
Abstract
B cells have a critical role in the initiation and acceleration of autoimmune diseases, especially those mediated by autoantibodies. In the peripheral lymphoid system, mature B cells are activated by self or/and foreign antigens and signals from helper T cells for differentiating into either memory B cells or antibody-producing plasma cells. Accumulating evidence has shown that epigenetic regulations modulate somatic hypermutation and class switch DNA recombination during B-cell activation and differentiation. Any abnormalities in these complex regulatory processes may contribute to aberrant antibody production, resulting in autoimmune pathogenesis such as systemic lupus erythematosus. Newly generated knowledge from advanced modern technologies such as next-generation sequencing, single-cell sequencing and DNA methylation sequencing has enabled us to better understand B-cell biology and its role in autoimmune development. Thus this review aims to summarize current research progress in epigenetic modifications contributing to B-cell activation and differentiation, especially under autoimmune conditions such as lupus, rheumatoid arthritis and type 1 diabetes.
Similar content being viewed by others


Introduction
Although increasing evidence has indicated a pivotal role of B cell in the initialization and acceleration of autoimmune disorders, the molecular mechanisms underlying dysregulated B-cell activation and differentiation are still poorly defined. Genome-wide association studies have identified hundreds of gene polymorphisms associated with B-cell functions and differentiation,1,2,3 which may increase the susceptibility to autoimmune development. As the concordance rate of autoimmune diseases is <50% in monozygotic (MZ) twins,4 the epigenetic differences in genomic distribution of 5-methylcytosine (5-mC) DNA and histone modifications among MZ twins can alter the gene expression profile and contribute to their disease susceptibilities.5,6,7 Moreover, these epigenetic differences appear to result from environmental factors, such as infection, diet, and drugs.8 Therefore, the synergistic effects of both genetically and environmentally induced epigenetic modifications may contribute to the etiopathogenesis of autoimmune diseases.
Epigenetic modifications mainly comprise DNA methylation/demethylation, histone modification and non-coding RNAs, which can ultimately determine gene expression and thereby have important roles in various biological processes, such as cell growth, apoptosis, development, differentiation, immune response and aging.8 It has been shown that DNA methylation/demethylation regulates T-cell differentiation and cytokine production.9,10,11,12 In addition, histone modifications and non-coding RNAs also contribute to this regulation. Here we focus on reviewing the epigenetic modifications in the activation and differentiation of B cells and their implications in the understanding of autoimmune pathogenesis.
The main epigenetic modifications
DNA methylation
DNA methylation is defined as a potentially heritable and stable epigenetic alteration, which is the first recognized and intensively investigated epigenetic modification of DNA. DNA methylation is a biochemical process in which a methyl group is added to a cytosine or adenine residue at the fifth position on the pyrimidine ring, locking the gene transcription in the ‘off’ status.13 Therefore, DNA methylation acts as a flag indicating the repression of gene transcription, which is a process involved in many important biological processes. DNA methylation is mediated by methyltransferases, including DNMT1, DNMT3a and DNMT3b. Notably, every methyltransferase shows distinguished capacities. During DNA replication, DNMT1 usually sustains the methylation status while other two methyltransferases participate in de novo methylation.14
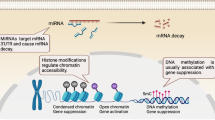
DNA demethylation occurs during the programmed failure in transmission of a methylation pattern, which results in re-activation of transcription of silenced genes.15 DNA demethylation occurs though the sequential iterative oxidation of 5-mC while the final modified group is removed by thymine DNA glycosylase (TDG) to yield cytosine instead of 5-mC.15 During this process, oxidation of 5-mC to 5-hydroxymethylcytosine (5-hmC) is mainly mediated by Ten-eleven translocation (TET) family dioxygenase enzymes, including TET1, TET2 and TET3,16 which can subsequently oxidize 5-hmC to 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-CaC), thereby displaying the order of 5-mC, 5-hmC, 5-fC and 5-CaC.17 In addition, both 5-fC and 5-CaC could be removed by TDG, which can further trigger base excision repair.18,19 (Figure 1)

DNA methylation and demethylation process.
