
Abstract
The tumor suppressor p53 is a multifunctional, highly regulated, and promoter-specific transcriptional factor that is uniquely sensitive to DNA damage and cellular stress signaling. The mechanisms by which p53 directs a damaged cell down either a cell growth arrest or an apoptotic pathway remain poorly understood. Evidence suggests that the in vivo functions of p53 seem to balance the cell-fate choice with the type and severity of damage that occurs. The concept of antirepression, or inhibition of factors that normally keep p53 at bay, may help explain the physiological mechanisms for p53 activation. These factors also provide novel chemotherapeutic targets for the reactivation of p53 in tumors harboring a wild-type copy of the gene.
Similar content being viewed by others

Introduction
The tumor suppressor p53 remains one of the single, most important transcription factors governing the genetic circuitry of cells. It represents a critical node for determining cell fate by specifically activating growth arrest, cellular senescence, or apoptotic pathways. Yet, recent studies have indicated that p53 has seemingly vast roles in other functions such as fertility, development, and glycolysis. Nevertheless, it has become increasingly clear that p53 has a critical role in protecting cells from uncontrolled growth, thus preventing tumorigenesis through the activation of specific cell growth-regulatory pathways.
The ability of p53 to choose cell survival or cell death in response to DNA damage and cellular stress has been the topic of a number of both recent and seminal studies. From the surface, it seems logical that p53 would have the capability of inducing cell death in cells that have sustained irreparable damage as a means to prevent unregulated growth and tumorigenesis. Cells that have been exposed to a low level of stress and sustained minimal damage could then be arrested, repaired, and allowed to survive. However, this hypothesized model is not straightforward. First, p53 has the ability to induce cellular senescence, a condition where a cell is kept in a permanent, inactive state. Cellular senescence has strong implications in longevity and the process of aging, but the exact mechanisms by which p53 functions to promote this cell fate remain poorly understood. Second, p53 has been shown to promote cell survival under specific stimuli through mechanisms that inhibit apoptosis and reduce the level of reactive oxygen species 1. Promotion of cell survival, particularly in damaged cells, seems counterintuitive for the overall scheme of prevention of tumorigenesis. However, in multicellular organisms, repair and survival of low to moderately damaged cells may provide a global benefit for the organism 2. Therefore, the mechanisms by which a cell undergoes a specific p53-mediated fate may be largely dependent on the severity of damage and the context of the cell environment 3.
The classical tumor suppression model for p53 includes stabilization and activation of the protein through a plethora of redundant post-translational modifications 4. p53 is maintained at low protein levels during times of homeostasis, when the cell is not exposed to stress or DNA-damaging events, by its predominant negative regulator Mdm2 through the ubiquitin-proteasome pathway. Initial stabilization of p53 occurs through the targeted disruption of the p53 and Mdm2 interaction. Subsequent acetylation of the protein activates its sequence-specific transactivation functions and leads to the transcription of a multitude of downstream regulatory factors. Still, recent in vivo evidence that has considered the extent of modification redundancy challenges us to step back and reconsider the simplicity of this classical model. Is it possible that p53 is a constitutively active transcription factor that requires complex and sometimes even subtle changes in its repressed state to become active? Recent data would suggest that it does, and the concept of an additional regulatory layer of antirepression may help explain how p53 functions dictate a particular cellular pathway in response to stress.
This review will discuss recent findings in the field of p53 regulation and the impact they have had on p53 regulation hypotheses. We will also discuss the complexity of this regulatory network and how recent data suggest the existence of an additional layer of in vivo antirepression.
