
Abstract
The defining features of embryonic stem cells (ESCs) are their self-renewing and pluripotent capacities. Indeed, the ability to give rise into all cell types within the organism not only allows ESCs to function as an ideal in vitro tool to study embryonic development, but also offers great therapeutic potential within the field of regenerative medicine. However, it is also this same remarkable developmental plasticity that makes the efficient control of ESC differentiation into the desired cell type very difficult. Therefore, in order to harness ESCs for clinical applications, a detailed understanding of the molecular and cellular mechanisms controlling ESC pluripotency and lineage commitment is necessary. In this respect, through a variety of transcriptomic approaches, ESC pluripotency has been found to be regulated by a system of ESC-associated transcription factors; and the external signalling environment also acts as a key factor in modulating the ESC transcriptome. Here in this review, we summarize our current understanding of the transcriptional regulatory network in ESCs, discuss how the control of various signalling pathways could influence pluripotency, and provide a future outlook of ESC research.
Similar content being viewed by others
Introduction
Embryonic stem cells (ESCs) are derived from the inner cell mass (ICM) of pre-implantation embryos1,2,3; and the two defining features of ESCs are their ability to self-renew, as well as give rise to all cell lineages of the organism. This unique feature of ESCs to form the various tissue types, termed 'pluripotency', besides enabling the in vitro study of early mammalian development, has also facilitated the advancement of regenerative medicine, whereby ESCs could be used to create clinically relevant cell types for the replacement of worn-out tissue. However, it is this same remarkable developmental plasticity of ESCs which poses a major challenge towards the efficient control of ESC differentiation into the desired lineage. Therefore, to better understand mammalian development, as well as to exploit the tremendous therapeutic potential of ESCs, it is necessary to identify the molecular mechanisms governing a pluripotent or differentiated ESC fate. Here in this review, we will summarize the current progress towards understanding the ESC transcriptional regulatory network, and also discuss how modulation of the various signalling pathways in ESCs could influence pluripotency.
Dissecting the ESC transcriptional regulatory network
The core ESC pluripotency factors: Oct4, Sox2 and Nanog
In ESCs, the pluripotent state is mainly regulated by the core transcription factor trio of Oct4, Sox2 and Nanog4.
Oct4 (encoded by the Pou5f1 gene), belongs to the Pit-Oct-Unc (POU) family of homeodomain proteins, and is exclusively expressed within the totipotent mouse blastomeres, pluripotent epiblast as well as primodial germ cells (PGCs)5,6. Importantly, Oct4 plays a critical role in the establishment and maintenance of pluripotency, as Pou5f1-null embryos do not form a pluripotent ICM, but rather, differentiate into trophectodermal tissue7. Similarly, Oct4 is also critical for maintaining mouse ESCs (mESCs) in an undifferentiated state and has to be tightly regulated. Depletion of Oct4 mRNA by 50% is sufficient to result in the formation of trophectoderm cells, while Oct4 overexpression by 50% will promote mesodermal and endodermal differentiation8.
Sox2, which contains the high-mobility group box DNA-binding domain, is expressed within the ICM and extraembryonic ectoderm of pre-implantation blastocysts9. Like Oct4 knockout mice, Sox2-null blastocysts fail to form a pluripotent ICM9, and mESCs deficient in Sox2 differentiated primarily into trophectoderm10. This similarity of phenotypes produced by Sox2 and Oct4 loss is attributed to the synergistic action of Oct4/Sox2 in the regulation of various ESC-specific genes11,12,13,14,15,16, including themselves17,18,19. Indeed, ectopic Oct4 expression was found to be sufficient in rescuing the differentiation phenotype of Sox2−/− mESCs10.
Nanog, the third member of the core ESC transcription factors, was discovered through a screen for pluripotency factors that could sustain mESC self-renewal in the absence of leukemia inhibitor factor (LIF)20,21. Although Nanog-null pre-implantation embryos do not possess a pluripotent ICM21,22, Nanog−/− mESCs could be established through the in vitro disruption of both endogenous Nanog alleles21,23. Importantly, these Nanog-null mESCs although prone to differentiation, could still be kept pluripotent21,23. Therefore, it is believed that Nanog, while necessary for the acquisition of pluripotency, is dispensable once pluripotency is achieved23.
Given the important roles of the core ESC transcription factors in establishing a pluripotent stem cell fate, chromatin immunoprecipitation (ChIP) technologies have been used to map the genomic-binding sites of these core ESC factors in mESCs and hESCs. Together, these studies found extensive Oct4, Sox2 and Nanog co-binding at numerous active, as well as silent genomic target sites24,25. Together, it is proposed that the core ESC transcription factors serve to establish a pluripotent state by: (1) activating the expression of other pluripotency-associated factors while simultaneously repressing lineage-specific genes, and (2) by activating their own gene expression and that of each other4. Importantly, this model may account for how ESCs can sustain self-renewal and pluripotency, while still remain poised for differentiation.
The expanded ESC pluripotency network
To uncover additional novel ESC regulators, one method is to perform RNA interference (RNAi)-mediated gene knockdown and to observe for any loss of pluripotency. Indeed, large-scale RNAi knockdown studies have led to the discovery of important mESC factors such as Esrrb, Tbx3 and Tcl126, as well as the chromatin regulators Tip60-p40027 and SetDB128. Similarly, unbiased genome-wide siRNA screens were able to identify Cnot3 and Trim2829, Paf1C30 and the mediator and cohesin complex31 as important mESC transcriptional cofactors. Extending this approach into hESCs, Chia et al.32 used a similar genome-wide siRNA screening to identify components of the INO80 chromatin remodeling complex, the mediator and TAF transcriptional regulatory complexes, and the COP9 signalosomes as important hESC factors. Importantly, the PRDM14 transcription factor was found to be an essential factor for hESCs, but not in mESCs, hence highlighting that critical species-specific differences exist between mouse and human ESCs (hESCs)32.
Determining the protein-binding partners of known pluripotency factors is another method of identifying novel ESC regulators. Through the coupling of affinity purification methods with mass spectrometry technology, numerous co-binding proteins of the core pluripotency transcription factors have been identified33,34,35,36,37,38,39,40. Taken together, these studies reveal an extensive protein-protein interaction network which includes other ESC transcription regulators, chromatin remodeling and modifying factors, DNA methyltransferases and Polycomb group proteins (PcG). This therefore suggests that the core ESC factors may regulate gene expression through the modulation of chromatin states. Importantly, this large and intricate network of protein interactions could suggest how small changes in the levels of core ESC factors, like Oct4 or Sox2, are sufficient to perturb the ESC self-renewal programme to trigger differentiation8,41.
