 days after inflammation. Genetic deletion of FXYD2 led to an elevation of NKA activity and corresponding membrane hyperpolarization in IB4-positive small DRG neurons, as well as a reduction of the inflammation-induced sEPSC frequency on day 4 post inflammation. Moreover, 14 days after inflammation, mechanical allodynia was alleviated in Fxyd2−/− mice but not in Fxyd2+/+ mice, although both the FXYD2/α1NKA interaction and the afferent transmission was also reduced in Fxyd2+/+ mice. Thus, the facilitation of excitatory afferent inputs on day 4 post CFA treatment, which was markedly reduced in the absence of FXYD2, may be required to induce the complex modulation of sensory circuits for further development of mechanical allodynia. Taken together, the increased FXYD2/α1NKA interaction in small DRG neurons may be an important mechanism for developing persistent mechanical allodynia, and day 4 post inflammation appears to be a critical time interval for the shift of acute pain to a chronic phase.
We conclude that peripheral inflammation increases the FXYD2/α1NKA interaction in a considerable number of small DRG neurons, particularly in IB4-positive ones, leading to the reduction in NKA activity, depolarization of membrane potential and facilitation of afferent synaptic transmission. Such a response contributes to the mechanism required for developing long-lasting mechanical allodynia during persistent inflammation, suggesting that FXYD2 could be a potential target for inflammatory pain therapy.
Materials and Methods
Animals
Experiments were approved by the Committee of Use of Laboratory Animals and Common Facility, Institute of Neuroscience, Chinese Academy of Sciences. Fxyd2−/− mice were purchased from Mutant Mouse Regional Resource Centers42. The heterozygotes were bred to obtain Fxyd2−/− mice and Fxyd2+/+littermates.
Animal model of inflammatory pain
Adult male mice were injected with 20 or 40 μl CFA in the plantar tissue of the bilateral hindpaws and were sacrificed at the indicated days after injection. L4 and L5 DRGs and spinal cords of mice were isolated for various experiments.
Real-time PCR
Total RNA (1 μg) extracted from the mouse tissues was reverse transcribed using oligo dT primers. Information about the primers for amplifying mRNAs encoding FXYD2, the α1 and β1 subunits of NKA, FSTL1 and GAPDH is provided in Supplementary information, Data S1. PCR was performed with equal amount of cDNA. The mRNA levels of FXYD2, the α1 and β1 subunits of NKA and FSTL1 were normalized to the GAPDH mRNA level. The Fxyd2 level in various tissues was normalized to that in the DRG.
In situ hybridization
Animals were fixed with 4% paraformaldehyde and 0.1% picric acid. L4 and L5 DRGs were dissected and post-fixed for 2 h. The tissue was cryo-protected and sectioned. Probes for the mRNAs encoding FXYD2, the α1 subunit of NKA, MRGPRB4, MRGPRD and TH are listed in Supplementary information, Data S1. The cRNA riboprobes were labeled with digoxigenin or FITC. The DRG sections were treated with proteinase K (10 μg/ml) in DEPC water for 10 min, acetylated and prehybridized in hybridization buffer for 3 h at 67 °C. Then, the sections were incubated with the hybridization buffer containing 1 μg/ml of the antisense probe for 16 h at 67 °C. After hybridization, the sections were incubated with alkaline phosphatase-conjugated sheep anti-digoxigenin antibody (anti-digoxigenin-AP; 1:2 000; Roche Molecular Biochemicals). The staining was developed in 1 μl/ml NBT and 3.5 μl/ml BCIP substrates in alkaline phosphatase buffer.
For in situ hybridization combined with immunostaining, the hybridized sections were incubated with anti-digoxigenin-AP (1:1 000) overnight at 4 °C. After being washed in TNT (TS7.5, 0.05% Tween 20) and TS8.0 (0.1 M Tris-HCl, pH 8.0, 0.1 M NaCl, and 10 mM MgCl2), the sections were incubated with HNPP/FR (1:100) in TS8.0. Once the signals were generated, the sections were washed and then processed for immunostaining.