Regulation of p53
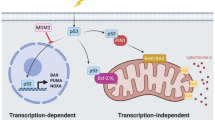
The importance of p53 in the regulation of cell survival and death pathways is emphasized by the seemingly endless upstream and downstream regulatory factors that continue to emerge. The Mdm2 E3 ubiquitin ligase represses p53 protein levels through continuous ubiquitination and degradation (Figure 1A) 5, 6. Targeted disruption of this interaction after stress induction occurs through numerous mechanisms, including post-translational modifications, physical sequestration, and degradation 7. The Mdm2-p53 interaction is inhibited by stress-induced phosphorylation of Ser395 and Tyr394 on Mdm2 by the kinases ATM and c-Abl, respectively 8, 9. A number of phosphorylation sites on p53 have been described and many serve to disrupt the Mdm2-p53 interaction as well. For example, phosphorylation of Thr18 in the transactivation domain of p53 significantly reduces Mdm2 binding 10. In addition, phosphorylation of Ser15 and Ser20 in the transactivation domain by stress-induced kinases ATM, ATR, Chk1, Chk2, and DNA-PK leads to p53 stabilization, presumably through the inhibition of Mdm2 interaction 11, 12, 13. However, in vivo evidence suggests a far more complicated regulatory picture than that elucidated from in vitro experiments. Studies using mice containing a Ser18Ala (human Ser15) mutation show no defects in cell stress-induced p53 stabilization 14, 15. Similar data were obtained from mutant knockin mice containing a Ser23Ala (human Ser20) substitution; however, the double mutant knockin (S18A/S23A) had a more pronounced p53 stabilization defect 16, 17, 18. Therefore, phosphorylation may be part of a series of post-translational events that need to occur in order for the p53 to be activated in response to cellular stress, but alone is likely not sufficient for p53 activation. Indeed, phosphorylation does not seem to be required for p53 to be activated in response to a number of specific types of cellular stress 18, 19, 20, 21. Despite these findings, an exhaustive analysis of all post-translational modification combinations has not been conducted in all cell types under all stress conditions; therefore, it may be too early to completely rule out phosphorylation as a mechanism for p53 activation. Nevertheless, a number of post-translational events combined with the release of repression may be needed for complete activation of p53 in response to cellular stress.

A model for p53 activation. (A) Classical model of p53 activation. DNA damage and oxidative stress signals lead to the rapid stabilization of p53 by blocking Mdm2 through various mechanisms described in the text. Once stabilized, p53 is activated through posttranslational modifications and binds to DNA, where it can interact with other transcriptional regulators for the induction of p53-responsive target genes. (B) A refined model of p53 activation. In addition to p53 stabilization, an additional step of antirepression may occur in vivo for full p53 activation. Mdm2 and MdmX repress p53 function and activation in vivo. Removal of this repression (i.e., antirepression) may be required for subsequent steps of p53 activation such as posttranslational modifications, DNA binding, and induction of p53-responsive target genes.
Acetylation of Mdm2 by CBP/p300 also disrupts the p53-Mdm2 interaction 22. Interestingly, in addition to transcriptional activation, acetylation of eight C-terminal lysine residues of p53 inhibits the p53-Mdm2 interaction in a mutually exclusive manner 23. In vitro experiments have also shown that purified, acetylated p53 cannot be ubiquitinated by Mdm2 and that ubiquitinated p53 levels drop upon induction of acetylation 24, 25. Mdm2 may therefore compete with acetyltransferases for access to the C-terminal lysines of p53. Although evidence suggests that six key C-terminal lysines are the predominant sites for Mdm2-mediated ubiquitination and subsequent degradation, mutant knockin mice carrying lysine to arginine mutations at these six sites (p53-6KR) have no changes in p53 protein levels 26, 27. In addition, p53 in cells derived from these mice can induce a DNA damage response and is sufficiently stabilized in response to cellular stress. This would suggest that other sites on p53 are ubiquitinated by Mdm2 or other E3 ligases. Indeed, specific lysine residues located in the DNA-binding domain have been shown to be ubiquitinated by Mdm2 in vitro 28. Furthermore, several other ubiquitin E3 ligases, including ARF-BP1/Mule, COP1, Pirh2, and MSL2, can ubiquitinate p53 and cause protein degradation or a change in its subcellular localization 29, 30, 31, 32. However, the biological importance of these findings in vivo remains to be elucidated. Taken together, in vivo evidence suggests that the p53-Mdm2 interaction is critical for maintaining p53 at low levels. Disruption of this interaction leads to p53 stabilization and activation. In addition, in vivo p53 ubiquitination may involve residues other than C-terminal lysines and depend on E3 ubiquitin ligases other than Mdm2. Recently, the structural mechanism for Pirh2-mediated regulation of p53 was described 33. The interaction between Pirh2 and p53 is dependent on the C-terminal zinc-binding motif of Pirh2, a domain that interacts with the tetramerization domain of p53. These data suggest that Pirh2 preferentially ubiquitinates the transcriptionally active, tetrameric form of p53, and therefore may provide a unique mechanism for regulating the protein in vivo. The multi-layered approach for regulating both p53 and Mdm2 levels could provide additional protection for the cell against inappropriate p53 signaling.
