
Abstract
Extracellular DNA is secreted from various sources including apoptotic cells, NETotic neutrophils and bacterial biofilms. Extracellular DNA can stimulate innate immune responses to induce type-I IFN production after being endocytosed. This process is central in antiviral responses but it also plays important role in the pathogenesis of a range of autoimmune diseases such as systemic lupus erythematosus. We discuss the recent advances in the understanding of the role of extracellular DNA, released from apoptotic and NETotic cells, in autoimmunity.
Similar content being viewed by others

Introduction
It was long discovered that cytosolic DNA was immune stimulatory and associated with autoimmunity but little was known about the mechanism. Over the last decade, a range of DNA sensors have been identified that recognize DNA, including viral DNA.1 Following sensor engagement, the adaptor protein stimulator of interferon genes (STING) is stimulated and proinflammatory cytokines released.2 This response is a key component of host defence against pathogens. However, when perturbed, this response can contribute to susceptibility to autoimmune diseases. Aberrant DNA sensing is implicated in the pathogenesis of systemic lupus erythematosus (SLE) and Aicardi–Goutieres syndrome (AGS).3 Unlike RNA sensors, which recognize pathogen-specific RNA, the majority of DNA sensors found to date recognize DNA in a sequence-independent manner.1,4 Under homeostatic conditions, DNA engagement is prevented by accessory proteins such as nucleases that cleave DNA and intracellular regulators for DNA receptors.5 Together, these prevent the development of an excessive immune response.6
DNA sensors and inflammation
DNA sensors can be generally divided into two groups: endosomal membrane receptors and intracellular receptors. Toll-like receptor 9 (TLR9) was the first DNA sensor that was identified. It is still the only endosomal expressed pathogen recognition receptor (PRR) for DNA.7,8 It is expressed mainly within plasmacytoid dendritic cells (pDC) and to a lesser extent on monocytes, B cells and dendritic cells. It was elegantly demonstrated that TLR9 is activated by CpG DNA (5′ cytosine-phosphate-guanine 3′), which in turn leads to type-I interferon (IFN) production via TLR9 adaptor protein, myeloid differentiation primary response gene 88 (MyD88), and interferon regulatory factor 7 (IRF7).9,10,11 Although TLR9 is able to recognize CpG DNA that is enriched in the microbial genome, DNA-protein complexes were also shown to induce proinflammatory cytokine production via TLR9.12
In contrast to endosomal PRR, cytosolic DNA sensors are widely expressed in mammalian cells.4 This enables them to recognize the invading pathogens in the affected cells and mount a prompt immune response. Cytosolic DNA sensors show great redundancy: 13 have been discovered so far. DNA-dependent activator of IFN-regulatory factors (DAI) was the first cytosolic DNA sensor identified. It is essential for viral clearance during cytomegalovirus infection in human fibroblasts. DAI engagement activates TANK-binding kinase 1 (TBK1) and the IRF3 complex to provoke type-I IFN production. The function of DAI in vivo is still under investigation.13 Pyrin and HIN domain-containing (PYHIN) family proteins, such as ‘absent in melanoma’ (AIM2) and interferon-inducible gene 16 (IFI16), function as DNA sensors. They have a DNA binding domain termed the carboxy-terminal HIN domain.14 AIM2 binds to cytosolic DNA and enables the assembly of the inflammasome with ASC and caspase 1 to induce interleukin-1 beta (IFN-β) production, which is essential for protection against pathogens such as vaccinia virus.15 The most important DNA sensor that has been found so far is the cyclic-di-GMP-AMP (cGAMP) synthetase (cGAS). The cGAS binds directly to dsDNA and the adaptor protein STING to activate TBK1 and IRF3–7. This in turn initiates type-I IFN production. cGAS was shown to be central for IFN-β production during DNA viral infection in mouse and human cells.16,17 Many other cytosolic DNA receptors including double-strand break repair protein MRE11 (encoded by MRE11A), DNA-dependent protein kinase (DNA-PK) and Leucine-rich repeat flightless interacting protein 2 (LRRFIP2) also recognize dsDNA and converge on STING to upregulate type-I IFN expression.18,19 Another interesting DNA sensor is DNA polymerase III. This converts AT-rich DNA to RNA which then activates RIG-I followed by type-I IFN production.17 However, the role of RNA polymerase III in DNA sensing is still unclear. The DExD/H-box helicases (DDX) are an emerging group of DNA sensors, which include DDX41, DDX9 and DHX36. DExD/H-box helicase 36 and 9 (DHX36 and DHX9) bind CpG DNA in pDC. This is followed by binding to MyD88 which triggers type-I IFN production.20 It was also reported that DDX41is an important signalling molecule for dsDNA-dependent responses in addition to its role as an innate DNA sensor.21 However, it is currently unclear how the DDX family proteins interact with nucleic acids and transduce signals downstream.