Using ChIP with massively parallel DNA sequencing (ChIP-seq)42, or in vivo biotinylated ChIP with DNA microarray43, two independent groups examined the context in which these additional pluripotency factors could play in the specification of an ESC identity. Together, these studies observed the binding of multiple transcription factors onto a similar genomic region; and importantly, these factors could also be grouped into either an Oct4- or Myc-centric module based on their genomic targets43. These data thus indicate that within ESCs, the Myc-cluster appears to function independently from the core pluripotency network29,42,43.
The role of Myc in ESC transcriptional regulation
The Myc module which consists of c-Myc, n-Myc, Rex1, Zfx and E2f1, is known to be involved in self-renewal and cell metabolism42,43,44. Although approximately one-third of all active ESC genes are bound by both c-Myc and the core ESC pluripotency factors45, the functions of these two modules in gene regulation appear to differ. For instance, Oct4, Sox2 and Nanog in concert with the Mediator complex are able to recruit RNA polymerase II (RNA Pol II) for gene transcription31, while c-Myc is believed to control the transcriptional pause release of RNA Poly II via the p-TEFb cyclin-dependant kinase45. It is thus believed that the core ESC factors will select ESC genes for expression through the recruitment of RNA Pol II, while c-Myc functions to control gene expression through the release of transcriptional pause4. Importantly, it should also be highlighted that initial reports of cancer cells possessing an ESC-like transcriptional program46,47 were later attributed to be a consequence of Myc-module activity, as opposed to the core ESC factors48.
Recent data by Lin et al.49 and Nie et al.50 suggest that rather than activating new genes, Myc acts only to amplify the existing transcriptional output of active genes. By increasing the levels of Myc, both groups found the loading of Myc proteins onto the promoters of active genes, but not for promoters of silent genes. This therefore indicates that Myc is unable to initiate de novo gene activation. Indeed, higher levels of p-TEFb occupancy with increased levels of elongation-associated RNA Polymerase II phosphorylation were found at Myc-bound sites49, consistent with the previously proposed idea that c-Myc controls transcriptional pause release45. Taken together, this ability to amplify existing active gene transcription may in part suggest how the addition of c-Myc during reprogramming is able to increase the efficacy of induced pluripotent stem cell (iPSC) formation51.
Transcriptional control of non-coding RNAs in ESCs
MicroRNAs (miRNAs), which post-transcriptionally regulate mRNA levels, are important for proper ESC function52,53. Importantly through ChIP-seq, Marson et al.54 found the binding of Oct4, Sox2, Nanog and Tcf3 transcription factors at ESC-related miRNA genes, as well as transcriptionally silent PcG-occupied tissue-specific miRNAs, suggesting that the core ESC transcriptional network is able to regulate miRNA expression. A notable example of these inactive miRNAs includes let-7, which is known to target c-Myc and the pluripotency factors Sall4 and Lin2855. Interestingly, the ESC-related miRNA-302/367 cluster which is also regulated by the ESC transcriptional circuit54, is able to reprogramme fibroblasts into iPSCs without the need for additional protein factors56.
Large intergenic non-coding RNAs (lincRNAs) expressed in mESCs are also known to be the targets of several pluripotency-associated transcription factors, and have been demonstrated to be essential for maintaining a pluripotent state and suppress lineage specification57. Upon knockdown, many of these ESC lincRNAs were shown to induce gene expression changes similar to the depletion of known ESC factors. Importantly, it was found that lincRNAs could associate with multiple chromatin complexes, hence suggesting the possibility that lincRNAs may serve as scaffolds for the recruitment of different protein complexes for specific functions. In that regard, it would be interesting to examine how the ESC-specific lincRNA expression signature, through the assembly of various protein complexes, is able to sustain ESC pluripotency.
Extrinsic signalling and ESC transcriptional regulation
Signalling in mouse ESCs
Traditionally, mESCs were cultured and kept pluripotent on a layer of mitotically inactivated feeder cells in serum-supplemented media1,2 (Figure 1). However, the undefined nature of feeder co-culture posed a significant challenge in mapping the specific extrinsic signalling factors, cellular pathways involved and their effects upon gene transcription which ultimately control ESC self-renewal and differentiation.

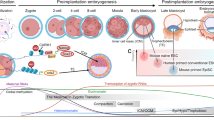
Interconversion between mouse pluripotent states. mESCs are derived from the inner cell mass (ICM) of mouse pre-implantation blastocysts, while EpiSCs are isolated from the epiblast compartment of post-implantation embryos. The conversion between the pluripotent mESC and EpiSC states is possible through cell culture; introduction of transcription factors (TFs), or application of chemical inhibitors. Unipotent PGCs are derived from the embryonic gonad and can also be differentiated from EpiSCs using BMP4; or with BMP4, BMP8b, LIF, and stem cell factor (SCF). Subsequently, PGCs can be restored to pluripotency through the culture with LIF, FGF2 and SCF to form mouse embryonic germ cells (EGCs). mESCs are also known to transiently cycle into a 2-cell (2C)-like state with expanded developmental potential, giving rise to both embryonic and extraembryonic tissues. The process and mechanism by which this 2C transition occurs are still not known, and whether 2C-like cells could be directly isolated from 2C embryos has not been established.
After the initial discovery of mESCs in 1981, work over the next 20 years then identified the IL-6 family cytokine LIF as the key active component produced by feeder cells which sustains mESC growth58,59, as well as Bone morphogenic protein 4 (BMP4) which could substitute for the use of serum in mESC culture60. Critically, the combinatorial use of LIF and BMP4 alone in a defined feeder- and serum-free culture was sufficient to derive and maintain germ-line transmittable mESCs60, hence delineating the signalling pathways controlling mESC self-renewal and pluripotency. At the transcriptional level, LIF and BMP4 signalling is able to induce phosphorylation and activation of their downstream transcription factors Stat3 and Smad1 respectively60,61. Importantly, genome-wide mESC transcription factor mapping studies reveal the co-binding of Stat3 and Smad1 transcription factors at Oct4, Sox2 and Nanog regions, thus demonstrating how LIF and BMP4 signalling may sustain the core ESC transcriptional network42.