The oncogenic stress-induced tumor suppressor ARF is a prominent regulator of p53 stabilization through the physical sequestration of Mdm2 34. Upon activation, ARF disrupts the p53-Mdm2 interaction and sequesters Mdm2 in the nucleolus, thereby halting cell proliferation through p53-dependent growth arrest or apopotsis 35, 36. Studies have shown that ARF can also directly inhibit the E3 ubiquitin ligase activity of Mdm2 37, 38. Despite this complexity, ARF is nevertheless a critical stress-induced activator of p53-dependent signaling pathways.
Degradation of Mdm2 is yet another mechanism for stabilizing p53 indirectly. As an E3 ligase, Mdm2 possesses inherent self-ubiquitination activity 39, 40, 41. Self-ubiquitination is inhibited during times of nonstress by the deubiquitase HAUSP, an enzyme that specifically interacts with and deubiquitinates both Mdm2 and p53 in a mutually exclusive manner 42, 43, 44. However, the HAUSP-Mdm2 interaction seems to be dependent on the proteins Daxx and RASSF1A, as cellular stress promotes the dissociation of DAXX and G(1)-S cell cycle progression promotes the dissociation of RASSF1A 45, 46. Dissociation of either of these proteins disrupts the stability of the Mdm2-HAUSP complex and promotes Mdm2 auto-ubiquitination and degradation. There also seems to be a delicate balance between these proteins, as a modest reduction of HAUSP levels prevents p53 deubiquitination but complete ablation of HAUSP causes robust p53 stabilization. This surprising contrast suggests that the level of Mdm2 is critically important in the temporal regulation of p53 function 47, 48.
Apoptosis vs cell growth arrest: how does p53 choose?
p53 imparts its tumor-suppressive and anti-proliferative effects through the induction of key downstream regulatory factors. For example, the cyclin-dependent kinase inhibitor p21 is a highly sensitive p53-response gene and a key mediator of permanent cell growth arrest or cellular senescence 49. However, given the large number of both growth arrest and apoptotic genes that are responsive to p53 induction, one question that arises is how does p53 activate one pathway over another? One assumption is that any cell harboring deleterious genetic abnormalities could be a liability and should activate an apoptotic pathway. Induction of numerous p53-responsive pro-apoptotic genes, such as Bax, PIG3, PUMA, and Pidd, gives a clear indication that p53 prevents a cell from aberrant growth in part by inducing an apoptotic response 50. However, specific lines of evidence suggest that induction of apoptosis in response to DNA damage and cellular stress may not be the only nor most prominent tumor-suppressive quality that p53 exhibits. Studies of mice with knockout of PUMA, a key pro-apoptotic p53-responsive gene, have shown that despite having a deficiency in a p53-dependent apoptotic response in fibroblasts and thymocytes, these mice do not show an increased susceptibility to cancer development 51, 52. If p53-mediated apoptosis is at least partially dependent on the induction of PUMA, the phenotype of these mice would suggest that other tumor-suppressive functions of p53 have an equal importance in preventing tumorigenesis. In addition, a particular point mutation in p53 has been described that seems to separate the two critical functions of p53: apoptosis and growth arrest 53. Mutant knockin mice that possess an arginine to proline mutation at amino-acid position 172 fail to mount a p53-dependent apoptotic response but can still induce cell cycle arrest. If apoptosis was the predominant tumor suppression mechanism of p53, these mice would presumably be predisposed to tumor development. However, Liu et al. 53 showed that mice harboring this mutation failed to develop the early-onset thymic lymphomas that p53-null mice did. These data suggest that in addition to apoptosis, the mechanisms of p53-mediated cell cycle arrest and cellular senescence may be equally important in the prevention of tumor development.