In addition to protection against pathogens, these DNA sensors also recognize self DNA. Recognition of self-DNA can initiate an inflammatory response (summarized in Figure 1). TLR9 and type-I IFN have been closely associated in SLE from studies in humans and murine models of SLE.22 Recently, cGAS and STING were identified as the key mediators in Aicardi–Goutieres syndrome.23 Genetic ablation of cGAS rescued the phenotype in a murine model of Aicardi–Goutieres syndrome.24 Expression of DAI and AIM2 have also been found to upregulated in patients with autoimmune diseases.25,26 In summary, cytosolic DNA is sensed by DNA sensors and subsequently activates adaptor proteins, such as STING and MyD88, and the inflammasome, resulting in proinflammatory cytokine production.

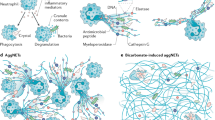
Model of extracellular DNA induced proinflammatory response. DNaseI digests extracellular DNA that is released from either neutrophils undergoing NETosis or apoptotic/necrotic cells. The undigested DNA may form immune complexes with anti-DNA antibodies, which are then phagocytosed following Fc receptor engagement. DNA can also be phagocytosed in the form of microparticles from apoptotic/necrotic cells. The DNA in the lysosome is degraded by DNase II. Undigested DNA in the lysosome may activate TLR9 to stimulate MyD88. This in turn assembles the Myddosome complex which is composed of MyD88, IRAK1, IRAK2 and IRAK4. TRAF6 is then recruited to the complex, leading to the activation of NF-kb and AP-1. RNA helicase DHX36 and DHX9 were found to identify CpG DNA in certain cell types and trigger MyD88 signalling. TREX1 is localized on the endoplasmic reticulum (ER), digests cytosolic DNA to prevent innate immune response. TREX1 mutations leads to DNA accumulation. This triggers the production of guanosine monophosphate- adenosine monophosphate (cGAMP) by cGAS upon intracellular DNA engagement. cGAMP activates the ER-resident STING, which then is shuttled to ER-Golgi intermediate compartment and the Golgi apparatus. STING initiates TBK1 activation, resulting in the expression of type-I IFNs. Other DNA sensors including double-strand break repair protein MRE11, DNA-dependent protein kinase (DNA-PK), IFNγ-inducible protein 16 (IFI16), protein kinase RNA-activated (PKR), the probable ATP-dependent RNA helicases DDX41 and DDX60, recognize DNA and signal through STING to initiate type-I IFN expression. DNA or RNA is sensed by Leucine-rich repeat flightless interacting protein 2 (LRRFIP2) to activate the β-catenin, resulting in the production of IFN- β. DAI directly recruits TBK1 which leads to type-I IFN production. AIM2 and NLRP3 trigger ASC caspase-1 inflammasome assembly upon dsDNA binding that promote the production of IL-1b and IL-18.
Role of type-I IFN
Type-I IFNs are critical anti-viral cytokines that are triggered by DNA sensors.27 The secreted type-I IFNs bind to IFN receptors to upregulate interferon-stimulated genes (ISGs) via Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway.28 This changes the cell-intrinsic status and production of interferon stimulated genes (ISGs) to fight against the microbial insult and limit the spread of the pathogens. It also facilitates antigen presentation and natural killer cell cytotoxicity. Furthermore, the adaptive immune system is activated resulting in the generation of activated T cells and high-affinity antibodies.29,30,31 The type-I IFN production is tightly regulated by multiple layers of mediators such as some of the ISGs to ensure appropriate immune response.5,32 In autoimmunity, inappropriate chronic type-I IFN production leads to autoreactive T cells and autoantibody production in some autoimmune animal models.33 However, it should be noted that type-I IFN also has a protective effect in some autoimmune diseases including inflammatory bowel disease and multiple sclerosis. This might be due to suppression of the production of other proinflammatory cytokine.34 The effect of type-I IFN in autoimmune disease is probably dependent on the stage of the disease; early type-I IFN production may initiate systemic autoimmunity, whilst later production might limit autoreactive T-cell function.
DNA sensing and autoimmunity
In mammalian cells, DNA is normally confined to the nucleus. Its presence in the cytosol can induce immune responses.35,36 DNA is predominantly released into the extracellular space by two mechanisms: cell death and bacterial biofilm. We discuss the role of DNA released from dead cells.
Abnormal response to DNA released from apoptotic cells
Cell death pathways
Death is inevitable but living is not. Multiple types of cell death exist but these can be broadly grouped into programed cell death (PCD) and passive cell demise. PCD plays a key role in development and homeostasis. It is also important in the pathogenesis of autoimmunity. Deficiency in the clearance of DNA/nucleosome derived from either PCD or passive cell death is associated with autoimmunity.