For double fluorescent in situ hybridization, the probe for FXYD2 mRNA was labeled with FITC, and the probe for the α1 subunit of NKA was labeled with digoxigenin. Both probes were added to the hybridization buffer. After being washed and blocked, the sections were incubated with HRP-conjugated anti-FITC antibody (1:4 000) overnight at 4 °C. After being washed with TNT, the sections were incubated with TSA-Plus (DNP, 1:50) for 10 min. Then, the sections were washed with TNT and incubated with anti-digoxigenin-AP (1:1 000) and Alexa488-conjugated anti-DNP antibody (1:500) in 1% blocking reagent overnight at 4 °C. Sections were washed with TNT and TS8.0, and HNPP/FR (1:100) in TS8.0 was added to develop the staining.
Cell culture and transfection
COS7 cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Biochrom). Then the COS7 cells were transiently transfected with plasmids pCDNA3.1-mFXYD2-Myc, pCDNA3.1-mATP1B1-HA and pIRES-EGFP-mATP1A1-Flag using Lipofectamine 2000 (Invitrogen). Two days after transfection, the cells were used for various assays.
Immunohistochemistry
Adult male mice were fixed with 4% paraformaldehyde and 1% picric acid. Cryostat sections of DRGs were processed for double-immunofluorescent staining or in situ hybridization combined with immunostaining. The sections were stained with rabbit antibodies against CGRP (1:2 000; 24112, Dia Sorin) or peripherin (1:500; AB1530, Chemicon), or 10 μg/ml fluorescein-conjugated IB4 (Vector Laboratories), or mouse antibodies against NF200 (1:2 000; N2912, Sigma), followed by incubation with Cy3-conjugated donkey secondary antibodies against rabbit (1:500; Jackson ImmunoResearch) and examined under a SP5 Laser Scanning Confocal Microscope (Leica).
Co-immunoprecipitation and immunoblotting
The spinal cord or DRGs or COS7 cells were lysed in ice-cold cell lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% Triton X-100, 10% glycerol, 0.5 mg/ml BSA and protease inhibitors). The suspended lysate was immunoprecipitated with 0.5 μg mouse antibodies against α1 subunit (MA3-929, ABR) or 0.5 μg rabbit antibodies against Flag (F7425, Sigma) overnight at 4 °C and then with protein G-Agarose beads for 1 h at 4 °C. Immunoprecipitates in the beads were collected. The sepharose was resuspended in ice-cold cell lysis buffer and incubated in sample buffer (50 mM Tris-HCl, pH 7.4, 2% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.01% bromophenol blue) for 20 min at 60 °C.
Immunoprecipitated samples and 3% of the lysate were run on SDS-PAGE and subjected to immunoblotting with goat antibodies against FXYD2 (1:500; sc-26100, Santa Cruz) or mouse antibodies against α1 subunit (1:2 000; MA3-929, ABR). Proteins were subsequently visualized with enhanced chemiluminescence. The lysate and immunoprecipitates were exposed separately. The intensity of immunoreactive bands was analyzed with the Image-Pro Plus 5.1 software (Media Cybernetics).
To examine the axonal transport of FXYD2, mice were subjected to surgery and L4-L5 dorsal roots were transected. Three days after the surgery, L4-L5 segment of the spinal cord was dissected, and the proteins collected from the spinal dorsal horn were processed for immunoblotting.
Microsome preparation
Plasma membrane was prepared as described previously43. In brief, resuspended COS7 cells or mouse DRG were homogenized on ice by 20 strokes with a tight-fitting Dounce homogenizer in 1 ml homogenization buffer (5 mM HEPES, 1 mM PMSF, 50 μM CaCl2, 10% sucrose and 1 mM DTT). Then the homogenate was centrifuged at 1 000× g for 10 min at 4 °C. The supernatant was centrifuged at 12 000× g for 20 min at 4 °C to yield the crude plasma membrane. The obtained pellet was washed, followed by resuspension and recentrifugation at 14 000× g for 20 min at 4 °C. The final pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.4) to obtain microsome. The protein concentration was determined with Lowry method44.