It is clear that p53 activates several downstream targets responsible for inducing cell cycle arrest, including GADD45, Reprimo, p21, and 14-3-3σ 54, 55. p53 has even been recently shown to induce microRNA expression as an additional mechanism for activating cell cycle arrest 56. In addition, p53 has transcription-independent functions in mitochondria that can induce apoptosis 50. Halting cell growth provides a relatively quick and non-committal way for the cell to assess DNA damage that has occurred and prevent the propagation of rogue genetic alterations. The next series of mechanistic steps that are needed for a cell to choose a particular fate, however, remain unclear. On one hand, providing additional time for the cell to repair damaged DNA through cell cycle arrest seems globally beneficial for the organism. On the other hand, if all damage is not repaired sufficiently and the cell is allowed to survive, propagation of these abnormalities could further promote the development of tumorigenesis. Therefore, there seems to be a fine balance between these two fates when cells are exposed to DNA damage and stress, and a third p53-dependent mechanism may be important for limiting the growth of damaged cells: the induction of cellular senescence. p53 is a potent activator of genes involved in cellular senescence, such as p21 and PAI-1, and p53-dependent activation of these genes has been shown to be a physiological, in vivo response to DNA damage events 35, 57, 58, 59, 60. Mutant knockin mice have also confirmed this arm of p53 function. p53R172P mice are incapable of inducing apoptosis but retain the partial ability to induce cell cycle arrest in response to stress 61. Nevertheless, these mice exhibit delayed tumor onset due to the maintenance of chromosome stability and p21-mediated cell cycle arrest 61. Separating the physiological functions of p53 through the use of an in vivo model indicates that a p53-dependent cellular senescent response is an equivalently and valid option for a cell experiencing genotoxic stress. It is possible that the type of p53 response elicited from a DNA damage event is highly dependent on the length and severity of the damage itself 1, 3. Cells experiencing low levels of cellular stress for a short period of time are more likely to sustain damage that is repairable for continued survival. Conversely, cells sustaining severe levels of DNA damage would elicit a p53-dependent apoptotic or cellular senescent response to eradicate the damage or prevent the cell from continuing to grow. In fact, a strong link exists between cellular senescence that is induced from exposure to low levels of oxidative stress and longevity 62, 63. In addition to being a potent tumor-suppressive mechanism, p53-dependent cellular senescence has a significant role in organismal aging. These mechanisms provide an interesting link between the prevention of cancer development and cellular aging. Interesting questions still remain as to how p53 regulates cellular fate in particular tissue and cell types upon exposure to cellular stress, as well as how the microenvironment conditions influence the type of p53 response that occurs. In vivo p53 modeling will surely offer further insight into these types of questions.
Antirepression
The regulation of p53 function and transcriptional activity requires multiple layers of signaling and post-translational control. As mentioned above, p53 levels are regulated at least in part by the negative regulator Mdm2. This E3 ubiquitin ligase directly ubiquitinates p53 and provides a signal for degradation by the 26S proteasome. Temporal control of Mdm2, as mentioned, is critically important for the proper function of p53. Another protein structurally related to Mdm2, MdmX, has had a perplexing history since its discovery as a novel p53-binding protein in 1996, but more recent evidence suggests that it may be as important as Mdm2 for governing p53 function 64, 65. Despite containing a RING domain and being a bonafide E3 ubiquitin ligase based on sequence homology, MdmX does not have intrinsic ubiquitin ligase activity towards p53. Rather, it has been shown that MdmX can directly inhibit p53-mediated transcriptional activation 66. MdmX and Mdm2 are also recruited to the promoters of p53-responsive genes and form a complex with p53 23, 67, 68. This recruitment directly inhibits p53-mediated transcription of several target genes 23. Interestingly, the importance of MdmX for regulating p53 function was further strengthened from several in vivo mouse studies 69, 70, 71. In one elegant set of experiments, double mutant knockout mice were created that possessed either p53/Mdm2−/− or p53/MdmX−/− and were then reintroduced with a temperature-sensitive p53 mutant 64. This approach allowed the assessment of p53 stabilization and activity in the absence of Mdm2 or MdmX. Interestingly, loss of Mdm2 promoted the expression of p53-induced apoptotic genes, while loss of MdmX promoted the expression of p53-induced cell cycle arrest genes. These data suggest that Mdm2 and MdmX both play important, albeit distinct, regulatory roles in the transactivation activity of p53.