Histone modification
Histone modification is a covalent posttranslational modification that regulates gene expression via changing chromatin structure or recruiting other modifications, thereby involving numerous biological processes. It has been well established that nucleosome is formed by 146 base pairs corresponding to two turns of DNA wrapped around a histone core, which displays two repeated sets of H2A, H2B, H3 and H4. These histones possess small protein ‘tails’ from the nucleosomes that are available for modifications, including acetylation, methylation, ubiquitination, phosphorylation and sumoylation.20 Acetylation and deacetylation can add or remove an acetyl group, which are mediated by histone acetyltransferases and histone deacetylases (HDACs), respectively.21 Histone methylation, defined as transferring 1, 2 or 3 methyl groups to arginine or lysine residues, is mainly mediated by histone methyltransferases and other enzymes, such as EZH2, G9a, SUV39-h1, ESET, SETDB1 and so on. The consequences of histone methylation depend on both modified residue and the number of methyl groups. Generally, acetylation promotes gene expression by opening the chromatin, while methylation switches the chromatin to the tight status, showing the opposite effects (Figure 2). For example, H3K4me3 activates gene transcription, whereas H3K27me3 and H3K9me3 result in gene silencing.22,23 Similar to methylation, ubiquitination on histones is implicated in both activation and repression of gene transcription.24 Many enzymes have been identified to control the addition and removal of ubiquitin. The ubiquitination on H2A and H2B has been found to have an essential role in numerous biological processes, such as transcription initiation, elongation and repression and DNA repair.25 Phosphorylation can occur at all four histone tails that contain acceptor sites, which is mediated by protein kinases.26 These four modifications can directly regulate histone–DNA interactions and also recruit non-histone proteins to chromatin. And the combinations of these modifications presenting on the same or the other histone tails have been reported as ‘histone codes’, which is deciphered by proteins that present specific binding motifs for each modification.26

Open and closed chromatin status by histone acetylation and methylation. White dots represent acetylation and red dots represent methylation.
MicroRNAs (miRNAs)
With a length of 21–25 base pairs, miRNAs belong to non-coding RNAs that regulate posttranscriptional and posttranslational gene expression. miRNAs bind to the 3′-untranslated region of specific target mRNA, resulting in mRNA cleavage, degradation or block translation27,28,29 (Figure 3). miRNAs, similar to other epigenetic regulations, involve numerous biological processes, including cell cycle, differentiation, apoptosis and innate and adaptive immune responses. Increasing evidence has shown that aberrant levels of miRNAs in different cell subtypes and tissues are associated with the pathogenesis of various diseases, facilitating them as potential non-invasive biomarkers for prediction and diagnosis, as well as potential treatment targets. To date, most work on non-coding RNAs in B-cell differentiation and antibody response has mainly focused on miRNAs. Moreover, miRNAs cross-talk with histone modification and DNA methylation,30 synergically regulating biological processes.

The pathway of miRNA regulation of gene expression. The maturation of miRNAs includes the production of the primary miRNA transcript (pri-miRNA) by RNA polymerase II or III and cleavage of the pri-miRNA by the microprocessor complex Drosha–DGCR8 (Pasha) in the nucleus. Then the pre-miRNA hairpin is exported from the nucleus by Exportin-5–Ran-GTP. In the cytoplasm, the RNase Dicer in complex with the double-stranded RNA-binding protein TRBP cleaves the pre-miRNA hairpin to its mature length. The functional strand of the mature miRNA is loaded together with Argonaute (Ago2) proteins into the RNA-induced silencing complex (RISC), where it guides the RISC to silence target mRNAs through mRNA cleavage, translational repression or deadenylation, whereas the passenger strand is degraded.