In contrast to the self-renewing effect of LIF and BMP4, mESC autocrine stimulation by fibroblast growth factor 4 (Fgf4)62,63, working through the Mek/Erk signalling pathway, is known to induce mESCs to exit self-renewal and initiate differentiation64,65. This pro-differentiation effect of Fgf/Mek/Erk signalling can be further inferred through complementary experiments, whereby chemical inhibition or genetic knockout of Fgf/Mek/Erk signalling cascade components caused impaired mESC differentiation64,65,66,67,68. The exact mechanism by which active Mek/Erk signalling induces mESCs to exit pluripotency is still not known. Given that terminal signalling kinases such as Erk have been previously shown in other cell types to regulate gene expression activity via transcription factor phosphorylation69,70, phosphorylation of chromatin remodelling complexes71, and direct binding onto DNA as a transcriptional repressor72, it would be interesting to determine if Erk may adopt similar mechanisms in regulating mESC pluripotency.
mESCs are also responsive to Wnt-signalling, but whether Wnt signalling promotes self-renewal or causes mESC differentiation is still being actively debated73,74. Active canonical Wnt-signalling leads to the stabilisation of β-catenin, hence allowing for the association of β-catenin with the TCF/LEF family of transcription factors for gene activation73. Tcf3, which is the most abundant Tcf protein in mESCs75, has been previously shown to co-localize with the core Oct4, Sox2 and Nanog-binding sites54,76, therefore suggesting that Wnt signalling, like LIF and BMP pathways, is integrated into the ESC transcriptional regulatory core. In mESCs, Tcf3 functions primarily as a transcriptional repressor75,77,78 and Tcf3-null mESCs are resistant towards differentiation75,79. However, as the process in which Tcf3 regulates gene expression is still not clear, and with multiple models being proposed to account for the mechanism of how β-catenin and Tcf3 interaction could initiate gene expression73, additional studies are clearly needed to define the role of Wnt-signalling in ESCs. Additionally, given that Wnt-signalling in hESCs has not been well explored, it would also be interesting to determine if similar Wnt-regulatory pathways are conserved in hESCs.
Signalling in hESCs
Although hESCs may share the same Oct4-Sox2-Nanog core ESC transcriptional regulatory circuit24,25; they differ from mESCs in their extrinsic signalling requirement to maintain self-renewal and pluripotency. Previous studies have shown that LIF is dispensable for hESC culture80, while the presence of BMP4 will cause hESC differentiation into trophoblast81. Rather, FGF2 and Activin/Nodal signalling is critical for the maintenance of an undifferentiated hESC state82,83,84.
The exact mechanism by which FGF2-signalling sustains hESC pluripotency is still not clear and is complicated by the fact that FGF-signalling could activate multiple signalling cascades85. Issues pertaining to signalling 'crosstalk' by other receptor kinases onto FGF-receptor-associated pathways86,87, as well as varying hESC culture conditions employed by different groups, have made the dissection of hESC FGF2-signalling more difficult. However, previous studies have indicated that FGF-signalling through its downstream protein ERK is necessary to prevent extra-embryonic differentiation of hESCs88,89, and that FGF2 may cooperate with other growth factors (like Activin signalling, through SMAD2/3 transcription factors), to upregulate the expression of the core pluripotency gene NANOG84,90.
There have already been several large-scale attempts at profiling the global hESC phosphoproteome via mass spectrometry techniques91,92,93,94, with two studies seeking to specifically address the dynamics of FGF2-dependent tyrosine and serine/threonine phosphorylation95,96. Together, FGF2-signalling in hESCs not only resulted in phosphorylation of proteins of various signalling cascades such as that of PI3K, MAPK, Wnt, but could also lead to phosphorylation of pluripotency-associated transcription regulators like OCT4, SOX2, SALL4 and DPPA495,96. These studies while informative in revealing a possible phospho-interactome downstream of FGF2-FGFR, unfortunately do not factor in signalling 'crosstalk' by other receptor kinases onto FGFR-associated pathways86,87. Therefore, future attempts at studying the hESC signalling pathways, aside from adopting defined culture conditions, should also seek to utilize specific kinase inhibitors, gene knockdown or gene deletion strategies to more accurately delineate the cell-signalling events.
Unlike the ambiguity surrounding FGF2 signalling, Smad2/3, the downstream effectors of Activin/Nodal signaling are previously known to directly bind and regulate the expression of NANOG97. Recent ChIP-seq in hESCs also found the binding of SMAD2/3 at OCT4, TERT, MYC and DPPA4 genes, with SMAD2/3 sharing approximately one-third overlap with NANOG genomic targets98. Importantly, the authors found that NANOG overexpression could sustain SMAD2/3 target gene expression even in the absence of Activin/Nodal signalling, thus suggesting that NANOG may function as a SMAD2/3 transcriptional co-factor during active Activin/Nodal signalling in hESCs98. In a separate study, ChIP-seq revealed extensive co-occupancy of SMAD3 along with the hESC genome with OCT4, although it was not verified if SMAD3 could bind alongside other hESC pluripotency factors99. Taken together, these data provide important insight into the potential mechanisms by which Activin/Nodal signalling helps sustain hESC pluripotency.
Transcriptional regulation of ground state mESCs
As previously mentioned, the conventional culture of mESCs requires the presence of LIF and BMP458,59,60. However, even in the presence of LIF/BMP4, the autocrine pro-differentiation Fgf4/Mek/Erk signalling in mESCs is still active60,100. This therefore suggests that pluripotency is sustained by LIF and BMP4 acting downstream of the Erk pathway to prevent mESC differentiation.
Expanding upon this observation, Ying et al.100 surprisingly found that the requirement of external LIF/BMP4 in mESC culture could be abrogated via the usage of small molecule kinase inhibitors. This two inhibitor (2i) culture, consisting of PD0325901 and CHIR99021 to respectively target Mek and Gsk3, serves to protect pluripotent mESCs from the pro-differentiation effect of Fgf4 stimulation64, as well as the transcriptional repressor activity of Tcf3101. More importantly, by using the 2i culture, germ-line transmitting ESCs could be generated from previously recalcitrant mouse strains100,102, and from the rat103,104.
When mESCs are cultured under conventional LIF conditions, there is a heterogeneous expression of pluripotency transcription factors such as Nanog, Rex1 and Stella23,105,106. Interestingly, after FACS-purification, these sorted cells rather than maintaining a pure cell population, will revert into a heterogeneous population. These data therefore suggest that gene transcription of mESCs under LIF culture conditions exists in a fluctuating and dynamic state. Upon transfer into 2i conditions, Nanog and Rex1 expression will become homogeneously high107. As Nanog-low cells are prone to differentiate, and Rex-low mESCs do not contribute to chimera formation upon blastocyst microinjection23,105, the capture of a Nanog/Rex1-high mESC state indicates that pluripotency may have been stabilised under 2i conditions107. Interestingly, under 2i or Mek-inhibition, the Nanog gene expression in mESCs switches into a biallelic expression mode, as opposed to the monoallelic expression status in conventional LIF/serum culture108. With these unique characteristics, 2i-cultured mESCs are said to reside in a novel and distinct 'ground state' pluripotency100.