Measurement of NKA activity
The NKA activity of membrane preparation from COS7 cells or mouse DRG was measured using NKA activity assay kit (Nanjing Jiancheng Bioengineering Institute) as described previously43. Briefly, a 100 μl microsome of COS7 cells or mouse DRG containing 20 μg of protein were incubated with 530 μl of reaction buffer (100 mM NaCl, 20 mM KCl, 2 mM MgCl2, 30 mM Histidine, pH 7.4) for 10 min at 37 °C. To measure the ouabain-insensitive ATPase, the same buffer was used but with 1 mM ouabain. The reaction was carried out by adding 50 μl of ATP disodium solution (final ATP concentration: 2.5 mM). After incubation for 10 min at 37 °C, the reaction was terminated by adding trichloroacetic acid (50%, 100 μl). The reacted solution was centrifuged at 4 000 rpm/min for 10 min at 4 °C. Then 1 ml of ammonium molybdate color reagent was added to 300 μl of the supernatant for 5 min. The absorbance was read at 636 nm in a spectrophotometer, using Na2HPO4 as a standard. NKA activity was measured by subtracting ouabain-insensitive Mg2+-ATPase activity from the total ATPase activity. The NKA activity levels in COS7 cells and DRG neurons were 23-87 and 123-310 nmol/min/mg protein, respectively. We also performed the time course of NKA activation by adding ATP into the reaction buffer with the purified microsome. To terminate the reaction, trichloroacetic acid was added after incubation for 1, 3, 5, 10, 20 or 30 min at 37 °C. NKA activity was plotted against the reaction time, which showed that NKA activity detected at 10 min fitted in the liner range of NKA activity time curve (Supplementary information, Figure S4).
Electrophysiological recording
L4 and L5 DRGs of mice were dissected and digested with collagenase (1 mg/ml), trypsin (0.4 mg/ml) and DNase (0.1 mg/ml) in DMEM for 30 min at 37 °C. After trituration, freshly dissociated DRG neurons were plated on coverslips and incubated in the ECS (150 mM NaCl, 5 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, pH 7.34) for at least 1 h. The freshly dissociated neurons were incubated with fluorescein-conjugated IB4 (1:1 000, FL-1201, Vector Laboratories) in ECS for 10 min. Cell recording was carried out in ECS with patch pipettes (resistance of 3-6 MΩ). The translucent and brilliant small DRG neurons labeled with fluorescein-conjugated IB4 were patched and recorded. The whole-cell configuration was obtained in voltage-clamp mode before proceeding to current-clamp recording. Whole-cell recordings were performed using the Axopatch 700B amplifier, digitized using the Digidata 1440A interface, and controlled using pCLAMP10.1 software. For detection of membrane potential, the DRG neuron was first subjected to whole-cell recording in voltage-clamp mode at −70 mV. After 5 min, the program was switched to current-clamp mode and no current was injected in the neuron. The membrane potential was recorded for at least 4 min. The values of membrane potential within 1-4 min of recording were averaged to be the mean value. During the recording, the variation of membrane potential was < 1 mV. The noise of membrane potential recording was < 0.2 mV. To induce a single AP for analysis of AHP, a single pulse (15 ms, 100 pA) was injected into the DRG neurons under current-clamp mode. To induce repetitive firing of AP, the current with an extended duration (200 ms) and increasing intensity was injected into the DRG neuron.
Transverse slices of spinal cord of adult mice were prepared for whole-cell recordings. Lamina II was a translucent band in the superficial dorsal horn of spinal cord under microscope. The pipette solution contained 135 mM K-gluconate, 0.5 mM CaCl2, 2 mM MgCl2, 5 mM KCl, 5 mM EGTA, 5 mM HEPES and 5 mM D-glucose. Resistance of patch pipettes was typically 4-10 MΩ. Currents were filtered at 2 kHz and digitized at 5 kHz using the Axopatch 200B and analyzed by pCLAMP10.1. Holding potential was −70 mV. sEPSCs were studied in the presence of 20 μM bicuculline and 2 μM strychnine. Frequency and amplitude of sEPSCs were analyzed.
