
Abstract
α-Galactosylceramide (α-GC) is widely known to activate invariant natural killer T (iNKT) cells to suppress myelin antigen-specific Th1 responses, protecting susceptible mice against experimental autoimmune encephalomyelitis (EAE). Here, we demonstrate an unexpected finding that high doses of α-GC exacerbated, rather than ameliorated, EAE. Similar results were observed when MOG35-55-specific T cells treated with high-dose α-GC were transferred into naïve syngeneic recipient mice. Further study showed that high doses of α-GC directly enhance the Th17 and Th1 response by activation of CD4+CD44+ memory T cells through phosphorylation of STAT3 and activation of NF-κB. Unlike the activation of iNKT cells by low doses of α-GC, high doses of α-GC directly interacted with CD1d expressed on T cells and activated Th17 and Th1 cells. Furthermore, antigen-presenting cells (APCs) predominantly express CD1d1, whereas the majority of CD4+ T cells express CD1d2. Knockdown of CD1d1 or CD1d2 gene expression by RNAi interfered with the activation of iNKT or Th17/Th1 cells, respectively. Therefore, α-GC treatment could improve or worsen EAE by engaging either APCs or Th17/Th1 cells depending on the dose used.
Similar content being viewed by others


Introduction
Experimental autoimmune encephalomyelitis (EAE) is considered to be a valuable model for the human demyelinating disease multiple sclerosis (MS) because they share many common pathological features and immune dysfunctions 1. Th1 cells are considered to be almost exclusively responsible for the autoimmune tissue damage. However, recent studies have uncovered a vital role for Th17 in the induction of EAE. Th17 cells were shown to be more potent than Th1 cells in transferring EAE to naïve wild-type animals 2. Many additional studies have supported the notion that Th17, rather than Th1 cells, are primarily responsible for the inflammatory pathology observed in EAE 2, 3, 4, 5. IL-17-deficient animals develop EAE with delayed onset and diminished severity 6. In addition, administration of an IL-17-blocking antibody (Ab) in mice immunized with a myelin antigen prevents chemokine expression in the brain and the subsequent development of EAE 2, 7.
The CD1 genes encode a family of glycoproteins that are related to major histocompatibility complex (MHC) molecules 8. CD1 molecules can be divided into two groups according to sequence homology and tissue distribution 9. The human CD1a, b and c genes are members of group I, whereas CD1d is a member of group II. Mice have only two CD1 genes, CD1d1 and CD1d2, which are 95% homologous and belong to group II 10. Mouse and human CD1 molecules present non-peptidic antigens to T cells. CD1a, b and c molecules present lipids or glycolipids to T cells. Human CD1d and mouse CD1d1 present α-galactosylceramide (α-GC) to invariant natural killer T (iNKT) cells 11, which are a unique and relatively small subset of T cells originally shown to share properties of both conventional T and NK cells. CD1d-restricted T cells are characterized by the expression of an invariant TCRα-chain (Vα14 in mice and Vα24 in humans) and markers of natural killer (NK) cells and are, thus, referred to as iNKT cells 12, 13, 14, 15. iNKT cells show a remarkable functional diversity, and elicit varying and opposing immune functions. On the one hand, they actively induce T-cell tolerance and are crucial for the prevention of autoimmune diseases in pre-clinical models of MS 16, 17, type 1 diabetes 18, 19, systemic lupus erythematosus 20, myasthenia gravis 21 and rheumatoid arthritis 22, 23. On the other hand, iNKT cells participate in the innate immune response to promote antimicrobial 24, 25, 26 and antitumor 27, 28, 29, 30 immunity.
It has been reported that α-GC can protect susceptible mice against EAE by activating iNKT to suppress myelin antigen-specific Th1 and/or Th2 responses. Unexpectedly, in our studies, we found that high doses of α-GC exacerbated, rather than ameliorated, EAE. We found that high doses of α-GC directly facilitated the differentiation and survival of Th17 and activation of Th17 and Th1. Depending on the dose used, α-GC treatment could improve or worsen EAE. Therefore, our findings caution against using α-GC as a treatment option for autoimmune diseases.