Together, these findings suggest a more refined and detailed model for p53 activation (Figure 1). A classical model would dictate that three fundamental events need to occur for sufficient p53 activation: protein stabilization, sequence-specific DNA binding, and transcription of target genes 12. However, in consideration of data suggesting that p53 is inherently active, a robust p53 response in vivo may require the removal of factors that are repressing the protein. Mdm2 and MdmX knockout mice are both embryonic lethal and can be rescued by crossing with p53 null mice 72, 73, 74, 75. The p53 cross-rescue indicates that the early developmental defects are largely p53 dependent and the presence of either Mdm2 or MdmX represses this phenotype. Furthermore, disruption of the p53-Mdm2 or p53-MdmX interaction is required for the activation of some p53-responsive genes 23. Since Mdm2 and MdmX form a protein complex with p53 on the promoters of target genes, removal of this repression may be required in addition to protein stabilization and other post-translational modifications (Figure 1). A knock-in mouse model harboring a mutation in p53 (p53QS), which disrupts the transactivation function of the protein and renders it almost completely incapable of activating p53-responsive genes, is embryonic lethal but does not cause p53 to lose DNA-binding ability, despite a loss of Mdm2 and MdmX interactions 76. The data from the p53QS study would suggest that Mdm2 and MdmX-mediated repression is required for full inhibition of p53 function in vivo, since Mdm2 and MdmX null mice are also embryonic lethal. Recent findings have also suggested that p53 has limited binding to target promoter regions of normal cells that are not undergoing a stress response 77. However, p53 in stressed cells as well as immortal cell lines seems to be constitutively bound to the promoters of target genes, suggesting that there may be differences in p53 function between primary cells and cells that are mounting a stress response. Nevertheless, it seems that additional steps for the relief of repression are still required even in primary cells that do not have promoter-bound p53 77. Release of repression may be a required step in the series of reactions that take place in a full p53 response to stress. In addition, placing an additional step of antirepression in the classical model of p53 activation may help bridge the findings from in vitro and in vivo experiments.
Therapeutics
The sheer number of studies to date on p53 showing its exquisite biochemical properties, combined with the daunting list of upstream regulators of the protein, would together indicate the magnitude of its importance in regulating cellular homeostasis. Indeed, the occurrence of p53 mutations in upward of 50% of all human tumors is quite humbling 78, 79, 80, 81. For tumors that do not possess mutations in the p53 gene, approximately 25% retain functional, wild-type p53 and possess other p53 pathway abnormalities such as Mdm2 overexpression 82. For these tumors, it may be interesting to explore chemotherapeutic approaches that target p53. Reactivation of p53 in several mouse tumor models has been shown to be a potent inhibitor of tumor growth 83, 84, 85. The development of specific activators of p53 would therefore be an interesting approach for stabilizing and activating the protein in these tumors. The small molecule, called reactivation of p53 and induction of tumor cell apoptosis (RITA), selectively binds p53 and strongly induces apoptosis in several tumor cell lines by blocking the p53-Mdm2 interaction 86. Reactivation of p53 could also occur by directly inhibiting critical negative regulators of the pathway. Nutlin 3A, a small-molecule antagonist that binds to the p53-binding pocket on Mdm2, potently blocks the p53-Mdm2 interaction and activates downstream p53-mediated transcriptional events 87. HIL98 also directly inhibits the ubiquitin E3 ligase activity of Mdm2 and activates p53 88. More recently, a potent and orally available small-molecule antagonist of Mdm2, MI-219, was described showing 10 000-fold higher selectivity for Mdm2 than MdmX 89. This inhibitor activated p53 and led to potent tumor inhibition in xenograft models with a favorable pharmacokinetic profile. The selective targeting and inhibition of Mdm2 as an approach for p53 activation is bolstered by genetic evidence showing that an Mdm2 RING-inactivating mutation (C462A) can also activate p53 in vivo 40. Optimized compounds of these leads are currently in pre-clinical and clinical development. Small-molecule inhibitors of SIRT1, another negative regulator of p53, have shown potent p53 activation effect as well. SIRT1 is a class III histone deacetlyase of the sirtuin family that specifically deacetylates and negatively regulates p53 63. SIRT1 inhibitors such as sirtonol, cambinol, EX-527, and Tenovin have all shown promising p53 activation function 63. In particular, Tenovin elicits potent and specific activation of p53 at single-digit micromolar concentrations 90. Similar to MI-219, Tenovin has a favorable pharmacokinetic profile with no genotoxic effects.