Epigenetic modifications in B-cell activation and differentiation
Epigenetic modification in B-cell activation
In the peripheral immune system, naive B cells display an inactive epigenetic status, showing genome-wide DNA hypermethylation and histone deacetylation,31 among which very few genes are expressed except for B-cell lineage genes such as Cd19, Pax5, Ebf1 and Spib, exhibiting active epigenetic status.32 Upon encountering antigens, naive matured B cells divide and then differentiate into germinal center (GC) B cells, and further differentiate into either plasma or memory B cells. During B-cell activation, the active epigenetic status of Igh, Cd19, Pax5, Ebf1 and Spib persists,33,34 while genome-wide DNA is hypomethylated, leading to increased levels of histone acetylation and miRNA expression.31,32
It has been well characterized that B-cell activation needs two major signals. Primary stimuli comprise dual B-cell receptor and Toll-like receptor binding to antigenic epitopes and pathogen-associated molecular patterns, respectively. Co-stimulatory signals are derived from CD40 and CD40L ligation, as well as signals from transmembrane activator and calcium-modulator and cyclophilin ligand interactor I (TACI) ligated with a proliferation-inducing ligand and B-cell-activating factor of the TNF family. The process induces several histone-modifying enzymes35 that activate H3K4me3, H3K9ac and H3K14ac in the promoter regions of activation-induced cytidine deaminase (AID) and miRNA host genes, as well as other somatic hypermutation (SHM)/class switch DNA recombination (SHM/CSR) factor genes. Moreover, removal of repressive H3K27me3 and H3K9me3 leads to chromatin decondensation.36,37,38 Recent evidence suggests that miRNAs, such as mir-16 and mir-155, decrease AID and Blimp expression in B cells.38,39 In contrast, AID regulates DNA methylation dynamics in GC B cells.40,41 For B-cell activation, secondary stimuli include cytokines such as interferon-γ, interleukin-4 and transforming growth factor-β, which activate transcription factors that interact with selected IH promoters and initiate germline IH-S-CH transcription, which then facilitate primary stimuli-induced histone modification-related enzymes to bind with RNA polymerase II to form a complex and then interact with the Sg1 region, catalyzing histone modifications in the S region for CSR targeting.42,43,44,45
Both DNA methylation and histone modification have an essential role in the SHM machinery, which targets V(D)J DNA through transcription.33,46,47,48 Remarkably, in comparable transcription of both alleles, only the demethylated allele can be hypermutated,33 indicating an essential role of DNA methylation in SHM. In an array-based genome-wide chromosomal imbalance and DNA methylation analysis, CREBBP and AID have been found to be possible modulators of both genetic and epigenetic co-evolution.49 DNA demethylation promotes H3K4me3, H3K9ac, H3K14ac and H4K8ac, which present enrichments in the V(D)J region, thereby leading to an ‘open’ chromatin status.50 In addition, histone modifications are capable of recruiting of DNA polymerases on the stage of DNA repair during SHM. For example, H2BK120 ubiquitination (ub) and H2AK119 (ub) are co-localized with error-prone translesion DNA polymerase η in AID-containing foci.44 H2BS14 phosphorylation has been found to mark the V(D)J region and this process is associated with AID regulation and perhaps recruit DNA repair-related factors.33
Epigenetic modification in B-cell differentiation
After the GC response, B cells ultimately differentiate into plasma cells. Although memory B cells are not capable of secreting antibodies, they can further experience SHM and/or CSR and then differentiate to plasma cells upon subsequent antigen exposure.51 Epigenetic modifications are involved in these processes, though how these stimuli and signals contribute to B-cell differentiation remains partially understood.