Results
High doses of α-GC potentiate whereas low doses ameliorate EAE
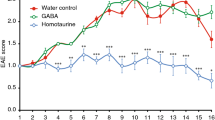
It has been reported that α-GC can protect susceptible mice against EAE 17, 31. However, treatment of mice with high dose of α-GC worsened EAE, whereas low doses reduced EAE severity (Figure 1A). These findings were consistent with histopathological analysis of the affected spinal cord (Figure 1B). When compared with the transfer of vehicle-treated (or low dose of α-GC-treated) MOG35-55-specific T cells, the transfer of high dose of α-GC-treated cells led to a more aggressive clinical course in terms of both disease onset and severity, which was confirmed by histopathological analysis (Figures 2A and 2B). When challenged with the eliciting MOG peptide, T cells derived from high dose of α-GC-treated EAE mice displayed a markedly enhanced proliferative response, which was accompanied by significantly increased levels of IL-17 and IFN-γ (Figures 3A and 3B). As shown in Figure 3C, flow cytometric analysis of Th17 and Th1 cells revealed that the high dose of α-GC markedly enhanced the development of Th17 and Th1 cells in the spleens and the spinal cords of EAE mice. The underlying molecular events involved in Th1 and Th17 cell differentiation were further analyzed in CD4+T cells derived from α-GC-treated or control EAE mice by immunoblot. As shown in Figure 3D, high dose of α-GC specifically enhanced the levels of STAT3 phosphorylation. These findings are consistent with the role of STAT3 in Th17 differentiation 32, 33. Although the expression and phosphorylation of STAT4 were only mildly increased, the upregulation of the Th1-specific transcription factor T-bet was greater in the high dose of α-GC-treated group than in the vehicle or low dose of α-GC treatment groups (Figure 3D). At the same time, we also investigated the expression of other transcription factors implicated in the generation of Th17 cells, such as RORα, RORγt and Ahr in different groups (vehicle, 0.5 μg and 5 μg), and found that α-GC treatment at high or low doses did not affect their expression (data not shown). Taken together, these data indicate that high-dose α-GC treatment on EAE was associated with enhanced Th17 and Th1 cell development.

High dose of α-GC potentiates, but low dose of α-GC ameliorates, EAE. (A) Mice were administered intraperitoneally with α-GC or vehicle at the day of EAE induction. Data are expressed as mean clinical score ± SEM. (B) Spinal cord sections were obtained from EAE mice treated with α-GC or vehicle at day 17 post immunization and analyzed for inflammation by H&E staining and demyelination by Luxol fast blue staining. Spinal cord samples derived from naïve mice were set as healthy control. Original magnification, × 100. Data are representative of three independent experiments.

Adoptive transfer of high dose of α-GC-treated MOG35-55-specific cells induces more severe EAE. (A) Splenocytes were isolated from EAE mice at day 13 post immunization and stimulated with MOG35-55 peptide (20 μg/ml) in the presence of various concentrations of α-GC or vehicle for 3 days. The resulting cells were adoptively transferred into naïve recipients. Data are expressed as mean clinical score ± SEM. (B) Spinal cord sections were obtained from EAE mice at day 13 post adoptive transfer and analyzed for inflammation by H&E staining and demyelination by Luxol fast blue staining. Spinal cord samples derived from naïve mice were set as healthy control. Original magnification, × 100. Data presented are representative of two independent experiments.

T-cell responses in EAE mice treated with α-GC or vehicle. Splenocytes and CNS-infiltrating mononuclear cells (MNCs) were isolated from mice treated with α-GC or vehicle at day 17 post immunization. (A) Splenocytes were examined for proliferation in the presence or absence of the MOG35-55 peptide. Data are expressed as mean c.p.m. ± SD. (B) Cell culture supernatants were analyzed for indicated cytokines by ELISA. Data are expressed as mean concentration ± SD. Asterisks represent statistical differences between groups; *P < 0.05. (C) Percentages of Th1 and Th17 cells in CD4 subsets were analyzed in mononuclear cells by intracellular staining of IFN-γ and IL-17, respectively. (D) Splenocytes were restimulated with MOG35-55 peptide for 24 h. CD4+ T-cell blasts were analyzed for the expression and phosphorylation levels of indicated proteins by immunoblotting. Data are representative of three independent experiments.
High doses of α-GC enhance Th17 differentiation and activation directly
We considered the possibility that high doses of α-GC can activate Th17 and Th1 cells rather than iNKT cells. As αGC-CD1d-Dimer is a reliable marker for iNKT cells 34, we sorted iNKT-depleted cells by excluding αGC-CD1d-Dimer+ population and stimulated these cells with α-GC. As described in Figure 4A, αGC-CD1d-Dimer− splenocytes and αGC-CD1d-Dimer−CD4+ T cells were highly reactive to high doses of α-GC. Moreover, the production of IL-17, IL-6 and IFN-γ was greatly enhanced in response to high dose, but not to low dose, of α-GC in αGC-CD1d-Dimer−CD4+ T cells, which was consistent with the results of IL-17 and IFN-γ intracellular staining (Figures 4B and 4C). When IL-6 neutralizing Ab was added into high dose of α-GC-induced Th17 cell culture, the level of Th17 cells was not affected by IL-6 antagonism (Figure 4C). Furthermore, high dose of α-GC promoted the differentiation of Th17 cells in vitro, which correlated with an increased phosphorylation of STAT3 (Figures 4D and 4E). Meanwhile, high dose of α-GC also activated Th17 and Th1 cells through activation of NF-κB by enhancing phosphorylation of p65 (Figure 4F). To further elucidate whether the α-GC-enhanced Th17 cell response was due to Th17 cell differentiation from naïve T cells or activation of differentiated memory cells, we stimulated sorted CD4+CD44− cells and CD4+CD44+ cells with high dose of α-GC, respectively. As shown in Figures 4G and 4H, in contrast to CD4+CD44− cells, only CD4+CD44+ cells produced IL-17 and IFN-γ when stimulated with high dose of α-GC. Thus, these data suggest that high doses of α-GC could directly activate CD4+CD44+ cells and enhance Th17 and Th1 cell response through STAT3 and NF-κB pathways.