Conclusion
Recent advances in understanding p53 regulation in vivo have added complexity to the model of its transcriptional activation and the series of events that take place for p53 to activate a particular stress-induced response. The studies of mouse knockin and knockout models have suggested that in vivo p53 activation is a multilayered, concerted effort that not only requires post-translational modifications of particular residues, but also needs further adaptations within the p53 pathway for a full transcriptional response. If we consider p53 to be a transcription factor that is in the constitutively active state, given the abundance of data that suggest so, then reversal of its repressed state may be required for in vivo p53 activation. Removal of the inhibitory pressure placed by Mdm2 and MdmX would allow for both protein stabilization and transcriptional activation to ensue. The level at which antirepression occurs may depend on the tissue type and specific microenvironment, but nevertheless it may have an impact on how p53 activates a particular signaling pathway. Antirepression may be a sufficient mechanism for the activation of particular target genes, such as p53-responsive genes that induce cell growth arrest, but additional post-translational mechanisms may be required for the activation of genes involved in apoptosis. The concept of in vivo antirepression may also help explain how p53 is activated in particular tissues and may connect some of the in vitro and in vivo studies presented to date. However, a number of specific mechanistic questions remain unsolved. How do specific post-translational modifications signal particular p53 responses? Do all p53 promoters respond in the same way to p53 signaling or do unique differences exist between subsets of genes? Are the in vivo responses of Mdm2 and MdmX tissue-dependent? How is p53-mediated cellular senescence linked to the aging process? These questions and others will continue to guide us in the way we view p53 regulation.
References
Bensaad K, Vousden KH . p53: new roles in metabolism. Trends Cell Biol 2007; 17:286–291.
Vousden KH, Lane DP . p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8:275–283.
Vousden KH, Prives C . Blinded by the light: the growing complexity of p53. Cell 2009; 137:413–431.
Kruse JP, Gu W . SnapShot: p53 posttranslational modifications. Cell 2008; 133:930–930.e1.
Haupt Y, Maya R, Kazaz A, Oren M . Mdm2 promotes the rapid degradation of p53. Nature 1997; 387:296–299.
Kubbutat MH, Jones SN, Vousden KH . Regulation of p53 stability by Mdm2. Nature 1997; 387:299–303.
Brooks CL, Gu W . p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21:307–315.
Mayo LD, Donner DB . The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci 2002; 27:462–467.
Meek DW . The p53 response to DNA damage. DNA Repair (Amst) 2004; 3:1049–1056.
Teufel DP, Bycroft M, Fersht AR . Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009; 28:2112–2118.
Appella E, Anderson CW . Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem 2001; 268:2764–2772.
Kruse JP, Gu W . Modes of p53 regulation. Cell 2009; 137:609–622.
Shieh SY, Ikeda M, Taya Y, Prives C . DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91:325–334.
Chao C, Hergenhahn M, Kaeser MD, et al. Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J Biol Chem 2003; 278:41028–41033.
Sluss HK, Armata H, Gallant J, Jones SN . Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol Cell Biol 2004; 24:976–984.
Chao C, Herr D, Chun J, Xu Y . Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J 2006; 25:2615–2622.
MacPherson D, Kim J, Kim T, et al. Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. EMBO J 2004; 23:3689–3699.