Although the protein levels of pluripotency regulators like Nanog and Rex1 are known to become uniformly-high when mESCs are switched from LIF/serum into 2i media107, Marks et al.109 however found through RNA-sequencing (RNA-seq) that apart from Tcl1, the mRNA levels of most other pluripotency genes like Pou5f1, Sox2, Nanog, Esrrb, Klf2, Klf4 and Tbx3 did not change between the two culture conditions. The reason behind this discrepancy is still unclear, and may have arisen due to the presence of LIF with the 2i media used in this study109. It is also possible that translational and/or post-translational regulatory mechanisms are involved.
This study by Marks et al.109 also found that lineage genes were suppressed under 2i/LIF conditions, and that 2i/LIF mESCs had higher expression of metabolic genes. Taken together, these data argue against the hypothesis proposed by Efroni et al.110 that an innate global transcriptional hyperactivity results in pluripotent developmental plasticity. Rather, it is believed that the presence of lineage gene expression in conventional mESC culture may be the result of serum stimulation109. Additionally, unlike cells cultured under LIF/serum conditions, 2i/LIF mESCs were found to contain more proximal-promoter pausing by RNA Polymerase II (RNA Pol II), especially at many lineage-specific genes. It was also proposed that this RNA Pol II pausing could be important towards establishing the ground state pluripotency.
Interconversion between alternative pluripotent states
Mouse epiblast stem cells
While the presence of LIF is necessary for mESCs to maintain pluripotency, the discovery of a novel LIF-independent pluripotent stem cell population derived from E5.5 to E7.5 post-implantation mouse embryos111,112, suggests that other states of pluripotency may exist (Figure 1). As these cells were isolated from the post-implantation epiblast tissue of the developing embryo, they were hence termed mouse epiblast stem cells (EpiSCs). While EpiSCs can self-renew and were demonstrated to be pluripotent through teratoma assays and through in vitro differentiation into germ cells111,112,113, it should be noted that EpiSCs exhibit limited developmental potential and are generally considered inefficient in the formation of chimeras111,112,114.
Similarly, while EpiSCs do express the core Oct4, Sox2 and Nanog pluripotency factors, they differ markedly from mESCs with regards to their gene expression profile, epigenetic status and usage of signalling pathways to maintain a stem cell identity106,111,112,115,116. Notably, EpiSC cultures like that of hESCs, require the addition of Activin and Fgf2, but not LIF or BMP4 for self-renewal117. The expression levels of pluripotency markers such as Rex1, Stella, Klf2, Klf4 are lower in EpiSCs, with a concomitant higher expression of differentiation markers112. It should also be highlighted that while female mESCs are in the pre-inactivation state with two active X-chromosomes (XaXa), EpiSCs have already undergone X-inactivation (XaXi), consistent with a more developmentally advanced stage117. Therefore, these findings indicate that EpiSCs have already been 'primed' for differentiation, as opposed to mESCs which exist in a more developmentally 'naive' state.
Overall, EpiSCs appear to partially resemble hESCs, and may potentially be the counterpart of hESCs. Therefore, EpiSCs could serve as a more relevant mouse model for the study of early human embryonic development, as compared with mESCs. However, despite the aforementioned similarities with hESCs, inhibition of Fgf2/Erk signalling in EpiSCs does not result in loss of Nanog expression, and no evidence of Oct4-mediated transcriptional regulation at Fgf2 promoters was observed for EpiSCs118. Similarly, certain key features of EpiSCs like the absence of Rex1, a mESC pluripotency marker, and the expression of the Fgf5 epiblast marker, are not shared by hESCs118. Further research is thus needed to establish whether these dissimilarities are due to species- or developmental-specific differences.
The transition between naive and primed pluripotency
As mESCs are derived from a developmentally earlier timepoint (pre-implantation embryos) as compared to EpiSCs (post-implantation embryos), the conversion of mESCs into EpiSCs would therefore correspond to a differentiation step along the normal developmental pathway (Figure 1). In this regard, culturing mESCs with FGF2 and Activin A readily results in the formation of EpiSCs114. Similarly, FGF treatment together with LIF/Stat3 blockade could also convert mESCs into EpiSCs118.
In contrast, the reverse transition of EpiSCs into mESCs is more difficult, occurring at an extremely low frequency through culture with LIF102,114. The reversion rates may be improved by 104–105 fold if the pluripotency factors Klf2, Klf4, Nanog, Nr5a2 or Esrrb were to be ectopically expressed22,102,114,119,120. Similarly, a chemical approach can also be adopted to convert EpiSCs into naive mESCs using a combination of LSD1, ALK5, MEK, FGFR and GSK3 inhibitors121 (Figure 1).
Recently, Prdm14 and Klf2 were found to synergize with each other to rapidly induce the reprogramming of EpiSCs into mESCs within 3-4 days122, in contrast to the usual process of 8-10 days. By itself, the introduction of Prdm14 does not have much effect on the reversion process. However, transcriptomic analysis revealed that Prdm14 actually serves to prime EpiSCs for conversion through the simultaneous repression of lineage-associated genes and activation of early epiblast genes. The synergistic effect of Prdm14-Klf2 can also be attributed to the ability of Prdm14 to enhance Klf2 recruitment onto key mESC pluripotency gene loci such as the Nr5a2 promoter and the Oct4 distal enhancer122.
Establishing a mESC-like hESC state
In addition to post-implantation embryos, EpiSCs can also be readily isolated from pre-implantation stage mouse blastocysts123, which have been traditionally used to derive mESCs (Figure 1). This meant that modulation of the signalling environment could influence the formation of naive or primed pluripotency states. Because hESCs resemble EpiSCs more than mESCs, despite also having been derived from pre-implantation blastocysts, it is believed that during the hESC derivation process, pre-hESCs may have progressed into a more developmentally advanced EpiSC-like state124. In support of this 'primed' pluripotent state of hESCs, non-human primate ESCs from rhesus monkeys, like mouse EpiSCs, are unable to contribute to chimera formation125.
Currently, hESCs suffer from very poor gene-targeting efficacy unlike mESCs, making the genetic manipulation of hESCs for research or therapeutic purposes extremely difficult126. Critically, the developmental stage differences between the two cell types imply that many of the protocols for mESC differentiation into various lineages may not work in a hESC system. In light of these problems, the creation of a mESC-like hESC state would not only make hESCs more amenable to gene targeting, but also enable the transfer of existing mouse differentiation protocols into hESCs.
There have been several attempts by various groups to generate mESC-like human pluripotent stem cells. The methods include either a direct conversion from conventional hESCs127,128 or the reprogramming of somatic cells into mESC-like hESCs128,129,130,131 (Figure 2). Unfortunately, these cells either were dependent upon transgene expression for long-term culture, or have not been thoroughly characterised for features such as naive mESC characteristics and complete transgene independence. Taken together, while these studies have demonstrated the feasibility of creating hESCs which exhibit characteristics of naive pluripotency, future efforts should focus on improving the culture conditions to enable transgene-free long-term maintenance of these mESC-like hESCs.