Apoptosis is the most familiar and well-studied PCD pathway. It can be stimulated by both intrinsic and extrinsic signals that activate the caspase protein family.37 Activated caspases cleave (1) nuclear lamins, resulting in nuclear shrinking and budding; (2) cytoskeleton proteins, resulting in loss of cell shape; and (3) PAK2, which mediates apoptotic body blebbing.38,39
During apoptosis, nucleases cleave condensed chromatin into oligonucleosomes.40 With an increase in the nuclear permeability, oligonucleosomes can migrate into the cytoplasm and fuse with the plasma membrane to form blebs. Apoptotic blebs are about one micron in size and contain DNA fragments (of 500–1000 base pairs), histones and ribonuclear proteins. These apoptotic blebs are then released from the apoptotic cells. Under normal conditions, the blebs are phagocytosed rapidly and under conditions that are anti-inflammatory in nature.41
Necrosis, unlike programmed cell death, is unplanned and typically associated with inflammation. Necrotic cells result from stress, heat shock or any stimulus that overwhelms the structural integrity of the cell. The process is morphologically different from apoptosis. The cell expands with consequent plasma membrane rupture. This is independent of cell fragmentation or DNA cleavage.42 Whereas apoptotic cell contents are packed in apoptotic bodies, necrotic cells release DAMPs, which promote inflammation. Necrotic cells are cleared by a process called macropinocytosis, which is slow and incomplete.43
Secondary necrosis occurs if the elimination of the apoptotic remnant is not cleared sufficiently. This phenomenon is easily induced in vitro but only observed in vivo under pathological conditions.44 Similar to primary necrosis, secondary necrosis is characterized by cytoplasmic swelling, plasma membrane rupture, mitochondria hyperpolarization, oxidative burst and the release of DAMPs as well as oxidized products.45 Unlike necroptosis, secondary necrosis does not rely on receptor-interacting serine/threonine-protein kinase 1 (RIP1) or mitochondrial dependent reactive oxidative species (ROS) production because neither necrostatin-1 (Nec-1), a RIP1 inhibitor, or knockdown of mitochondrial complex I chaperone affects secondary necrosis.46 DNA released from necrosis and secondary necrosis is not specifically cleaved in a manner seen during apoptosis. DNA appears as a smear on gel electrophoresis instead of the ladder-like pattern characteristic of apoptosis. Phagocytosis of secondary necrotic cells by phagocytes is enhanced by proteins such as C1q, mannose-binding lectin (MBL) and polyreactive IgM.47,48
DNA accumulation and autoimmunity: lessons from mouse models
When the quantity of apoptotic cells overwhelms the regulatory system, autoimmunity can develop. Considerable data support the idea that defective clearance of apoptotic cells leads to autoimmunity.49,50 For example, a subset of SLE patients show defective apoptotic cell clearance ability with morphologically different macrophages that have impaired phagocyte function.51,52 In addition, accumulated apoptotic cells were observed in the bone marrow of SLE patients and skin of patients with cutaneous lupus.53 These findings are further confirmed in mice models. Mice lacking milk fat globule-EGF factor 8 protein (Mfge8), proto-oncogene tyrosine-protein kinase (Mer), or C1q accumulated apoptotic cells and developed an SLE-like disease with anti-nuclear antibodies and glomerulonephritis.54,55 However, mice lacking CD14 have accumulated apoptotic cells without autoimmunity. And mice that have received apoptotic cell by injection display only transient autoantibody production.56,57
DNaseII
DNA released from dead cells is processed by multiple regulatory mechanisms to prevent inflammation. DNA from the engulfed apoptotic cells is normally transported to the lysosome of phagocytes for degradation. DeoxyribonucleaseII (DNaseII) is the nuclease in the lysosome that degrades DNA. Because huge numbers of cells undergo apoptosis, almost one gram of DNA needs to be cleaved by DNaseII per day. Not surprisingly, DNaseII knockout mice die before or shortly after birth with numerous undigested DNA in the macrophages.58 DNaseII and IFN-I receptor (IFN-IR) double knockout mice are born normally although they still have lots of undigested DNA.59 These double-knockout mice, and mice with induced DNaseII knockout after birth, develop an age-dependent rheumatoid arthritis-like disease.60 Similar to the human rheumatoid arthritis, proinflammatory cytokines including TNF-a, IL-6 and IL-1 are upregulated in the inflamed joints. They stimulate macrophage and fibroblast in the affected joints, followed by pannus formation and tissue destruction.61
TREX
Three-prime repair exonuclease 1 (Trex1) is a mammalian 3′-DNA exonuclease. It cleaves cytosolic DNA which might have leaked from the lysosome.62 Unlike DNase II-deficient mice, TREX-deficient mice develop inflammatory myocarditis, while a TREX mutation in human leads to Aicardi–Goutieres syndrome.63 A strong proinflammatory cytokine profile is found in the heart, joints and cerebrospinal fluid in Aicardi–Goutieres syndrome possibly due to the cytosolic DNA accumulation.64 Because both DNase II and TREX-deficient mice display high IFN-α expression, it is plausible that undigested DNA initiates the inflammatory process. Why are different tissues affected in the two deficiencies? This is most likely because DNase II only works in acidic conditions and is expressed in specific organs including bone marrow and spleen. Conversely, TREX1 works in the cytosol and is expressed ubiquitously.65