Epigenetic modifications in plasma cell formation
B lymphocyte-induced maturation protein 1 (Blimp-1, encoded by Prdm1) has a central role in the differentiation of plasma cells. Overexpression of Blimp-1 in peripheral mature B cells promotes J-chain upregulation and antibody production. Moreover, the knockdown of Blimp-1 expression in plasma cells retains plasma cell-related transcriptional markers but loses the capacity to produce antibodies.52 Prior to differentiation, Prdm1 is suppressed by Bcl-6. The increased expression of Prdm1 may result from the release of Bcl-6-bound HDACs, thereby increasing the histone acetylation levels on the promoter region of Prdm1.53,54 Furthermore, the HDAC inhibitor trichostatin A is capable of enhancing the expression of Blimp-155 and CD138, suggesting a critical role of histone acetylation in B-cell differentiation.55 Moreover, the expression of Blimp is regulated by several miRNAs, such as mir-125b,56 mir-127,57 mir-9,58 mir-30,57 mir-146a59 and let7b.60 In contrast, Blimp-1 can also regulate miRNAs such as mir-21.61 Additionally, mir-155, highly expressed by GC B cells, has been found to be associated with B-cell differentiation. In mir-155 knockout mice, reduced GC B cells and memory B cells are found with decreased high-affinity IgG1 antibodies.62,63,64
Blimp-1 is the transcription repressor of Bcl6, Pax5 and Spib, which, in return, suppress Blimp-1 expression and B-cell differentiation.65 Blimp-1 induces histone deacetylation in the promoter region of these genes, which display low histone acetylation levels in plasma cells.66 Blimp-1 decreases c-Myc expression to maintain the stable status of plasma cells via similar epigenetic mechanisms.66 Furthermore, Blimp-1 has been found to bind to H3K9 methyltransferase G9a, therefore recruiting this enzyme to the promoter regions of Spib and Pax5 and leading to gene silencing.67
Epigenetic modifications in memory B-cell formation
Epigenetic modifications also contribute to the differentiation of memory B cells. The hallmark genes of memory B cells, such as CD38 in mouse and CD27 in human, seem to be controlled by histone modifications.68,69 In quiescent memory B cells, histone lysine methylation levels are reduced compared with active memory B cells.70 Enhancer of zeste homolog 2 (Ezh2), with the ability of catalyzing H3K27me3, displays high levels in human GC B cells. The inhibition of Ezh2 activation in GC B cells can result in a reduction of memory B-cell percentage, GC reactions and antibody response,71 indicating an important role for histone methylation in GC reactions and memory B-cell differentiation, which might be associated with suppression of Prdm-1 and Irf4 transcription by Ezh2. In addition, histone acetyltransferase monocytic leukemia zinc finger protein has been revealed as a modulator in memory B-cell formation, by affecting the primary and secondary antibody responses.72
DNA methylation contributes to memory B-cell differentiation, a notion supported by the evidence that DNMTs are highly expressed by memory B cells while immune-related genes display distinctive DNA methylation patterns.73 Furthermore, mir-125b and let-7 negatively regulate Blimp-1 expression.56,60 Both mir-16 and mir-15a have been observed to regulate memory B cell via targeting Bcl-2;74 mir-223 contributes to B-cell differentiation though targeting LMO2, an important transcription factor in this process; mir-155 regulates AID expression and has an vital role in the differentiation of memory B cells.75
Therefore, epigenetic modifications coordinate with transcription factors in the activation of SHM and CSR during B-cell differentiation, determining cell fate and homeostasis (Figure 4).

The involvement of epigenetic modifications in B-cell differentiation.
Dysregulated epigenetic mechanisms in B cells contributing to autoimmune diseases
As described above, epigenetic modifications are closely involved in the activation and differentiation of B cells. Thus dysregulated epigenetic modifications in B cells may result in autoimmune pathogenesis.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a multi-organ autoimmune disease characterized by abundant autoantibodies in the circulation, which predominately affects females during their reproductive years.76 Numerous lines of evidence have shown a key role of abnormal epigenetic regulations in its pathogenesis.8,77,78,79,80,81 As the main source of pathogenic autoantibodies, B cells have been well documented as a major player in the pathogenesis of SLE. Recent clinical trials of B-cell-targeting therapies prove to be effective. DNA hypomethylation has been investigated in B cells from lupus patients,82 which contribute to B-cell auto-reactivity. Altered expression of HRES1/p28 in lupus B cells is found to be mediated by DNA methylation.83 A decreased methylation level of LINE-1 has been reported in B cells from SLE patients.84 A role of DNA demethylation in B cells is supported by the findings that adoptive transfer of DNMT1 inhibitor-treated B cells into syngeneic mice resulted in increased production of antinuclear antibodies.85 Although it is clear that DNA demethylation in V(D)J region and Igh 3′-LCR contributes to antibody production,86 little is known of this process during SLE. In addition, a lower level of DNA methylation has been observed in auto-reactive B cells, which might be a consequence of reduced DNMT1 and DNMT3b expression, or AID-mediated active DNA demethylation.87