Wu Z, Earle J, Saito S, et al. Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol 2002; 22:2441–2449.
Ashcroft M, Kubbutat MH, Vousden KH . Regulation of p53 function and stability by phosphorylation. Mol Cell Biol 1999; 19:1751–1758.
Ashcroft M, Taya Y, Vousden KH . Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 2000; 20:3224–3233.
Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ, Herrlich P . DNA damage induced p53 stabilization: no indication for an involvement of p53 phosphorylation. Oncogene 1999; 18:1723–1732.
Wang X, Taplick J, Geva N, Oren M . Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett 2004; 561:195–201.
Tang Y, Zhao W, Chen Y, Zhao Y, Gu W . Acetylation is indispensable for p53 activation. Cell 2008; 133:612–626.
Ito A, Lai CH, Zhao X, et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 2001; 20:1331–1340.
Li M, Luo J, Brooks CL, Gu W . Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 2002; 277:50607–50611.
Feng L, Lin T, Uranishi H, Gu W, Xu Y . Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol 2005; 25:5389–5395.
Krummel KA, Lee CJ, Toledo F, Wahl GM . The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci USA 2005; 102:10188–10193.
Chan WM, Mak MC, Fung TK, et al. Ubiquitination of p53 at multiple sites in the DNA-binding domain. Mol Cancer Res 2006; 4:15–25.
Chen D, Kon N, Li M, et al. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005; 121:1071–1083.
Dornan D, Wertz I, Shimizu H, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004; 429:86–92.
Kruse JP, Gu W . MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J Biol Chem 2009; 284:3250–3263.
Leng RP, Lin Y, Ma W, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003; 112:779–791.
Sheng Y, Laister RC, Lemak A, et al. Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat Struct Mol Biol 2008; 15:1334–1342.
Lowe SW, Sherr CJ . Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev 2003; 13:77–83.
Sherr CJ . Divorcing ARF and p53: an unsettled case. Nat Rev Cancer 2006; 6:663–673.
Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D . Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol 1999; 1:20–26.
Honda R, Yasuda H . Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBOJ 1999; 18:22–27.
Llanos S, Clark PA, Rowe J, Peters G . Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat Cell Biol 2001; 3:445–452.
Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM . Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 2000; 275:8945–8951.
Itahana K, Mao H, Jin A, et al. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 2007; 12:355–366.
Stommel JM, Wahl GM . Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J 2004; 23:1547–1556.
Brooks CL, Li M, Hu M, Shi Y, Gu W . The p53--Mdm2--HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 2007; 26:7262–7266.
Hu M, Gu L, Li M, et al. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol 2006; 4:e27.
Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002; 416:648–653.
Song MS, Song SJ, Kim SY, Oh HJ, Lim DS . The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBOJ 2008; 27:1863–1874.
Tang J, Qu LK, Zhang J, et al. Critical role for Daxx in regulating Mdm2. Nat Cell Biol 2006; 8:855–862.
Cummins JM, Rago C, Kohli M, et al. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 2004; 428:1 p following 486.
Li M, Brooks CL, Kon N, Gu W . A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell 2004; 13:879–886.
Brown JP, Wei W, Sedivy JM . Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997; 277:831–834.
Green DR, Kroemer G . Cytoplasmic functions of the tumour suppressor p53. Nature 2009; 458:1127–1130.
Michalak EM, Villunger A, Adams JM, Strasser A . In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ 2008; 15:1019–1029.
Yu J, Zhang L . No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell 2003; 4:248–249.
Liu G, Parant JM, Lang G, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 2004; 36:63–68.
el-Deiry WS . Regulation of p53 downstream genes. Semin Cancer Biol 1998; 8:345–357.
Yu J, Zhang L . The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun 2005; 331:851–858.
He L, He X, Lowe SW, Hannon GJ . microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer 2007; 7:819–822.
Deng Y, Chan SS, Chang S . Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer 2008; 8:450–458.
Halazonetis TD, Gorgoulis VG, Bartek J . An oncogene-induced DNA damage model for cancer development. Science 2008; 319:1352–1355.
Kortlever RM, Higgins PJ, Bernards R . Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 2006; 8:877–884.