In vitro activation of Th17 and Th1 cells by α-GC. (A) Total splenocytes, purified αGC-CD1d-Dimer− splenocytes or αGC-CD1d-Dimer−CD4+ T cells were isolated from naïve mice and cultured in the absence or presence of various concentrations of α-GC for 72 h. Cell proliferation was determined by [3H]-thymidine incorporation. Data are expressed as mean c.p.m. ± SD. (B) Purified αGC-CD1d-Dimer−CD4+ T cells were stimulated with vehicle or α-GC at 10 ng/ml or 400 ng/ml for 60 h. Supernatants were then collected for determination of indicated cytokines by ELISA. Data are expressed as mean concentration ± SD. Asterisks represent statistical differences between groups; *P < 0.05, **P < 0.01. (C) Purified αGC-CD1d-Dimer−CD4+ T cells derived from naïve mice were cultured with various concentrations of α-GC in the absence or presence of anti-IL6 mAb (10 μg/ml) for 72 h. Cells were then collected for analysis of intracellular IL-17 and IFN-γ. (D) Purified αGC-CD1d-Dimer−CD4+ T cells from naïve mice were activated with plate-bound anti-CD3 and soluble anti-CD28 in the presence of various concentrations of α-GC for 4 days under Th17 polarizing conditions. Intracellular staining of IL-17 and IFN-γ were analyzed in CD4+ T cells. (E and F) Purified αGC-CD1d-Dimer−CD4+ T cells derived from naïve mice were cultured with vehicle (medium) or α-GC at indicated concentrations for 24 h. Cells were then collected for immunoblotting. (G and H) FACS-sorted CD4+CD44+ and CD4+CD44−T cells were cultured in the absence or presence of α-GC for 72 h; cell proliferation and the concentration of IL-17 and IFN-γ were determined as mentioned above. Data are representative of three independent experiments.
To determine whether CD1d was involved in the Th17 and Th1 responses to high doses of α-GC, we examined the α-GC-induced proliferation and cytokine production in the presence of CD1d-blocking Ab. The data showed that CD1d monoclonal Ab, and not its isotype-matched control mAb, could abrogate the proliferation of and IL-17/IFN-γ production by αGC-CD1d-Dimer−CD4+ T cells (Figure 5A). Both high- and low-dose α-GC-pulsed antigen-presenting cells (APCs) failed to stimulate αGC-CD1d-Dimer−CD4+ T cells to proliferate and produce IL-17 and IFN-γ (Figure 5A). In contrast, sorted iNKT (αGC-CD1d-Dimer+) cells only proliferated and produced IFN-γ and IL-4 in response to α-GC loaded onto APCs (Figure 5B). Collectively, these data suggest that high doses of α-GC directly interact with CD1d expressed on CD4+ T cells and enhance Th1 and Th17 responses through activation of memory cells.

Activation of αGC-CD1d-Dimer−CD4+ T cells and iNKT cells by different doses of α-GC. Sorted αGC-CD1d-Dimer−CD4+ T cells (A) and iNKT cells (α-GC-CD1d-Dimer+) (B) were stimulated with α-GC at the indicated concentrations or with α-GC-pulsed APCs for proliferation and cytokine production. Data represent mean c.p.m. ± SD or mean concentration (pg/ml ± SD) of triplicates, and are representative of three individual experiments. CD1d-blocking Ab 1B1 or matching isotype control was added at 10 μg/ml, 30 min before the addition of α-GC. Proliferation was measured by [3H]thymidine uptake after a 72-h culture. Supernatants from the same culture were measured for the concentration of IL-4, IFN-γ and IL-17 using ELISA. Values represent mean concentration (pg/ml ± SD) of three mice from one of the three individual experiments with similar results.
Pathogenic cells are significantly increased in transfer EAE in response to MOG-specific T cells treated with high doses of α-GC
To correlate EAE disease onset with the migration of transferred pathogenic cells, we evaluated the time course of disease onset using GFP+ MOG-specific T cells as a tracer. Cytofluorometric analysis of total MOG-T cell, CD4+ or CD8+ cell trafficking in immune organs and in the spinal cord on days 4, 8, 13 and 35 revealed that the number of α-GC-treated GFP+ cells was significantly increased as compared to the vehicle-treated cells, particularly in the central nervous system (CNS), which correlated with the disease course (Figure 6). These data further confirmed that high doses of α-GC-enhanced pathogenic cells, namely, Th17 and Th1 cells, in transfer EAE.

In vivo migration of high-dose α-GC-treated MOG35-55-specific cells. α-GC-treated MOG35-55-specific cells (400 ng/ml) from GFP-Tg mice were transferred in naïve B6 mice. GFP+ cells in the spleen, lymph nodes and the CNS were determined by FACS on 4, 9, 13 and 35 days post injection. Number of total GFP+ cells (upper) and CD4+ and CD8+ subsets (lower) per 105 cells were enumerated.