When regulatory mechanisms are impaired, undigested DNA released from dead cells can stimulate proinflammatory cytokine production via an array of mechanisms. Macrophages lacking Dnase II or TREX1 can engulf apoptotic cells. But DNA accumulates within the macrophages. It was observed that TLR9 deficiency did not rescue the DnaseII deficient mouse phenotype, indicating the DNA-stimulated immune response in DnaseII deficiency is TLR9 independent66 It is likely that undigested DNA leaks into the cytosol and engages cytosolic DNA sensors. For example, Eyes absent 4 (EYA4) stimulates IFN-β and CXCL10 expression in response to undigested DNA associated with apoptotic cells.67 And STING is critical for the cytosolic DNA-induced cytokine production. STING deficiency rescues DnaseII-dependent embryonic lethality.68
DNaseI
DNA may also be released into the extracellular space during cell death in the form of apoptotic bodies, microparticles or naked nucleosomes.69,70 DNaseI is the predominant nuclease in serum. It was reported that DNaseI deficient mice develop an SLE-like disease with anti-DNA antibodies and glomerulonephritis.71 It should be noted that only 38% of the DNaseI deficient mice develop full-blown glomerulonephritis, indicating the presence of compensatory mechanisms such as C1q-mediated apoptotic cell clearance.71 These observations are consistent with human data. Heterozygous DNase I mutations are associated with an SLE-like illness but the DNase I level is not correlated with disease activity.72
C1q-deficient mice also develop autoimmunity but this is influenced by genetic background.55 Deoxyribonuclease I-like 3 (DNaseIL3) is homologous to DNaseI. It digests extracellular DNA. A frameshift mutation of DNaseIL3 was found to be associated with autosomal recessive SLE.73 Dnase1l3-deficient mice display similar phenotype to Dnase1-deficient mice: high anti-DNA antibody titres, splenomegaly and strain-dependent glomerulonephritis. Unlike DNaseI, DNaseIL3 can digest membrane-coated DNA such as microparticles that are released from secondary necrotic cells. Taken together, DNaseI and DNaseIL3 act to digest extracellular DNA released from apoptotic cells.74
DNA modification
The quantity and nature of DNA can trigger autoimmunity. Extracellular DNA is poorly immunogenic but certain modifications, such as oxidation and unmethylated CpG, make it a more potent activator.75,76 Oxidized DNA is commonly found in the ultraviolet (UV)-induced apoptotic cells and is resistant to TREX1 digestion.77,78 Hence, oxidized DNA could accumulate in the cytosol and engages cGAS to stimulate STING and initiate type-I IFN production. This phenomenon is relevant to SLE and rheumatoid arthritis (RA) patients as high levels of oxidized DNA have been observed in lupus-specific skin lesions after UV exposure and synovial fluid (SF).79,80 Notably, oxidative DNA damage was abundant in UV-exposed cutaneous lesions in SLE patients and injection of oxidized DNA into the skin of a lupus mouse model (MRL/lpr strain) induced skin lesions.81
The accumulated DNA may also form complexes with nuclear proteins to mediate immune responses. The best studied DAMP that is complexed with DNA is high mobility group box 1 (HMGB1), a non-histone nuclear protein that is composed of two DNA binding domains called the A box and B box as well as C terminal tail.45 HMGB1 is not normally tightly bound to DNA and has considerable mobility inside the cell, translocating from nucleus to cytoplasm depending on the state of the cell. HMGB1 is released from primary necrotic and secondary necrotic cells, either by itself or bound to chromatin.82 HMGB1 complexed to nucleosome is able to induce IL-β, IL-6 and TNF-α secretion from DC and macrophages. Neither HMGB1 nor nucleosome alone mediates these effects.83 These processes may be relevant to tissue injury in scleroderma. In this condition, excessive damage of the fibroblast is thought to initiate the fibrosis, the pathological hallmark of the condition.84 DAMPs released from apoptotic fibroblasts can trigger inflammation.85 Nucleic acids alone or in complex with autoantibodies against topoisomerase I can stimulate TLRs to produce type-I IFN.86,87 However, the role of type-I IFN in scleroderma remains unclear.
Anti-DNA antibody
Excessive extracellular nucleosome production can promote the production of anti-DNA antibodies that might further amplify the inflammatory responses. The level of anti-dsDNA antibodies broadly correlates with disease activity in SLE and they can bind to apoptotic cells.88 Some of these anti-DNA antibodies were demonstrated to inhibit apoptotic cell uptake via Fc receptors.89 However, polyclonal anti-dsDNA antibodies from SLE patients may form complexes with secondary necrotic cells and promote phagocytosis and IL-8 and IL-1 secretion from granulocytes and monocytes, respectively.90 The isotype of the anti-dsDNA antibody is important. Anti-DNA IgM antibodies (T15-Nab) form complexes with apoptotic cells and facilitate uptake. This process suppresses the inflammatory cytokine production by macrophages.91 The seemingly contradictory results are probably caused by two main factors. First, anti-DNA antibodies are highly heterogeneous. They have distinct antigen reactivity as well as antibody isotype. Second, results from studies are likely influenced by the different experimental conditions and their effects on phagocytosis. An improvement would be to focus on the study of individual antibodies derived from SLE patients.