Methods to create mESC-like hESCs. (A) hESCs with mESC-like characteristics can be generated directly from conventional hESCs using OCT4 with SOX2, or, KLF4 with KLF2, in the culture with LIF/PD/CH128. Similarly, the generation of hESCs which resemble mESC colonies could be achieved through culture with LIF/PD/SB127. (B) Human fibroblasts can also be reprogrammed into mESC-like human iPSCs through expression of OCT4, SOX2, NANOG, LIN28 with LIF/PD/CH/A83129; through OCT4, SOX2, KLF4, c-MYC, NANOG with LIF130; using OCT4, SOX2, KLF4 with LIF/PD/CH128; or OCT4, SOX2, KLF4, c-MYC, NR5A2, RAR-γ with LIF/PD/CH131. PD: MEK inhibitor PD0325901. CH: GSK3 inhibitor CHIR99021. SB: p38 inhibitor SB203580. A83: ALK4,5,7 inhibitor A83-01.
Several studies have also explored the possibility of generating mESC-like hESCs directly from pre-implantation human embryos. Female hESCs derived from the culture of human blastocysts in hypoxic (5% O2) conditions were found to be in an XaXa status, a characteristic of mouse naive pluripotency132. However, these cells were maintained in conventional FGF-containing media and were not tested further for features of naive pluripotency.
The generation of naive hESCs from embryos using 2i media was reported to be unsuccessful133. In mice, culturing 8-cell stage embryos under MEK inhibition will result in the failure to form the hypoblast compartment with a reciprocal expansion of the pluripotent epiblast134. However, culturing early human embryos with MEK inhibitor does not block hypoblast formation, and neither would the development of the NANOG-positive epiblast compartment be affected133,135. Thus taken together, these studies suggest that additional signalling pathways could be involved in the segregation of the human ICM into the epiblast and hypoblast, or that species-specific differences exist between mouse and human embryos in the biological functions of FGF/MEK signalling.
The totipotential of 2C-like mESCs?
Recently, Macfarlan et al.136 reported the discovery of a rare transient population of mESCs, which can give rise to both embryonic and extraembryonic tissues. By comparing the gene expression signature between mouse oocytes and 2-cell (2C)-staged embryos using RNA-seq, the authors found that the 2C transcriptome contained many genes, which were driven by retroviral elements. Among these repeats, the MuERV-L family of retroelements were found to be the most abundant. Indeed, by combining this MuERV-L regulatory sequence with a tdTomato red fluorescence gene, the authors demonstrated through zygotic injection that strong MuERV-L reporter fluorescence was detected at the 2C stage, which would then gradually decrease and become undetectable by the blastocyst stage.
Surprisingly, stable integration of this MuERV-L tdTomato reporter into mESCs led to the detection of a similar red fluorescence within a very small population of cells (approximately 0.2%-1.5%); and gene expression profiling of these tdTomato+ mESCs revealed that they resembled 2C embryos (Figure 1). Like Nanog, Rex1 and Stella23,105,106, MuERV-L tdTomato expression was also found to exist in a dynamic state. Subsequent genetic labelling experiments suggested that all mESCs within the culture could transiently pass through this 2C-like state. Intriguingly, these 2C-like mESCs do not contain any Oct4, Sox2 and Nanog proteins, even though the transcript levels of these genes were unaffected. Perhaps the most fascinating aspect of this study, is the demonstration that injection of these 2C-like mESCs into morula stage embryos could result in the contribution of these 2C-like donor cells towards both embryonic and extraembryonic tissues, hence suggesting a totipotent-like capability.
Together, this study raises several new interesting questions. First, what is the significance of this 2C-like stage, and why do mESCs transiently enter this phase? Second, as these 2C-like mESCs do not possess Oct4, Sox2 and Nanog proteins, how would the transcriptional network of these 2C-like mESCs be regulated? Third, as only a very rare population of mESCs are expressing MuERV-L at a given time, what would be the signalling pathways or cellular mechanisms that regulate the entry or exit of this phase? And lastly, do hESCs possess a similar transient 2C-like phase, and if so, would it also be regulated by endogenous retroviral elements?
Future outlook
Deciphering the ESC transcriptional network is essential towards understanding the cellular mechanisms that govern pluripotency. In this regard, the different studies involving various experimental approaches have now enabled researchers to appreciate the processes by which the core ESC transcription factors establish an overall ESC identity.
To bring the potential of hESCs one step closer towards its application in regenerative medicine, future research should likely focus upon dissecting the pathways that regulate lineage commitment. Recently, by probing the temporal gene expression and chromatin changes during the directed differentiation of ESCs into cardiac lineages, several novel regulators of cardiac tissue formation have been identified137,138. This is achieved either by (1) determining the stage-specific activation of gene enhancers, and applying a DNA binding motif search to predict the transcription factors that are involved during cardiac differentiation137; or (2) by predicting key regulatory genes based on the induction of RNA expression, loss of repressive H3K27me3 marks and reciprocal increase of active H3K4me3 modification138. Therefore, it would be interesting to test if other novel tissue-specific regulators could be identified for different somatic lineages through analysis of temporal chromatin changes. In this regard, the ability to genetically modify and introduce hESCs reporter genes into that specify certain tissue lineages would be of tremendous value for the purpose of studying directed differentiation. For instance, by coupling these lineage-specific hESC reporters with high-throughput genome-wide siRNA screens or miRNA mimic libraries, numerous factors that regulate differentiation into specific lineages could be identified.
Improving the efficacy of directed ESC differentiation into the desired cell-type, as well as overcoming the immature phenotype of hESC-derived differentiated cells139 represent key issues to be tackled in the future. Resolution of these problems likely will require the optimization of important cell culture parameters such as the extracellular matrix, growth factor and cytokine-signalling environment, three-dimensional (3D) cell organization and cell culture duration. Similarly, the capture of ESC-differentiated cells at the progenitor stage may enable easier expansion and more efficient differentiation into the desired cell type of choice. This can be achieved either through the co-culture of ESC-differentiated tissue with the appropriate mesenchymal cells140, or through the isolation of self-renewing progenitors within a heterogenous cell population141.
While directed differentiation is useful for the derivation of a single, or a few cell types for tissue replacement, the complete generation of complex organs comprising many cell types that work in a coordinated fashion presents a greater challenge. Most remarkably, it was recently demonstrated that pluripotent stem cells retain a self-organising ability to differentiate into 3D organoids, which resemble optic cups142,143 and adenohypophysis tissues144. Similarly, intestinal organoids have been reported to develop from hESC-derived posterior endoderm monolayers upon exposure to the appropriate signalling factors145, and functional thyroid gland tissue has been generated from mESCs146. Therefore, if one were to harness this powerful self-developing property of pluripotent stem cells for the generation of novel complex organ types for research or medical purposes, it would be important to first determine the mechanisms underlying these processes. In this regard, studying the temporal transcriptional changes occurring at the whole-organoid, or at the single-cell level, may be a good starting point to dissect the pathways involved.