Activation of iNKT requires CD1d1, whereas activation of Th17 and Th1 requires CD1d2
The iNKT is activated by low doses of α-GC, whereas Th17 and Th1 cells are activated by high doses of α-GC. It is conceivable that the activation of iNKT and Th17/Th1 cells by different doses of α-GC may be due to the different CD1d isoforms (for example, CD1d1 or CD1d2) expressed on APCs, iNKT and αGC-CD1d-Dimer−CD4+ T cells. Interestingly, αGC-CD1d-Dimer−CD4+ T cells mainly express CD1d2 mRNA, whereas APCs mainly express CD1d1 mRNA, but both of the molecules are expressed at very low levels in iNKT cells (Figure 7A). We further confirmed by FACS that CD1d expression on APCs is predominantly CD1d1, while that on αGC-CD1d-Dimer−CD4+ T cells is predominantly CD1d2 (Figure 7B). The activation of iNKT cells by α-GC-pulsed APCs, as measured by IFN-γ production, was greatly inhibited by RNAi-mediated knockdown of CD1d1 in APCs, but not CD1d2 (Figure 7C). Meanwhile, activation of Th17 cells by α-GC was significantly reduced by knockdown of CD1d2, but not CD1d1, in T cells as measured by reduced IL-17 production (Figure 7C). These results collectively support the fact that the activation of iNKT and Th17 by different concentrations of α-GC is associated with the CD1d isoforms expressed on indicated cells (Figure 8).

The role of CD1d1 and CD1d2 on the activation of iNKT and αGC-CD1d-Dimer−CD4+ T cells. (A) Relative expression of CD1d1 and CD1d2 by APCs, αGC-CD1d-Dimer−CD4+ T cells and iNKT cells from splenocytes of naïve mice was determined by real-time PCR. (B) Purified APCs, sorted αGC-CD1d-Dimer−CD4+ T cells and iNKT were incubated with the blocking mAb anti-CD1d 1B1 for 30 min and then washed for transfection. The resulting cells cultured for 4 days were analyzed by FACS for CD1d expression after CD1d1 and CD1d2 knockdown by RNAi. (C) Supernatants from the iNKT cells cultured with α-GC-pulsed APCs were assessed for the concentration of IFN-γ and supernatants from sorted αGC-CD1d-Dimer−CD4+ T cells cultured with α-GC were analyzed for the concentration of IL-17 using ELISA after CD1d1 and CD1d2 knockdown by RNAi. Asterisks represent statistical differences between groups; **P < 0.01.

The schematic of activation of iNKT and αGC-CD1d-Dimer−CD4+ T cells by α-GC. In the presence of low doses or high doses of α-GC, CD1d1 on APCs binds α-GC for presentation to iNKT by means of TCR recognition and activates IFN-γ and IL-4 release. Th17 and Th1 cells can be activated only in high doses of α-GC through its binding with CD1d2 on Th17 and Th1 directly and release IL-17 and IFN-γ.
Discussion
In this study, we provide compelling evidence that, in addition to its activation of iNKT, high doses of α-GC directly enhance Th1 and Th17 responses through activating memory cells, which are critically involved in autoimmune pathologies such as MS and its animal model, EAE. It has been reported that α-GC can protect susceptible mice against EAE through activation of iNKT to suppress myelin antigen-specific Th1 responses and/or to promote myelin antigen-specific Th2 responses in vivo. Meanwhile, here we demonstrate that, when administered at high doses, α-GC activates Th17 and Th1 cells, leading to a more severe autoimmune disease. These findings have important implications in our understanding of the doses used in α-GC treatment of autoimmune diseases. Our study reveals the underlying mechanism of the exacerbation of EAE by high-dose α-GC. First, we found that high doses of α-GC could upregulate STAT3 phosphorylation and activate the NF-κB pathway in CD4+ encephalitogenic T cells, leading to enhanced Th17 cell development and earlier onset of EAE. On the other hand, it is clear that Th1 cells are also involved in the development of autoimmune responses. The T-box transcription factor T-bet is a key inducer of IFN-γ and Th1 cell differentiation, and STAT4 is essential for the stabilization of IFN-γ production and the development of terminally differentiated Th1 cells. Although high doses of α-GC only mildly influenced expression and phosphorylation of STAT4, the expression of T-bet was greatly upregulated by high doses of α-GC. Second, high doses of α-GC also activated Th17 and Th1 cells through activation of the NF-κB pathway. In particular, α-GC was capable of promoting Th1 and Th17 response mainly through activating CD4+CD44+ memory cells, which were critically involved in the systemic and CNS inflammation in EAE. We also investigated the effect of α-GC on Treg cell development, both in vivo and in vitro, and found that the levels of Treg cells were not affected by high or low doses of α-GC (data not shown).