Neutrophil extracellular trap and autoimmunity
NET kills pathogens
Neutrophil extracellular trap (NET) secretion is a key mechanism through which neutrophils neutralize pathogens. The pathway is determined by the size of the pathogen. If the pathogen is small enough to be phagocytosed, neutrophil elastase (NE) will be trafficked away from the nucleus, preventing chromatin decondensation, and therefore inhibiting NETosis.92
If the pathogen is too big to be phagocytosed, NETosis is initiated. The NET is composed of decondensed neutrophil DNA. DNA in the NET mediates intracellular signal transduction and immune responses that not only contribute to pathogen destruction, but under certain conditions can contribute to inflammation in the setting of autoimmunity.93 It is still unclear which receptor transduces the signal but reactive oxygen species are critical for NETosis. It drives the protease to migrate from primary granules to the nucleus to degrade histones.94,95 The chromatin is then further decondensed by NE and myeloperoxidase and released into the cytoplasm.96,97 Finally, NET are released after plasma membrane rupture. The NET release via cell death is slow while NET release from viable neutrophils is about two to three fold quicker. The decondensed DNA and antimicrobial proteins are wrapped in vesicles that are secreted from viable neutrophils in response to, for example, Staphylococcus aureus.98
Immunological effects of NET clearance
Like DNA released from apoptotic cells, DNase digests DNA in the NET. Nevertheless, DNase alone may not be sufficient as they cannot degrade the insoluble aggregates formed by DNA complexed with LL37/human neutrophil peptide (HNP). This insoluble aggregate induces type-I IFN production in a TLR9-dependent manner.99 NETs can be engulfed by professional scavenger cells like monocytes/macrophages, a process facilitated by DNase and complement. Engulfed NETs can be digested in the lysosome without proinflammatory cytokine production.100 This finding, however, is challenged by other studies that demonstrate that NETs initiate proinflammatory signalling pathways in PBMC, monoctyes and DC.77 These studies suggest that the DNA content of the NET activates cGAS and TLR9 in PBMC and pDC, respectively, which subsequently initiates type-I IFN production.101 This makes the clearance of NETs remarkably different from apoptotic cell clearance, which upregulate TGF-β and IL-10 production. One possible explanation for this disparity is that NETs are predominantly produced to combat infection. Hence, in this setting it might be favorable to promote type-I IFN secretion. Many aspects of NET processing/degradation mechanisms are still elusive and remain an active field of research (Table 1).
NET and autoimmune diseases
Serum from SLE patients protected NETs from DNase degradation: undegraded NETs were observed in kidney tissue and correlated with active lupus nephritis.102 Glomerular NETs were also observed in patients with ANCA vasculitis.103 Undigested NETs in SLE appear to promote inflammatory responses through TLR9 dependent and independent pathways. TLR9 recognizes both CpG DNA and NET-associated DNA.104 The DNA complex in NETs was shown to induce type-I IFN production from pDC in a TLR9-dependent manner, driven by the TLR adaptor MydD88 and IRF7. Only the NET DNA–protein complex but not naked DNA is able to activate TLR9 in pDC due to the stability of the complex.99 NETs are mainly composed of dsDNA therefore it is not surprising that NET stimulates intracellular DNA sensors. In SLE patients, a group of granulocyte population called low-density granulocytes (LDGs) were found to simulate the production of type-I IFNs.105,106 LDGs secrete NETs containing LL-37 (a cathelicidin-related antimicrobial peptides), matrix metallopeptidase 9 (MMP9), dsDNA and histones. The dsDNA released from LDGs was noted to be oxidized because of its mitochondrial origin. Oxidized DNA is more resistant to cytosolic DNase and augments type-I IFN production from PBMC.107,108 DNA in endocytosed NETs is sensed by cGAS, which activates the adaptor protein STING to simulate type-I IFNs. IFN-α secreted in response to NETs enhanced the autoreactivity of the autoreactive B cells.104,109 In addition to DNA, cathelicidin and other NET-associated proteins engage P2X7 receptor on LPS-primed macrophages. This activates the NLRP3 inflammasome, leading to IL-1 and IL-18 release.15,110,111 This proinflammatory response appears to be augmented by the lack of NET clearance in SLE patients. A proportion of SLE patients have defective DNase and/or DNase inhibitors that impair NET degradation. The presence of anti-NET antibodies in some patients might also block the access of DNase to the NET-associated DNA.102
NETs are also detected in synovial fluid, skin and rheumatoid nodules of RA patients.80,112 NETs have direct effects on fibroblast-like synoviocytes promoting IL-6 and IL-8 production. This process is diminished with DNase I treatment. These data suggest a role for extracellular DNA in the inflammatory response in RA.