Behavior tests
For behavior testing, Fxyd2−/− mice and littermates were habituated, and tests were performed blind to genotype. Procedures for rotarod test, radiant heat test, hot plate test and formalin test are provided in Supplementary information, Data S1. Mechanical threshold of the hindpaw was determined with von Frey fibers. Mechanical stimuli were applied using ascending graded individual monofilaments. A von Frey filament was applied 5 times (several seconds for each stimulus) to each testing site of the hindpaw. The bending force of the von Frey filament to evoke paw withdrawal with over 50% occurrence frequency was determined as the mechanical threshold.
After intraplantar injection with 20 μl CFA, the thermal hyperalgesia only persisted for 1 week, whereas the mechanical allodynia persisted for 2 weeks. After injection with 40 μl CFA, the thermal hyperalgesia persisted for 2 weeks and the mechanical allodynia persisted for 4-5 weeks. To avoid the ceiling effect, we chose both the low dose (20 μl) and high dose (40 μl) of CFA for detection of thermal hyperalgesia. The measurements were performed before and 1, 2, 4 and 7 days after injection. We chose 40 μl CFA for detection of the mechanical sensitivity using the von Frey test, and these measurements were performed before and 1, 2, 4, 7, 14 and 21 days after injection.
Statistical analysis
Statistical analysis for enzyme activity, behavior tests, and electrophysiology was performed by Student's t-test or paired t-test. Comparisons between multiple groups were performed using ANOVA with a post hoc Bonferroni's test. Statistical analysis for firing frequency was performed by χ2 test. Data are presented as mean ± SEM. Differences were considered statistically significant when P < 0.05.
Details of the materials and experimental procedures are provided in Supplementary information, Data S1.
References
Geering K . The functional role of the β-subunit in the maturation and intracellular transport of Na,K-ATPase. FEBS Lett 1991; 285:189–193.
Noguchi S, Mishina M, Kawamura M, Numa S . Expression of functional (Na+ + K+)-ATPase from cloned cDNAs. FEBS Lett 1987; 225:27–32.
Jewell EA, Lingrel JB . Comparison of the substrate dependence properties of the rat Na,K-ATPase 1, 2, and 3 isoforms expressed in HeLa cells. J Biol Chem 1991; 266:16925–16930.
Therien AG, Blostein R . Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol 2000; 279:C541–566.
Charnock JS, Post RL . Evidence of the mechanism of ouabain inhibition of cation-activated adenosine triphosphate. Nature 1963; 199:910–911.
Nesher M, Shpolansky U, Rosen H, Lichtstein D . The digitalis-like steroid hormones: new mechanisms of action and biological significance. Life Sci 2007; 80:2093–2107.
Li KC, Zhang FX, Li CL, et al. Follistatin-like 1 suppresses sensory afferent transmission by activating Na+,K+-ATPase. Neuron 2011; 69:974–987.
Li KC, Wang F, Zhong YQ, et al. Reduction of follistatin-like 1 in primary afferent neurons contributes to neuropathic pain hypersensitivity. Cell Res 2011; 21:697–699.
Crambert G, Geering K . FXYD proteins: new tissue-specific regulators of the ubiquitous Na,K-ATPase. Sci STKE 2003; 2003:RE1.
Sweadner KJ, Rael E . The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics 2000; 68:41–56.
Geering K . FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol 2006; 290:F241–F250.
Forbush B 3rd, Kaplan JH, Hoffman JF . Characterization of a new photoaffinity derivative of ouabain: labeling of the large polypeptide and of a proteolipid component of the Na, K-ATPase. Biochemistry 1978; 17:3667–3676.
Arystarkhova E, Wetzel RK, Asinovski NK, Sweadner KJ . The γ subunit modulates Na+ and K+ affinity of the renal Na,K-ATPase. J Biol Chem 1999; 274:33183–33185.