Abbreviations
- 2C:
-
(2-cell)
- 2i:
-
(two inhibitor)
- BMP4:
-
(bone morphogenic protein 4)
- ChIP:
-
(chromatin immunoprecipitation)
- ChIP-seq:
-
(chromatin immunoprecipitation and sequencing)
- ESCs:
-
(embryonic stem cells)
- EpiSCs:
-
(Epiblast-derived stem cells)
- Fgf4:
-
(fibroblast growth factor 4)
- hESCs:
-
(human embryonic stem cells)
- ICM:
-
(inner cell mass)
- iPSCs:
-
(induced pluripotent stem cells)
- LIF:
-
(leukemia inhibitory factor)
- lincRNA:
-
(large intergenic non-coding RNA)
- mESCs:
-
(mouse embryonic stem cells)
- miRNA:
-
(micro RNA)
- ncRNA:
-
(non-coding RNA)
- PcG:
-
(Polycomb group proteins)
- PGCs:
-
(primodial germ cells)
- POU:
-
(Pit-Oct-Unc)
- RNAi:
-
(RNA interference)
- RNA-seq:
-
(RNA sequencing)
- SCF:
-
(stem cell factor)
- siRNA:
-
(short interfering RNA)
- XaXa:
-
(double X-chromosome active)
- XaXi:
-
(single X-chromosome inactivated)
References
Evans MJ, Kaufman MH . Establishment in culture of pluripotential cells from mouse embryos. Nature 1981; 292:154–156.
Martin GR . Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA 1981; 78:7634–7638.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282:1145–1147.
Young RA . Control of the embryonic stem cell state. Cell 2011; 144:940–954.
Rosner MH, Vigano MA, Ozato K, et al. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature 1990; 345:686–692.
Scholer HR, Dressler GR, Balling R, Rohdewohld H, Gruss P . Oct-4: a germline-specific transcription factor mapping to the mouse t-complex. EMBO J 1990; 9:2185–2195.
Nichols J, Zevnik B, Anastassiadis K, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 1998; 95:379–391.
Niwa H, Miyazaki J, Smith AG . Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet 2000; 24:372–376.
Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R . Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 2003; 17:126–140.
Masui S, Nakatake Y, Toyooka Y, et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 2007; 9:625–635.
Yuan H, Corbi N, Basilico C, Dailey L . Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev 1995; 9:2635–2645.
Nishimoto M, Fukushima A, Okuda A, Muramatsu M . The gene for the embryonic stem cell coactivator UTF1 carries a regulatory element which selectively interacts with a complex composed of Oct-3/4 and Sox-2. Mol Cell Biol 1999; 19:5453–5465.
Tokuzawa Y, Kaiho E, Maruyama M, et al. Fbx15 is a novel target of Oct3/4 but is dispensable for embryonic stem cell self-renewal and mouse development. Mol Cell Biol 2003; 23:2699–2708.
Nakatake Y, Fukui N, Iwamatsu Y, et al. Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol Cell Biol 2006; 26:7772–7782.
Kuroda T, Tada M, Kubota H, et al. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol 2005; 25:2475–2485.
Rodda DJ, Chew JL, Lim LH, et al. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem 2005; 280:24731–24737.
Okumura-Nakanishi S, Saito M, Niwa H, Ishikawa F . Oct-3/4 and Sox2 regulate Oct-3/4 gene in embryonic stem cells. J Biol Chem 2005; 280:5307–5317.
Tomioka M, Nishimoto M, Miyagi S, et al. Identification of Sox-2 regulatory region which is under the control of Oct-3/4-Sox-2 complex. Nucleic Acids Res 2002; 30:3202–3213.
Chew JL, Loh YH, Zhang W, et al. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol 2005; 25:6031–6046.
Chambers I, Colby D, Robertson M, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003; 113:643–655.
Mitsui K, Tokuzawa Y, Itoh H, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003; 113:631–642.
Silva J, Nichols J, Theunissen TW, et al. Nanog is the gateway to the pluripotent ground state. Cell 2009; 138:722–737.
Chambers I, Silva J, Colby D, et al. Nanog safeguards pluripotency and mediates germline development. Nature 2007; 450:1230–1234.
Loh YH, Wu Q, Chew JL, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 2006; 38:431–440.
Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005; 122:947–956.
Ivanova N, Dobrin R, Lu R, et al. Dissecting self-renewal in stem cells with RNA interference. Nature 2006; 442:533–538.
Fazzio TG, Huff JT, Panning B . An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 2008; 134:162–174.
Bilodeau S, Kagey MH, Frampton GM, Rahl PB, Young RA . SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev 2009; 23:2484–2489.
Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ . A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev 2009; 23:837–848.
Ding L, Paszkowski-Rogacz M, Nitzsche A, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell 2009; 4:403–415.
Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010; 467:430–435.
Chia NY, Chan YS, Feng B, et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 2010; 468:316–320.
Wang J, Rao S, Chu J, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006; 444:364–368.
Liang J, Wan M, Zhang Y, et al. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat Cell Biol 2008; 10:731–739.
Wu Q, Chen X, Zhang J, et al. Sall4 interacts with Nanog and co-occupies Nanog genomic sites in embryonic stem cells. J Biol Chem 2006; 281:24090–24094.
van den Berg DL, Snoek T, Mullin NP, et al. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 2010; 6:369–381.
Pardo M, Lang B, Yu L, et al. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell 2010; 6:382–395.
Ding J, Xu H, Faiola F, Ma'ayan A, Wang J . Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res 2012; 22:155–167.
Gao Z, Cox JL, Gilmore JM, et al. Determination of protein interactome of transcription factor Sox2 in embryonic stem cells engineered for inducible expression of four reprogramming factors. J Biol Chem 2012; 287:11384–11397.
Mallanna SK, Ormsbee BD, Iacovino M, et al. Proteomic analysis of Sox2-associated proteins during early stages of mouse embryonic stem cell differentiation identifies Sox21 as a novel regulator of stem cell fate. Stem Cells 2010; 28:1715–1727.
Kopp JL, Ormsbee BD, Desler M, Rizzino A . Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 2008; 26:903–911.
Chen X, Xu H, Yuan P, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008; 133:1106–1117.
Kim J, Chu J, Shen X, Wang J, Orkin SH . An extended transcriptional network for pluripotency of embryonic stem cells. Cell 2008; 132:1049–1061.