NET and anti-NET antibody
NETs were also found to form immune complexes with anti-NET antibodies. Autoantibodies against DNA are specific to SLE and anti-dsDNA antibodies may form immune complexes with NETs. These immune complexes are able to activate pDC to secrete IFN-α and TNF-α in a TLR9-dependent manner.113 The engagement of Fcgamma receptors by the immune complexes recruits cytosolic endoplasmic reticulum to the phagosome thereby triggering the downstream interferon production.113 In addition to pDC, other TLR9- and CD32-positive cells including monocytes, B cells and GM-CSF-treated PMNs also respond to DNA–antibody immune complexes.114 pDC also produce increased amounts of IFN-α in patients with active but not inactive small vessel vasculitis,103 a phenomenon that correlates with circulating chromatin levels.103 Hence, it is possible that, in small vessel vasculitis, NETs contribute to IFN-α production by pDC.
NET in animal models
The pathogenic role of NET in vivo has been studied in various animal models. Nakazawa and colleagues demonstrated that NETs induced by propylthiouracil and phorbol myristate acetate were relatively resistant to DNase I degradation and had a more compact conformation. When rats were immunised with these abnormal NETs they developed anti-myeloperoxidase antibodies and pulmonary capillaritis.115 In addition to the direct tissue damage, intact NETs can also act as a source of autoantigens. Myleoid dendritic cell (mDC) can interact and take up NETs. Injection of mDC loaded with NETs to wild-type mice promoted the production of anti-MPO and anti-dsDNA autoantibodies. However, the pathological consequence of the NET uptake remains incompletely understood.116,117
Evidence from lupus-prone mice strains remains incomplete. New Zealand mixed 2328 (NZM) mice have enhanced NEtosis which might lead to the NET deposition in the kidney. The inhibition of NET formation by peptidylarginine deiminase (PAD) inhibitor abolished the NET production and reduced anti-dsDNA antibody level in kidney 118 However, it should be noted that the PAD inhibitor did not ameliorate the kidney inflammation. By contrast, disease activity of MRL/lpr lupus-prone mouse disease is exacerbated by the Nox2 deficiency, which inhibits NET formation.119 The Nox2-deficient mice had defective NET formation but higher titres of anti-nuclear antibodies, proteinuria and more severe glomerulonephritis. More studies using inhibitors within the different NET forming pathways are needed to elucidate the role of NETs in SLE.
Concluding and perspectives
The fundamental function of DNA is to store information in a heritable manner. Foreign DNA is recognized by DNA sensors which can elicit an immune response as part of host defence against pathogens. But inappropriate response to self DNA is associated with autoimmune diseases, such as SLE and AGS. The discovery of DNA-sensing pathways and their contribution to autoimmunity has revealed potential new therapeutic targets. Anti-INF-α and anti-IFNR treatments are under clinical trials. For example, sifalimumab showed promising results in SLE.120,121 NET inhibition and manipulation of apoptotic cell removal remains in the pre-clinical study area: inhibitors of NADPH oxidase; mitochondrial ROS production; actin cytoskeleton; and PAD enzymes have been studied in lupus models. For example, PAD deficiency prevents NET formation and ameliorate arthritis in an induced arthritis model.118,122,123. And, since PPARβ and LXR coordinate apoptotic cell engulfment and induce tolerance in APCs, agonists against PPARβ or LXR, at least in theory, might be beneficial in autoimmunity.124,125,126 Clearly, there is a complex regulatory network to prevent an aberrant immune response to host DNA and all the pathways described in our review are important in homeostatic processes and/or response to pathogens and tissue injury. Manipulating these in vivo is a major challenge both in terms of expected efficacy (for example, disease-modifying outcomes in systemic autoimmunity) and in terms of safety (for example, infection risk, impaired homeostatic tissue turnover).
References
Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 2016; 14: 360–373.
Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 2016; 17: 1142–1149.
Nagata S. Apoptosis and autoimmune diseases. Ann N Y Acad Sci 2010; 1209: 10–16.
Paludan SR, Bowie AG. Immune sensing of DNA. Immunity 2013; 38: 870–880.
Liu J, Cao X. Cellular and molecular regulation of innate inflammatory responses. Cell Mol Immunol 2016; 13: 711–721.
Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol 2016; 16: 35–50.
Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol 2006; 7: 49–56.
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000; 408: 740–745.
Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005; 434: 772–777.
Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 2005; 434: 1035–1040.
Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 2004; 5: 1061–1068.
Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 2005; 115: 407–417.
Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007; 448: 501–505.
Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 2010; 11: 997–1004.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009; 458: 514–518.
Wu J, Sun L, Chen X, Du F, Shi H, Chen C et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013; 339: 826–830.
Cao X. New DNA-sensing pathway feeds RIG-I with RNA. Nat Immunol 2009; 10: 1049–1051.
Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci USA 2013; 110: 2969–2974.
Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y et al. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat Immunol 2010; 11: 487–494.
Kim T, Pazhoor S, Bao M, Zhang Z, Hanabuchi S, Facchinetti V et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc Natl Acad Sci USA 2010; 107: 15181–15186.
Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 2011; 12: 959–965.
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 2002; 416: 603–607.
Gray EE, Treuting PM, Woodward JJ, Stetson DB. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres syndrome. J Immunol 2015; 195: 1939–1943.
Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci USA 2015; 112: E5699–E5705.