Beguin P, Crambert G, Guennoun S, Garty H, Horisberger JD, Geering K . CHIF, a member of the FXYD protein family, is a regulator of Na,K-ATPase distinct from the γ-subunit. EMBO J 2001; 20:3993–4002.
Beguin P, Wang X, Firsov D, et al. The γ subunit is a specific component of the Na,K-ATPase and modulates its transport function. EMBO J 1997; 16:4250–4260.
Maxwell DJ, Réthelyi M . Ultrastructure and synaptic connections of cutaneous afferent fibres in the spinal cord. Trends Neurosci 1987; 10:117–123.
Woolf CJ, Ma Q . Nociceptors-noxious stimulus detectors. Neuron 2007; 55:353–364.
Gold MS, Gebhart GF . Nociceptor sensitization in pain pathogenesis. Nat Med 2010; 16:1248–1257.
Fink DJ, Fang D, Li T, Mata M . Na,K-ATPase β subunit isoform expression in the peripheral nervous system of the rat. Neurosci Lett 1995; 183:206–209.
Mata M, Siegel GJ, Hieber V, Beaty MW, Fink DJ . Differential distribution of (Na,K)-ATPase α isoform mRNAs in the peripheral nervous system. Brain Res 1991; 546:47–54.
Dobretsov M, Hastings SL, Stimers JR . Non-uniform expression of α subunit isoforms of the Na+/K+ pump in rat dorsal root ganglia neurons. Brain Res 1999; 821:212–217.
Dobretsov M, Hastings SL, Stimers JR . Functional Na+/K+ pump in rat dorsal root ganglia neurons. Neuroscience 1999; 93:723–729.
Venteo S, Bourane S, Mechaly I, et al. Regulation of the Na,K-ATPase gamma-subunit FXYD2 by Runx1 and Ret signaling in normal and injured non-peptidergic nociceptive sensory neurons. PLoS One 2012; 7:e29852.
Vrontou S, Wong AM, Rau KK, Koerber HR, Anderson DJ . Genetic identification of C fibres that detect massage-like stroking of hairy skin in vivo. Nature 2013; 493:669–673.
Li L, Rutlin M, Abraira VE, et al. The functional organization of cutaneous low-threshold mechanosensory neurons. Cell 2011; 147:1615–1627.
Ataka T, Kumamoto E, Shimoji K, Yoshimura M . Baclofen inhibits more effectively C-afferent than Aδ-afferent glutamatergic transmission in substantia gelatinosa neurons of adult rat spinal cord slices. Pain 2000; 86:273–282.
Bird GC, Han JS, Fu Y, Adwanikar H, Willis WD, Neugebauer V . Pain-related synaptic plasticity in spinal dorsal horn neurons: role of CGRP. Mol Pain 2006; 2:31.
Masocha W, Horvath G, Agil A, et al. Role of Na+, K+-ATPase in morphine-induced antinociception. J Pharmacol Exp Ther 2003; 306:1122–1128.
Woolcock K, Specht SC . Modulation of Na, K-ATPase activity by prostaglandin E1 and [D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin. Life Sci 2006; 78:1653–1661.
Feraille E, Doucet A . Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol Rev 2001; 81:345–418.
Aizman O, Aperia A . Na+,K+-ATPase as a signal transducer. Ann NY Acad Sci 2003; 986:489–496.
Albers RWS, Siegel GJ . The ATP-Dependent Na+, K+ Pump. in: Basic neurochemistry: molecular, cellular, and medical aspects. 6th Edition. Philadelphia: Lippincott-Raven 1999.
Scuri R, Lombardo P, Cataldo E, Ristori C, Brunelli M . Inhibition of Na+/K+ ATPase potentiates synaptic transmission in tactile sensory neurons of the leech. Eur J Neurosci 2007; 25:159–167.