Furthermore, we explored the mechanisms underlying the activation of iNKT and Th17/Th1 cells in response to different doses of α-GC. There are two main differences in the activation of iNKT and Th17/Th1 in response to α-GC. First, although both iNKT cells and αGC-CD1d-Dimer−CD4+ T cells function in a CD1d-dependent manner, Th17 and Th1 cells, unlike iNKT, do not require exogenous CD1d-expressing cells to respond to α-GC. iNKT cells recognize α-GC presented by the MHC class I-related glycoprotein CD1d, whereas Th17/Th1 cells are activated directly by high doses of α-GC. Second, the α-GC concentration required for the activation of iNKT is very low, whereas that required for the activation of Th17 and Th1 cells is much higher. In mouse, CD1 molecules are constitutively expressed by nearly all hematopoietic cells, including B and T lymphocytes, macrophages and dendritic cells 35, 36, 37. However, the physiological role of CD1d expression on peripheral murine T cells is currently unknown. In this study, our data suggest that interaction with CD1d by α-GC at high doses may directly activate T cells. The mouse CD1 system is formed by two closely related genes, CD1d1 and CD1d2 9, 38. CD1d1 encodes a molecule that presents antigens to iNKT cells. However, the function of the CD1d2 gene has not been elucidated. Our results reveal that αGC-CD1d-Dimer−CD4+ T cells predominantly express CD1d2, whereas APCs predominantly express CD1d1, and both CD1d1 and CD1d2 are expressed at very low levels in iNKT cells. Furthermore, we demonstrated that CD1d1 on APCs could present α-GC to iNKT and activate iNKT cells to produce IFN-γ and IL-4, while high doses of α-GC directly interact with CD1d2 on T cells and promote Th1/Th17 response. Several recent reports have demonstrated the existence of a functionally competent subpopulation of iNKT cells, which preferentially produce IL-17 39, 40, 41, 42, 43. In our study, however, the production of IL-17 and IFN-γ was confined to αGC-CD1d-Dimer−CD4+ T cells, since we employed the method of FACS sorting to remove iNKT and APCs, which may confound interpretation of the data.
Taken together, our study has revealed a novel function of α-GC: high dose α-GC directly enhances Th1 and Th17 development, altering the clinical outcome of autoimmune disease. In this context, our results caution against the use of high doses of α-GC, as the risk of activating the Th17 and Th1 response could lead to a potential worsening of the disease.
Materials and Methods
Mice
Male C57BL/6 (B6) mice aged 6-12 weeks were purchased from the Shanghai Laboratory Animal Center, Chinese Academy of Sciences. GFP-Tg mice of the same C57BL/6 background were a kind gift from Dr XF Zhou (Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences). Mice were maintained under pathogen-free conditions.
Reagents
α-GC was purchased from Alexis Biochemicals and was reconstituted in phosphate-buffered saline (PBS) containing 0.5% polysorbate-20 (Sigma-Aldrich). Purified rat anti-mouse CD1d Ab 1B1 and its isotype-matched mAb (IgG2b,κ), FITC-conjugated anti-TCRβ mAb, mouse CD1d dimer, PE-conjugated anti-IgG1, FITC-conjugated anti-IgG1, purified mouse IgG1 isotype control mAb, anti-CD16/32, anti-IL6 mAb, PE-conjugated anti-IL-17 mAb, APC-conjugated anti-IFN-γ mAb and the Cytofix/Cytoperm Plus fixation/permeabilization kit with GolgiPlug were purchased from BD Pharmingen. APC-conjugated anti-CD4 mAb and FITC-conjugated anti-CD4 mAb were purchased from eBioScience. Stat3 and Phospho-Stat3 (Tyr705) Abs were purchased from Cell Signaling Technology. T-bet, Stat4 and Phospho-Stat4 Abs were purchased from Santa Cruz Technology. Mouse CD4+ T-cell isolation kit and MicroBead-conjugated anti-PE Ab were purchased from Miltenyi Biotec. RNeasy Mini Kit, Sensiscript RT kit and RNase-free DNase were purchased from Qiagen. SYBR Green PCR master mix was purchased from ABI. The mouse T-cell Nucleofector kit was purchased from Amaxa.
Induction of EAE by active immunization or by passive transfer
The encephalitogenic peptide MOG used to induce EAE corresponds to residues 35-55 (Met-Glu-Val-Gly-Trp-Tyr-Arg-Ser-Pro-Phe-Ser-Arg-Val-Val-His-Leu-Tyr-Arg-Asn-Gly-Lys). The peptide was purchased from BioAsia Biotechnology and had 95% purity. Active EAE was induced by subcutaneous immunization with 300 μg of the MOG35-55 peptide in CFA containing 5 mg/ml of heat-killed H37Ra strain of Mycobacterium tuberculosis (Difco Laboratories) in the back region. Pertussis toxin (200 ng per mouse; List Biological Laboratories Inc) dissolved in PBS was administered intravenously on the day of immunization and 48 h later. For the treatment of mice with α-GC, the indicated dose of α-GC dissolved in 200 μl PBS was given intraperitoneally at the time of EAE induction. To induce EAE by passive transfer, mononuclear cells (MNC) were isolated from draining lymph nodes and spleens of MOG-immunized B6 mice and were cultured in vitro with MOG35-55 (20 μg/ml) for 84 h. The cells were pulsed with either the indicated doses of α-GC or vehicle during the last 12 h of culture prior to harvest. The resulting cells were separated on a Ficoll gradient and injected intraperitoneally into naïve B6 recipients (1 × 107 cells per mouse). Pertussis toxin (200 ng per mouse) dissolved in PBS was administered intravenously on the day of adoptive transfer and 48 h later. The mice were weighed and examined daily for disease symptoms. They were scored for disease severity using the EAE scoring scale: 0, no clinical signs; 1, limp tail; 2, paraparesis (weakness, incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with fore limb weakness or paralysis; 5, moribund state or death. The animal protocols were approved by the institutional review board of the Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences.