Zhang W, Zhou Q, Xu W, Cai Y, Yin Z, Gao X et al. DNA-dependent activator of interferon-regulatory factors (DAI) promotes lupus nephritis by activating the calcium pathway. J Biol Chem 2013; 288: 13534–13550.
Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Goss C, Anz D et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med 2011; 3: 82ra38.
Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 1957; 147: 258–267.
Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014; 14: 36–49.
Kalliolias GD, Ivashkiv LB. Overview of the biology of type I interferons. Arthritis Res Ther 2010; 12 (Suppl 1): S1.
Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 1993; 75: 83–97.
Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001; 14: 461–470.
Jiang LQ, Xia T, Hu YH, Sun MS, Yan S, Lei CQ et al. IFITM3 inhibits virus-triggered induction of type I interferon by mediating autophagosome-dependent degradation of IRF3. Cell Mol Immunol 2017. https://doi.org/10.1038/cmi.2017.15.
Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 2006; 25: 383–392.
Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 2008; 28: 675–686.
Isaacs A, Cox RA, Rotem Z. Foreign nucleic acids as the stimulus to make interferon. Lancet 1963; 2: 113–116.
Rotem Z, Cox RA, Isaacs A. Inhibition of virus multiplication by foreign nucleic acid. Nature 1963; 197: 564–566.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell 2011; 147: 742–758.
Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 2015; 16: 329–344.
Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407: 770–776.
Iglesias-Guimarais V, Gil-Guinon E, Sanchez-Osuna M, Casanelles E, Garcia-Belinchon M, Comella JX et al. Chromatin collapse during caspase-dependent apoptotic cell death requires DNA fragmentation factor, 40-kDa subunit-/caspase-activated deoxyribonuclease-mediated 3'-OH single-strand DNA breaks. J Biol Chem 2013; 288: 9200–9215.
Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz HM. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ 2008; 15: 183–191.
Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature 2000; 407: 784–788.
Krysko DV, Denecker G, Festjens N, Gabriels S, Parthoens E, D'Herde K et al. Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ 2006; 13: 2011–2022.
Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 2007; 32: 37–43.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008; 29: 21–32.
Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 2010; 17: 922–930.
Gullstrand B, Martensson U, Sturfelt G, Bengtsson AA, Truedsson L. Complement classical pathway components are all important in clearance of apoptotic and secondary necrotic cells. Clin Exp Immunol 2009; 156: 303–311.
Poon IK, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ 2010; 17: 381–397.
Vaux DL, Flavell RA. Apoptosis genes and autoimmunity. Curr Opin Immunol 2000; 12: 719–724.
Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol 2006; 27: 244–250.
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum 1998; 41: 1241–1250.
Licht R, Dieker JW, Jacobs CW, Tax WJ, Berden JH. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J Autoimmun 2004; 22: 139–145.
Casciola-Rosen L, Rosen A, Petri M, Schlissel M. Surface blebs on apoptotic cells are sites of enhanced procoagulant activity: implications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc Natl Acad Sci USA 1996; 93: 1624–1629.
Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 2004; 304: 1147–1150.
Botto M, Dell'Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet 1998; 19: 56–59.
Devitt A, Parker KG, Ogden CA, Oldreive C, Clay MF, Melville LA et al. Persistence of apoptotic cells without autoimmune disease or inflammation in CD14(−/−) mice. J Cell Biol 2004; 167: 1161–1170.
Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med 1998; 188: 387–392.
Kawane K, Fukuyama H, Kondoh G, Takeda J, Ohsawa Y, Uchiyama Y et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 2001; 292: 1546–1549.
Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol 2005; 6: 49–56.
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 2007; 446: 102–102.
Zwerina J, Redlich K, Polzer K, Joosten L, Kronke G, Distler J et al. TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA 2007; 104: 11742–11747.
Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 2007; 131: 873–886.
Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet 2007; 80: 811–815.
Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M et al. Mutations in the gene encoding the 3 '-5 ' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 2006; 38: 917–920.
Evans CJ, Aguilera RJ. DNase II: genes, enzymes and function. Gene 2003; 322: 1–15.
Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med 2005; 202: 1333–1339.
Okabe Y, Sano T, Nagata S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature 2009; 460: 520–524.
Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA 2012; 109: 19386–19391.
Pisetsky DS. The origin and properties of extracellular DNA: from PAMP to DAMP. Clin Immunol 2012; 144: 32–40.
Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD et al. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001; 61: 1659–1665.
Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 2000; 25: 177–181.
Bodano A, Gonzalez A, Balada E, Ordi J, Carreira P, Gomez-Reino JJ et al. Study of DNASE I gene polymorphisms in systemic lupus erythematosus susceptibility. Ann Rheum Dis 2007; 66: 560–561.
Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet 2011; 43: 1186–1188.
Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell 2016; 166: 88–101.
Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995; 374: 546–549.
Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2001; 2: 1144–1150.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012; 36: 401–414.
Esteve JM, Mompo J, Garcia de la Asuncion J, Sastre J, Asensi M, Boix J et al. Oxidative damage to mitochondrial DNA and glutathione oxidation in apoptosis: studies in vivo and in vitro. FASEB J 1999; 13: 1055–1064.
Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 1994; 179: 1317–1330.
Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A, Collins LV. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res Ther 2003; 5: R234–R240.
Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 2013; 39: 482–495.
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195.
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 2008; 205: 3007–3018.
Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol 2011; 8: 42–54.
Tanjore H, Lawson WE, Blackwell TS. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Bba-Mol Basis Dis 2013; 1832: 940–947.
Kim D, Peck A, Santer D, Patole P, Schwartz SM, Molitor JA et al. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I. Arthritis and Rheumatism 2008; 58: 2163–2173.
Svegliati S, Marrone G, Pezone A, Spadoni T, Grieco A, Moroncini G et al. Oxidative DNA damage induces the ATM-mediated transcriptional suppression of the Wnt inhibitor WIF-1 in systemic sclerosis and fibrosis. Sci Signal 2014; 7.
Richardson C, Chida AS, Adlowitz D, Silver L, Fox E, Jenks SA et al. Molecular basis of 9G4 B cell autoreactivity in human systemic lupus erythematosus. J Immunol 2013; 191: 4926–4939.
Reefman E, Horst G, Nijk MT, Limburg PC, Kallenberg CGM, Bijl M. Opsonization of late apoptotic cells by systemic lupus erythematosus autoantibodies inhibits their uptake via an Fc gamma receptor-dependent mechanism. Arthritis Rheum 2007; 56: 3399–3411.
Munoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum 2009; 60: 1733–1742.
Chen Y, Khanna S, Goodyear CS, Park YB, Raz E, Thiel S et al. Regulation of dendritic cells and macrophages by an anti-apoptotic cell natural antibody that suppresses TLR responses and inhibits inflammatory arthritis. J Immunol 2009; 183: 1346–1359.
Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 2014; 15: 1017–1025.
Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 2012; 189: 2689–2695.
Keshari RS, Verma A, Barthwal MK, Dikshit M. Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J Cell Biochem 2013; 114: 532–540.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532–1535.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 2010; 191: 677–691.
Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 2011; 117: 953–959.
Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 2010; 185: 7413–7425.
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 2011; 3: 73ra19.
Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol 2013; 191: 2647–2656.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016; 22: 146–153.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abu Abed U, Brinkmann V et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA 2010; 107: 9813–9818.
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 2009; 15: 623–625.
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu ZH et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011; 3.
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol 2010; 185: 3779–3779.
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 2011; 187: 538–552.
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol 2012; 188: 3522–3531.
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016; 22: 146–153.
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 2011; 187: 538–552.
Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol 2013; 190: 1217–1226.
Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol 2007; 179: 6933–6942.
Tarner IH, Muller-Ladner U, Gay S. Emerging targets of biologic therapies for rheumatoid arthritis. Nat Clin Pract Rheumatol 2007; 3: 336–345.
Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 2012; 37: 986–997.
Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT. Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 2005; 115: 407–417.
Nakazawa D, Tomaru U, Suzuki A, Masuda S, Hasegawa R, Kobayashi T et al. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012; 64: 3779–3787.
Knight JS, Carmona-Rivera C, Kaplan MJ. Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front Immunol 2012; 3: 380.
Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012; 120: 3007–3018.
Knight JS, Zhao WP, Luo W, Subramanian V, O'Dell AA, Yalavarthi S et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 2013; 123: 2981–2993.
Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med 2012; 4: 157ra141.
Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis 2016; 75: 1909–1916.
Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol 2017; 69: 376–386.
Seri Y, Shoda H, Suzuki A, Matsumoto I, Sumida T, Fujio K et al. Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose-6-phosphate isomerase-induced arthritis model. Sci Rep-Uk 2015; 5: 13041.
Knight JS, Subramanian V, O'Dell AA, Yalavarthi S, Zhao WP, Smith CK et al. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis 2015; 74: 2199–2206.
Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med 2009; 15: 1266–U1259.
A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009; 31: 245–258.
Uderhardt S, Herrmann M, Aschermann S, Ipseiz N, Frey B, Rothe T et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunology 2012; 137: 40–40.
Acknowledgements
MCP is a Wellcome Trust Senior Fellow in Clinical Science (WT082291MA). Hantao Lou is PHD candidate funded by Prof Gavin Screaton supported by Wellcome Trust (G.R.S).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Lou, H., Pickering, M. Extracellular DNA and autoimmune diseases. Cell Mol Immunol 15, 746–755 (2018). https://doi.org/10.1038/cmi.2017.136
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2017.136
Keywords
This article is cited by
-
Extracellular vesicles package dsDNA to aggravate Crohn’s disease by activating the STING pathway
Cell Death & Disease (2021)
-
mRNA level of ROCK1, RHOA, and LIMK2 as genes associated with apoptosis in evaluation of effectiveness of adalimumab treatment
Pharmacological Reports (2020)
-
Type I Interferons in the Pathogenesis and Treatment of Autoimmune Diseases
Clinical Reviews in Allergy & Immunology (2020)