Kidder BL, Yang J, Palmer S . Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One 2008; 3:e3932.
Rahl PB, Lin CY, Seila AC, et al. c-Myc regulates transcriptional pause release. Cell 2010; 141:432–445.
Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008; 40:499–507.
Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY . Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008; 2:333–344.
Kim J, Woo AJ, Chu J, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010; 143:313–324.
Lin CY, Loven J, Rahl PB, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012; 151:56–67.
Nie Z, Hu G, Wei G, et al. c-Myc Is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012; 151:68–79.
Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 2008; 26:101–106.
Kanellopoulou C, Muljo SA, Kung AL, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 2005; 19:489–501.
Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R . DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet 2007; 39:380–385.
Marson A, Levine SS, Cole MF, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008; 134:521–533.
Melton C, Judson RL, Blelloch R . Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature 2010; 463:621–626.
Anokye-Danso F, Trivedi CM, Juhr D, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011; 8:376–388.
Guttman M, Donaghey J, Carey BW, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477:295–300.
Williams RL, Hilton DJ, Pease S, et al. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 1988; 336:684–687.
Smith AG, Heath JK, Donaldson DD, et al. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 1988; 336:688–690.
Ying QL, Nichols J, Chambers I, Smith A . BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003; 115:281–292.
Niwa H, Burdon T, Chambers I, Smith A . Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev 1998; 12:2048–2060.
Rathjen J, Lake JA, Bettess MD, Washington JM, Chapman G, Rathjen PD . Formation of a primitive ectoderm like cell population, EPL cells, from ES cells in response to biologically derived factors. J Cell Sci 1999; 112 (Part 5):601–612.
Ma YG, Rosfjord E, Huebert C, et al. Transcriptional regulation of the murine k-FGF gene in embryonic cell lines. Dev Biol 1992; 154:45–54.
Kunath T, Saba-El-Leil MK, Almousailleakh M, Wray J, Meloche S, Smith A . FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development 2007; 134:2895–2902.
Stavridis MP, Lunn JS, Collins BJ, Storey KG . A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development 2007; 134:2889–2894.
Cheng AM, Saxton TM, Sakai R, et al. Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell 1998; 95:793–803.
Burdon T, Stracey C, Chambers I, Nichols J, Smith A . Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev Biol 1999; 210:30–43.
Chen S, Do JT, Zhang Q, et al. Self-renewal of embryonic stem cells by a small molecule. Proc Natl Acad Sci USA 2006; 103:17266–17271.
Johnson GL, Lapadat R . Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002; 298:1911–1912.
Ramos JW . The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol 2008; 40:2707–2719.
Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL . p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet 2004; 36:738–743.
Hu S, Xie Z, Onishi A, et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009; 139:610–622.
Sokol SY . Maintaining embryonic stem cell pluripotency with Wnt signaling. Development 2011; 138:4341–4350.
Wray J, Hartmann C . WNTing embryonic stem cells. Trends Cell Biol 2012; 22:159–168.
Pereira L, Yi F, Merrill BJ . Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol Cell Biol 2006; 26:7479–7491.
Cole MF, Johnstone SE, Newman JJ, Kagey MH, Young RA . Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev 2008; 22:746–755.
Yi F, Pereira L, Merrill BJ . Tcf3 functions as a steady-state limiter of transcriptional programs of mouse embryonic stem cell self-renewal. Stem Cells 2008; 26:1951–1960.
Tam WL, Lim CY, Han J, et al. T-cell factor 3 regulates embryonic stem cell pluripotency and self-renewal by the transcriptional control of multiple lineage pathways. Stem Cells 2008; 26:2019–2031.
Guo G, Huang Y, Humphreys P, Wang X, Smith A . A PiggyBac-based recessive screening method to identify pluripotency regulators. PLoS One 2011; 6:e 18189.
Daheron L, Opitz SL, Zaehres H, et al. LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells 2004; 22:770–778.
Xu RH, Chen X, Li DS, et al. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat Biotechnol 2002; 20:1261–1264.
Xu RH, Peck RM, Li DS, Feng X, Ludwig T, Thomson JA . Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat Methods 2005; 2:185–190.
James D, Levine AJ, Besser D, Hemmati-Brivanlou A . TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 2005; 132:1273–1282.
Vallier L, Alexander M, Pedersen RA . Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J Cell Sci 2005; 118:4495–4509.
Turner N, Grose R . Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010; 10:116–129.
Katoh M . Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther 2006; 5:1059–1064.
Guo X, Wang XF . Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res 2009; 19:71–88.
D'Amour KA, Gage FH . Genetic and functional differences between multipotent neural and pluripotent embryonic stem cells. Proc Natl Acad Sci USA 2003; 100 Suppl 1:11866–11872.
Li J, Wang G, Wang C, et al. MEK/ERK signaling contributes to the maintenance of human embryonic stem cell self-renewal. Differentiation 2007; 75:299–307.
Vallier L, Mendjan S, Brown S, et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 2009; 136:1339–1349.
Swaney DL, Wenger CD, Thomson JA, Coon JJ . Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci USA 2009; 106:995–1000.
Van Hoof D, Munoz J, Braam SR, et al. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell 2009; 5:214–226.
Brill LM, Xiong W, Lee KB, et al. Phosphoproteomic analysis of human embryonic stem cells. Cell Stem Cell 2009; 5:204–213.
Rigbolt KT, Prokhorova TA, Akimov V, et al. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal 2011; 4:rs3.
Ding VM, Boersema PJ, Foong LY, et al. Tyrosine phosphorylation profiling in FGF-2 stimulated human embryonic stem cells. PLoS One 2011; 6:e 17538.
Zoumaro-Djayoon AD, Ding V, Foong LY, Choo A, Heck AJ, Munoz J . Investigating the role of FGF-2 in stem cell maintenance by global phosphoproteomics profiling. Proteomics 2011; 11:3962–3971.
Xu RH, Sampsell-Barron TL, Gu F, et al. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 2008; 3:196–206.
Brown S, Teo A, Pauklin S, et al. Activin/Nodal signaling controls divergent transcriptional networks in human embryonic stem cells and in endoderm progenitors. Stem Cells 2011; 29:1176–1185.
Mullen AC, Orlando DA, Newman JJ, et al. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 2011; 147:565–576.
Ying QL, Wray J, Nichols J, et al. The ground state of embryonic stem cell self-renewal. Nature 2008; 453:519–523.
Wray J, Kalkan T, Gomez-Lopez S, et al. Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol 2011; 13:838–845.
Hanna J, Markoulaki S, Mitalipova M, et al. Metastable pluripotent states in NOD-mouse-derived ESCs. Cell Stem Cell 2009; 4:513–524.