Leal JF, Fominaya J, Cascon A, et al. Cellular senescence bypass screen identifies new putative tumor suppressor genes. Oncogene 2008; 27:1961–1970.
Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G . p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci USA 2006; 103:19842–19847.
Bordone L, Guarente L . Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol 2005; 6:298–305.
Brooks CL, Gu W . How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer 2009; 9:123–128.
Barboza JA, Iwakuma T, Terzian T, El-Naggar AK, Lozano G . Mdm2 and Mdm4 loss regulates distinct p53 activities. Mol Cancer Res 2008; 6:947–954.
Marine JC, Jochemsen AG . Mdmx and Mdm2: brothers in arms? Cell Cycle 2004; 3:900–904.
Marine JC, Jochemsen AG . Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun 2005; 331:750–760.
Minsky N, Oren M . The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol Cell 2004; 16:631–639.
Ohkubo S, Tanaka T, Taya Y, Kitazato K, Prives C . Excess HDM2 impacts cell cycle and apoptosis and has a selective effect on p53-dependent transcription. J Biol Chem 2006; 281:16943–16950.
Iwakuma T, Lozano G . Crippling p53 activities via knock-in mutations in mouse models. Oncogene 2007; 26:2177–2184.
Marine JC, Francoz S, Maetens M, et al. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ 2006; 13:927–934.
Wahl GM . Mouse bites dogma: how mouse models are changing our views of how P53 is regulated in vivo. Cell Death Differ 2006; 13:973–983.
Jones SN, Roe AE, Donehower LA, Bradley A . Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378:206–208.
Migliorini D, Lazzerini Denchi E, Danovi D, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol 2002; 22:5527–5538.
Montes de Oca Luna R, Wagner DS, Lozano G . Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378:203–206.
Parant J, Chavez-Reyes A, Little NA, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 2001; 29:92–95.
Johnson TM, Hammond EM, Giaccia A, Attardi LD . The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet 2005; 37:145–152.
Shaked H, Shiff I, Kott-Gutkowski M, et al. Chromatin immunoprecipitation-on-chip reveals stress-dependent p53 occupancy in primary normal cells but not in established cell lines. Cancer Res 2008; 68:9671–9677.
Caron de Fromentel C, Soussi T . TP53 tumor suppressor gene: a model for investigating human mutagenesis. Genes Chromosomes Cancer 1992; 4:1–15.
Greenblatt MS, Bennett WP, Hollstein M, Harris CC . Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54:4855–4878.
Hainaut P, Hollstein M . p53 and human cancer: the first ten thousand mutations. Adv Cancer Res 2000; 77:81–137.
Hollstein M, Sidransky D, Vogelstein B, Harris CC . p53 mutations in human cancers. Science 1991; 253:49–53.
Toledo F, Wahl GM . Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 2006; 6:909–923.
Martins CP, Brown-Swigart L, Evan GI . Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006; 127:1323–1334.
Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445:661–665.
Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656–660.
Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004; 10:1321–1328.
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844–848.
Yang Y, Ludwig RL, Jensen JP, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005; 7:547–559.
Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA 2008; 105:3933–3938.
Lain S, Hollick JJ, Campbell J, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008; 13:454–463.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brooks, C., Gu, W. New insights into p53 activation. Cell Res 20, 614–621 (2010). https://doi.org/10.1038/cr.2010.53
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2010.53
Keywords
This article is cited by
-
A role for bioinorganic chemistry in the reactivation of mutant p53 in cancer
JBIC Journal of Biological Inorganic Chemistry (2022)
-
Methylglyoxal induces p53 activation and inhibits mTORC1 in human umbilical vein endothelial cells
Scientific Reports (2021)
-
Puma, noxa, p53, and p63 differentially mediate stress pathway induced apoptosis
Cell Death & Disease (2021)
-
c-Myc inactivation of p53 through the pan-cancer lncRNA MILIP drives cancer pathogenesis
Nature Communications (2020)
-
Mutagenic assessment of chemotherapy and Smac mimetic drugs in cells with defective DNA damage response pathways
Scientific Reports (2018)