Arystarkhova E, Wetzel RK . Gamma structural variants differentially regulate Na,K-ATPase properties. Ann NY Acad Sci 2003; 986:416–419.
Arystarkhova E, Sweadner KJ . Splice variants of the gamma subunit (FXYD2) and their significance in regulation of the Na, K-ATPase in kidney. J Bioenerg Biomembr 2005; 37:381–386.
Bibert S, Liu CC, Figtree GA, et al. FXYD proteins reverse inhibition of the Na+-K+ pump mediated by glutathionylation of its β1 subunit. J Biol Chem 2011; 286:18562–18572.
Ferrari LF, Bogen O, Levine JD . Nociceptor subpopulations involved in hyperalgesic priming. Neuroscience 2010; 165:896–901.
Ferrari LF, Bogen O, Levine JD . Role of nociceptor αCaMKII in transition from acute to chronic pain (hyperalgesic priming) in male and female rats. J Neurosci 2013; 33:11002–11011.
Joseph EK, Levine JD . Mu and delta opioid receptors on nociceptors attenuate mechanical hyperalgesia in rat. Neuroscience 2010; 171:344–350.
Liu XJ, Zhang FX, Liu H, et al. Activin C expressed in nociceptive afferent neurons is required for suppressing inflammatory pain. Brain 2012; 135:391–403.
Zhang FX, Liu XJ, Gong LQ, et al. Inhibition of inflammatory pain by activating B-type natriuretic peptide signal pathway in nociceptive sensory neurons. J Neurosci 2010; 30:10927–10938.
Jones DH, Li TY, Arystarkhova E, et al. Na,K-ATPase from mice lacking the γ subunit (FXYD2) exhibits altered Na+ affinity and decreased thermal stability. J Biol Chem 2005; 280:19003–19011.
Wu ZQ, Li M, Chen J, Chi ZQ, Liu JG . Involvement of cAMP/cAMP-dependent protein kinase signaling pathway in regulation of Na+,K+-ATPase upon activation of opioid receptors by morphine. Mol Pharmacol 2006; 69:866–876.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ . Protein measurement with the folin phenol reagent. J Biol Chem 1951; 193:265–275.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31130066) and the Strategic Priority Research Program (B) of Chinese Academy of Sciences (XDB01020300).
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Recording of membrane potential in IB4-negative small DRG neurons. (PDF 39 kb)
Supplementary information, Figure S2
Recording of AP and AHP in IB4-positive small DRG neurons. (PDF 39 kb)
Supplementary information, Figure S3
Expression of α1 and β1 subunits of NKA, FSTL1 and FXYD2 in DRGs after peripheral inflammation. (PDF 135 kb)
Supplementary information, Figure S4
Time course of NKA activity in ATP-treated microsomes. (PDF 36 kb)
Supplementary information, Data S1
Materials and Methods (PDF 42 kb)
Rights and permissions
About this article

Cite this article
Wang, F., Cai, B., Li, KC. et al. FXYD2, a γ subunit of Na+,K+-ATPase, maintains persistent mechanical allodynia induced by inflammation. Cell Res 25, 318–334 (2015). https://doi.org/10.1038/cr.2015.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2015.12
Keywords
This article is cited by
-
Single cell multiomic analysis reveals diabetes-associated β-cell heterogeneity driven by HNF1A
Nature Communications (2023)
-
FXYD2 mRNA expression represents a new independent factor that affects survival of glioma patients and predicts chemosensitivity of patients to temozolomide
BMC Neurology (2021)
-
Effect of a purine derivative containing selenium to improve memory decline and anxiety through modulation of the cholinergic system and Na+/K+-ATPase in an Alzheimer’s disease model
Metabolic Brain Disease (2021)
-
The spinal microglial IL-10/β-endorphin pathway accounts for cinobufagin-induced mechanical antiallodynia in bone cancer pain following activation of α7-nicotinic acetylcholine receptors
Journal of Neuroinflammation (2020)
-
High-dose maternal folic acid supplementation before conception impairs reversal learning in offspring mice
Scientific Reports (2017)