Histology
Spinal cords were removed from the mice 17 days after active immunization or 13 days after adoptive transfer and were immediately fixed in 4% paraformaldehyde. Paraffin-embedded 5- to 10-μm sections were stained with Luxol fast blue or hematoxylin/eosin and then examined by light microscopy.
Preparation of splenic APCs, αGC-CD1d-Dimer−CD4+ T cells and iNKT cells and isolation of MNCs from mouse CNS tissue
APCs were positively selected from mouse splenocytes using anti-MHC class II MicroBeads. iNKT (αGC-CD1d-Dimer+) cells were sorted by the binding of CD1d dimer loaded with αGC and TCRβ by BD FACSAria cell sorter. αGC-CD1d-Dimer−CD4+ T cells were further sorted from the Dimer− population based on CD4 expression. iNKT and αGC-CD1d-Dimer−CD4+ T cells were enriched by MACS before sorting. Final purity was 95%∼99%. For preparation of CNS-infiltrating MNCs, mice were perfused with 30 ml of PBS via the heart to eliminate peripheral blood. The brain and spinal cord tissues were harvested and dissociated by passing through a wire mesh. The CNS cell suspension was centrifuged in a Percoll gradient. MNCs at the interface between the two gradients (37% and 70% Percoll) were collected and washed by centrifugation in RPMI 1640 medium.
Proliferative responses and cytokine secretion
For MOG35-55-induced proliferation assays, spleens were removed from EAE mice on day 17 after active immunization and were prepared for single-cell suspensions. Splenocytes (2 × 105 per well) were cultured in triplicate in complete RPMI 1640 (RPMI 1640 with 10% fetal calf serum, 10 mM HEPES, 50 μM β-mercaptoethanol, 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 100 U/ml penicillin and 100 μg/ml streptomycin, GIBCO) in 96-well round-bottomed plates. The cells were cultured in the presence or absence of the MOG peptide (20 μg/ml) at 37 ºC in 5% CO2 for 72 h. For α-GC-induced proliferation assays, splenic APCs, sorted αGC-CD1d-Dimer−CD4+ T cells and iNKT cells (2 × 105) were cultured with the indicated doses of α-GC or with α-GC-pulsed APCs (1 × 105, the splenic APCs had been pre-treated with 10 ng/ml of α-GC for 3 h, irradiated and washed extensively) for 72 h. The cells were pulsed with 1 μCi [3H]-thymidine during the last 16-18 h of culture prior to harvest. [3H]-thymidine incorporation was measured as c.p.m. using a Betaplate counter. For cytokine secretion assays, the indicated cells (1 × 106 per well) were cultured in complete RPMI 1640 in 96-well flat-bottomed plates. Supernatants were collected from the cell cultures at 60 h and diluted for the measurement of IFN-γ, TNF-α, IL-17, IL-2, IL-4 and IL-10 by ELISA (R&D Systems), according to the manufacturer's instructions. A standard curve was prepared for each plate and used to calculate the absolute concentrations of the indicated cytokines. Cytokine levels are expressed as mean ± SD of triplicate cell cultures.
Flow cytometry
Cells were resuspended in PBS containing 1% BSA (Sigma-Aldrich) for cell surface staining. After preincubation with anti-mouse CD16/32 mAb for 10 min, the cells were stained at 4 ºC for 20 min with fluorescence-labeled mAb. For intracellular staining, the cells were restimulated with 500 ng/ml ionomycin and 50 ng/ml of PMA (Sigma-Aldrich) in the presence of GolgiPlug for 5 h. The cells were then washed, blocked with anti-mouse CD16/32 mAb and stained for surface markers. After three washes, the cells were fixed and permeabilized using Cytofix/Cytoperm Plus fixation/permeabilization kit with GolgiPlug in accordance with the manufacturers instructions. The cells were then stained with PE-anti-mouse IL-17 and APC-anti-mouse IFN-γ. The stained cells were analyzed on a FACSCalibur (Becton Dickinson) using CellQuest software (BD Biosciences).
Immunoblot analysis
Protein extracts were loaded onto 10% or 12% SDS-polyacrylamide gels and subjected to electrophoresis. Immunoblot analysis was performed by initial transfer of the proteins onto nitrocellulose filters using Mini Trans-Blot (Bio-Rad) followed by a blocking step using Tris-buffered saline supplemented with 0.1% Tween-20 and 5% nonfat dried milk for 1 h at room temperature. After blocking, the membranes were incubated overnight at 4 °C with the specific primary Abs. After washing and subsequent incubation with a goat anti-rabbit (Sigma-Aldrich) or anti-mouse (Cell Signaling Technology) Ab conjugated with HRP for 1 h at room temperature and extensive washing, the signals were visualized with ECL substrate (Pierce). β-Actin was used as an endogenous control to normalize for differences in the amount of total protein in each sample.