Buehr M, Meek S, Blair K, et al. Capture of authentic embryonic stem cells from rat blastocysts. Cell 2008; 135:1287–1298.
Li P, Tong C, Mehrian-Shai R, et al. Germline competent embryonic stem cells derived from rat blastocysts. Cell 2008; 135:1299–1310.
Toyooka Y, Shimosato D, Murakami K, Takahashi K, Niwa H . Identification and characterization of subpopulations in undifferentiated ES cell culture. Development 2008; 135:909–918.
Hayashi K, Lopes SM, Tang F, Surani MA . Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell 2008; 3:391–401.
Wray J, Kalkan T, Smith AG . The ground state of pluripotency. Biochem Soc Trans 2010; 38:1027–1032.
Miyanari Y, Torres-Padilla ME . Control of ground-state pluripotency by allelic regulation of Nanog. Nature 2012; 483:470–473.
Marks H, Kalkan T, Menafra R, et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell 2012; 149:590–604.
Efroni S, Duttagupta R, Cheng J, et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2008; 2:437–447.
Brons IG, Smithers LE, Trotter MW, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007; 448:191–195.
Tesar PJ, Chenoweth JG, Brook FA, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 2007; 448:196–199.
Hayashi K, Surani MA . Self-renewing epiblast stem cells exhibit continual delineation of germ cells with epigenetic reprogramming in vitro. Development 2009; 136:3549–3556.
Guo G, Yang J, Nichols J, et al. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 2009; 136:1063–1069.
Bao S, Tang F, Li X, et al. Epigenetic reversion of post-implantation epiblast to pluripotent embryonic stem cells. Nature 2009; 461:1292–1295.
Sun B, Ito M, Mendjan S, et al. Status of genomic imprinting in epigenetically distinct pluripotent stem cells. Stem Cells 2012; 30:161–168.
Hanna JH, Saha K, Jaenisch R . Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell 2010; 143:508–525.
Greber B, Wu G, Bernemann C, et al. Conserved and divergent roles of FGF signaling in mouse epiblast stem cells and human embryonic stem cells. Cell Stem Cell 2010; 6:215–226.
Hall J, Guo G, Wray J, et al. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 2009; 5:597–609.
Festuccia N, Osorno R, Halbritter F, et al. Esrrb is a direct Nanog target gene that can substitute for Nanog function in pluripotent cells. Cell Stem Cell 2012; 11:477–490.
Zhou H, Li W, Zhu S, et al. Conversion of mouse epiblast stem cells to an earlier pluripotency state by small molecules. J Biol Chem 2010; 285:29676–29680.
Gillich A, Bao S, Grabole N, et al. Epiblast stem cell-based system reveals reprogramming synergy of germline factors. Cell Stem Cell 2012; 10:425–439.
Najm FJ, Chenoweth JG, Anderson PD, et al. Isolation of epiblast stem cells from preimplantation mouse embryos. Cell Stem Cell 2011; 8:318–325.
De Los Angeles A, Loh YH, Tesar PJ, Daley GQ . Accessing naïve human pluripotency. Curr Opin Genet Dev 2012; 22:272–282.
Tachibana M, Sparman M, Ramsey C, et al. Generation of chimeric rhesus monkeys. Cell 2012; 148:285–295.
Zwaka TP, Thomson JA . Homologous recombination in human embryonic stem cells. Nat Biotechnol 2003; 21:319–321.
Xu Y, Zhu X, Hahm HS, et al. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc Natl Acad Sci USA 2010; 107:8129–8134.
Hanna J, Cheng AW, Saha K, et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci USA 2010; 107:9222–9227.
Li W, Wei W, Zhu S, et al. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell 2009; 4:16–19.
Buecker C, Chen HH, Polo JM, et al. A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell 2010; 6:535–546.
Wang W, Yang J, Liu H, et al. Rapid and efficient reprogramming of somatic cells to induced pluripotent stem cells by retinoic acid receptor gamma and liver receptor homolog 1. Proc Natl Acad Sci USA 2011; 108:18283–18288.
Lengner CJ, Gimelbrant AA, Erwin JA, et al. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell 2010; 141:872–883.
Roode M, Blair K, Snell P, et al. Human hypoblast formation is not dependent on FGF signalling. Dev Biol 2012; 361:358–363.
Nichols J, Silva J, Roode M, Smith A . Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development 2009; 136:3215–3222.
Kuijk EW, van Tol LT, Van de Velde H, et al. The roles of FGF and MAP kinase signaling in the segregation of the epiblast and hypoblast cell lineages in bovine and human embryos. Development 2012; 139:871–882.
Macfarlan TS, Gifford WD, Driscoll S, et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012; 487:57–63.
Wamstad JA, Alexander JM, Truty RM, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012; 151:206–220.
Paige SL, Thomas S, Stoick-Cooper CL, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 2012; 151:221–232.
Cohen DE, Melton D . Turning straw into gold: directing cell fate for regenerative medicine. Nat Rev Genet 2011; 12:243–252.
Sneddon JB, Borowiak M, Melton DA . Self-renewal of embryonic-stem-cell-derived progenitors by organ-matched mesenchyme. Nature 2012; 491:765–768.
Cheng X, Ying L, Lu L, et al. Self-renewing endodermal progenitor lines generated from human pluripotent stem cells. Cell Stem Cell 2012; 10:371–384.
Eiraku M, Takata N, Ishibashi H, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011; 472:51–56.
Nakano T, Ando S, Takata N, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012; 10:771–785.
Suga H, Kadoshima T, Minaguchi M, et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature 2011; 480:57–62.
Spence JR, Mayhew CN, Rankin SA, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011; 470:105–109.
Antonica F, Kasprzyk DF, Opitz R, et al. Generation of functional thyroid from embryonic stem cells. Nature 2012; 491:66–71.
Acknowledgements
We would like to thank Winston Chan and Kevin Andrew Uy Gonzales from the Genome Institute of Singapore for their comments on this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yeo, JC., Ng, HH. The transcriptional regulation of pluripotency. Cell Res 23, 20–32 (2013). https://doi.org/10.1038/cr.2012.172
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2012.172
Keywords
This article is cited by
-
Review on kidney diseases: types, treatment and potential of stem cell therapy
Renal Replacement Therapy (2023)
-
A multi-omics integrative analysis based on CRISPR screens re-defines the pluripotency regulatory network in ESCs
Communications Biology (2023)
-
Intracellular metabolic reprogramming mediated by micro-RNAs in differentiating and proliferating cells under non-diseased conditions
Molecular Biology Reports (2021)
-
Global translation during early development depends on the essential transcription factor PRDM10
Nature Communications (2020)
-
Zygotic genome activation in the chicken: a comparative review
Cellular and Molecular Life Sciences (2020)