In vitro stimulation with α-GC and Ab-blocking studies
Total splenocytes, sorted αGC-CD1d-Dimer− splenocytes and αGC-CD1d-Dimer−CD4+ T cells isolated from naïve mice were cultured with a titrated dose of α-GC or with vehicle control (0.0025% polysorbate) in complete RPMI 1640 medium for 72 h. In the CD1d-blocking experiments, anti-CD1d mAb 1B1 or its isotype-matched control mAb was added at 10 μg/ml 30 min before the addition of α-GC. In the IL-6 neutralizing experiments, anti-IL-6 mAb was added at 10 μg/ml with 400 ng/ml α-GC.
Th17 differentiation in vitro
For Th17 differentiation, sorted αGC-CD1d-Dimer−CD4+ T cells were activated with plate-bound anti-CD3 and soluble anti-CD28 under Th17-polarizing conditions (IL-6, 20 ng/ml; TGF-β, 2 ng/ml; anti-IL-4, 10 μg/ml; anti-IFN-γ, 10 μg/ml) in the presence or absence (control) of different concentrations of α-GC (10 or 400 ng/ml). After 4 days of culture, cells were restimulated with 50 ng/ml PMA and 500 ng/ml ionomycin in the presence of GolgiPlug for 5 h; IL-17 and IFN-γ producing cells were analyzed using intracellular staining techniques, as described above.
Analysis of CD1d1 and CD1d2 mRNA expression
Total RNA was isolated from cell pellets using an RNeasy mini kit. Genomic DNA was removed from total RNA by DNase digestion during RNA purification and the resulting RNA was stored at −80 °C . First-strand cDNA synthesis was performed using a Sensiscript RT kit with random hexamers (Qiagen). The mRNA expression of mouse CD1d1 and CD1d2 was determined by real-time PCR using SYBR Green Master Mix (Applied Biosystems). The thermocycler conditions included an initial holding at 50 °C for 2 min and then at 95 °C for 10 min, which was followed by a two-step PCR program: 95 °C for 15 s and 60 °C for 60 s for 40 cycles. Data were collected and quantitatively analyzed on an ABI PRISM 7900 sequence detection system (Applied Biosystems). The PCR primer pairs were as follows: β-actin, forward 5′-GTG AAG GTC GGA GTC AAC G-3′ and reverse 5′-TGA GGT CAA TGA AGG GGT C-3′; CD1d1, forward 5′-GGC CTG ATC GTC TTC ATA GTA CTG-3′ and reverse 5′-AAC TAT GGA AAC TGA CGA AAC AAC TC-3′; CD1d2 forward 5′-CGG GGA ACA TCT TGG CAG AGG-3′ and reverse 5′-CCT CTA GGA GAC CAC GGG CAA ACT-3′.
CD1d1 and CD1d2 knockdown by small interfering RNA (RNAi)
Purified APCs, sorted αGC-CD1d-Dimer−CD4+ T cells and iNKT were incubated with the blocking mAb anti-CD1d 1B1 for 30 min and then washed for transfection. Annealed siRNA constructs were obtained from Shanghai GenePharma. The sense sequences were as follows: CD1d1, 5′-CAC UGA ACU AGA AAG CAU ATT-3′, CD1d2, 5′-UCU UGG CAG AGG GUC CUA GTT-3′; 5′-UUC UCC GAA CGU GUC ACG UTT-3′ was used as a negative control. Each RNAi at 300 nM was mixed with 100 μl of Nucleofector solution (Amaxa) and transfected into 5 × 106 cells using the Nucleofector electroporator device. The cells were then transferred into 0.5 ml of culture medium containing 20% FBS and cultured for 4 days at 37 °C and 5% CO2 before performing the assays.
Statistics
Student's t-test was used to analyze for differences between groups. One-way ANOVA was initially performed to determine whether an overall statistically significant change existed before using the two-tailed paired or unpaired Student's t-test. A P < 0.05 was considered statistically significant.
Abbreviations
- α-GC:
-
(α-galactosylceramide)
- EAE:
-
(experimental autoimmune encephalomyelitis)
- MS:
-
(multiple sclerosis)
- CFA:
-
(complete Freund's adjuvant)
- MOG:
-
(myelin oligodendrocyte glycoprotein)
- APC:
-
(antigen-presenting cell)
- CNS:
-
(central nervous system)
- PMA:
-
(phorbol 12-myristate 13-acetate)
References
Paterson PY . Autoimmune diseases of myelin. Prog Clin Biol Res 1980; 49:19–36.
Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233–240.
Chen Y, Langrish CL, McKenzie B, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest 2006; 116:1317–1326.
Cua DJ, Sherlock J, Chen Y, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003; 421:744–748.
Park H, Li Z, Yang XO, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6:1133–1141.
Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 2006; 177:566–573.
Hofstetter HH, Ibrahim SM, Koczan D, et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol 2005; 237:123–130.
Porcelli SA, Modlin RL . The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol 1999; 17:297–329.
Porcelli SA . The CD1 family: a third lineage of antigen-presenting molecules. Adv Immunol 1995; 59:1–98.
Balk SP, Bleicher PA, Terhorst C . Isolation and expression of cDNA encoding the murine homologues of CD1. J Immunol 1991; 146:768–774.
Kawano T, Cui J, Koezuka Y, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997; 278:1626–1629.
Bendelac A, Rivera MN, Park SH, Roark JH . Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu Rev Immunol 1997; 15:535–562.
Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG . NKT cells: facts, functions and fallacies. Immunol Today 2000; 21:573–583.
Kronenberg M, Gapin L . The unconventional lifestyle of NKT cells. Nat Rev Immunol 2002; 2:557–568.
Kronenberg M . Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol 2005; 23:877–900.
Miyamoto K, Miyake S, Yamamura T . A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature 2001; 413:531–534.
Singh AK, Wilson MT, Hong S, et al. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med 2001; 194:1801–1811.
Hong S, Wilson MT, Serizawa I, et al. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med 2001; 7:1052–1056.
Sharif S, Arreaza GA, Zucker P, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med 2001; 7:1057–1062.
Zeng D, Lee MK, Tung J, Brendolan A, Strober S . Cutting edge: a role for CD1 in the pathogenesis of lupus in NZB/NZW mice. J Immunol 2000; 164:5000–5004.
Liu R, La Cava A, Bai XF, et al. Cooperation of invariant NKT cells and CD4+CD25+ T regulatory cells in the prevention of autoimmune myasthenia. J Immunol 2005; 175:7898–7904.
Kojo S, Adachi Y, Keino H, Taniguchi M, Sumida T . Dysfunction of T cell receptor AV24AJ18+, BV11+ double-negative regulatory natural killer T cells in autoimmune diseases. Arthritis Rheum 2001; 44:1127–1138.
Kaieda S, Tomi C, Oki S, Yamamura T, Miyake S . Activation of invariant natural killer T cells by synthetic glycolipid ligands suppresses autoantibody-induced arthritis. Arthritis Rheum 2007; 56:1836–1845.
Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB . Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat immunol 2003; 4:1230–1237.
Nieuwenhuis EE, Matsumoto T, Exley M, et al. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat Med 2002; 8:588–593.
Ronet C, Darche S, Leite de Moraes M, et al. NKT cells are critical for the initiation of an inflammatory bowel response against Toxoplasma gondii. J Immunol 2005; 175:899–908.
Crowe NY, Coquet JM, Berzins SP, et al. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med 2005; 202:1279–1288.
Godfrey DI, Kronenberg M . Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest 2004; 114:1379–1388.
Shimizu K, Goto A, Fukui M, Taniguchi M, Fujii S . Tumor cells loaded with alpha-galactosylceramide induce innate NKT and NK cell-dependent resistance to tumor implantation in mice. J Immunol 2007; 178:2853–2861.
Chung Y, Qin H, Kang CY, Kim S, Kwak LW, Dong C . An NKT-mediated autologous vaccine generates CD4 T-cell dependent potent antilymphoma immunity. Blood 2007; 110:2013–2019.
Jahng AW, Maricic I, Pedersen B, et al. Activation of natural killer T cells potentiates or prevents experimental autoimmune encephalomyelitis. J Exp Med 2001; 194:1789–1799.
Mathur AN, Chang HC, Zisoulis DG, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol 2007; 178:4901–4907.
Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 2007; 282:9358–9363.
Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L . NKT cells: what's in a name? Nat Rev Immunol 2004; 4:231–237.
Brossay L, Jullien D, Cardell S, et al. Mouse CD1 is mainly expressed on hemopoietic-derived cells. J Immunol 1997; 159:1216–1224.
Mandal M, Chen XR, Alegre ML, et al. Tissue distribution, regulation and intracellular localization of murine CD1 molecules. Mol Immunol 1998; 35:525–536.
Park SH, Roark JH, Bendelac A . Tissue-specific recognition of mouse CD1 molecules. J Immunol 1998; 160:3128–3134.
Jullien D, Brossay L, Sieling PA, Modlin RL, Kronenberg M . CD1: clues on a new antigen-presenting pathway. Res Immunol 1996; 147:321–328.
Lee KA, Kang MH, Lee YS, et al. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell Immunol 2008; 251:50–55.
Michel ML, Keller AC, Paget C, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med 2007; 204:995–1001.
Pichavant M, Goya S, Meyer EH, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med 2008; 205:385–393.
Rachitskaya AV, Hansen AM, Horai R, et al. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol 2008; 180:5167–5171.
Yoshiga Y, Goto D, Segawa S, et al. Invariant NKT cells produce IL-17 through IL-23-dependent and -independent pathways with potential modulation of Th17 response in collagen-induced arthritis. Int J Mol Med 2008; 22:369–74.
Acknowledgements
This work was supported by Grants NSF-30430650 and NSF-30571731 from the National Natural Science Foundation of China and by Grant 04DZ14902 from the Shanghai Commission of Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Qian, G., Qin, X., Zang, Y. et al. High doses of α-galactosylceramide potentiate experimental autoimmune encephalomyelitis by directly enhancing Th17 response. Cell Res 20, 480–491 (2010). https://doi.org/10.1038/cr.2010.6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2010.6
Keywords
This article is cited by
-
Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE
Cellular & Molecular Immunology (2019)
-
Toll-like Receptor agonists and alpha-galactosylceramide synergistically enhance the production of interferon-gamma in murine splenocytes
Scientific Reports (2013)