In SLE patients, increased levels of miR-30a have been reported in B cells. The level of miR-30 negatively correlates with Lyn, a key negative regulator of B-cell activation.88 Both miR-155 and miR-181b have been found to negatively regulate the expression of AID, thereby affecting antibody diversity.89,90 In regulatory B cells, the expression level of miR-15a has been found to show positive correlation with the serum level of anti-dsDNA antibodies in lupus mice.91 Our recent studies have demonstrated that increased expression of miR-1246 in B cells from lupus patients affects EBF1 expression and therefore promotes costimulatory molecule expression and antibody production by B cells.92 In addition, increased levels of mir-21 and mir-17-92 have been observed in lupus B cells, which may contribute to autoimmune development.93,94 Recently, the microRNA profiling of B-cell subset has been proposed as biomarkers in lupus.95 Conversely, mir-150 is decreased in MRL-lpr mouse B cells, which may result from reduced acetylation status and repression of the mir-150 host gene.96
Rheumatoid arthritis
Rheumatoid arthritis (RA) has been defined as a chronic inflammatory autoimmune disorder that immune system primarily attacks the joints,97 in which synovial fibroblasts are believed to initiate RA.98,99 Epigenetic regulation has become an intensive research field in the studies of the pathogenesis of RA.100,101,102,103,104,105,106 Several epigenetic abnormalities have been reported in RA, such as increased DNA methylation,107 aberrant histone acetylation108 and differentially expressed miRNAs.109
B cells are recognized to be involved in RA via two major mechanisms: antigen presentation and autoantibody production. Autoantibodies against type II collagen, rheumatoid factor and citrullinated proteins have been found in the blood and synovial fluid of 70% of patients with early RA.110 As described before, epigenetic modifications tightly regulate antibody production, indicating that epigenetically regulated B-cell activation and differentiation have an important role in RA. However, few studies have revealed the association of epigenetic regulations in B cells in RA. Increased levels of mir-155 are found in B cells from the synovium and affect B-cell function by targeting PU.1.111,112 Moreover, mir-29a has been demonstrated to regulate B-cell proliferation and antibody secretion in mice with collagen-induced arthritis and contribute to the disease pathology,113 indicating that mir-29a is a potential therapeutic target in RA. In recent studies, histone deacetylases and their inhibitors have shown therapeutic effects in RA mouse models via immune suppression and inflammatory regulation.114,115,116,117,118,119 T cells and synovial fibroblasts are the main targets for these therapies, but other cells, such as B cells and neutrophils, might also be altered by these epigenetic drugs, which need to be further investigated.
Type 1 diabetes
Type 1 diabetes (T1D) is an organ-specific autoimmune disorder in which aberrantly activated immune cells target pancreatic beta cells. T1D was believed to be a T-cell-mediated disorder. However, recent studies have suggested a pathogenic role of B cells in T1D.120,121,122 Two main mechanisms are involved in the pathogenesis of T1D by B cells: one is antigen presentation by B cells,123 whereas the other one is autoantibody production by islet antigen-specific B cells.121 T1D usually occurs in genetically susceptible individuals and is triggered by environmental factors.124 Epigenetic mechanisms might partially exert the influences of environmental factors, especially diet, on T1D.125
In a recent epigenome-wide association study, 406 365 CpGs in 52 MZ twin pairs discordant for T1D in CD4+ T cells, CD19+ B cells and monocytes were analyzed. A substantial enrichment of differentially variable CpG positions was observed in these three different cell types from T1D twins,126 suggesting the contribution of DNA methylation of B cells to T1D development. Although there is little evidence showing the association of epigenetic modifications in B cells with the pathogenesis of T1D, epigenetic drugs such as 5-Aza,127 HDACs128,129 and HDAC inhibitors130,131 may exert their therapeutic effects on T1D via modifying B-cell activation and differentiation. Thus further studies on the potential link between abnormal epigenetic regulation of B cells and T1D may broaden our understanding of T1D pathogenesis (Table 1).
Concluding remarks
Recent findings of epigenetic regulations enable us to better understand the complex processes of B-cell activation and differentiation. However, further epigenetic studies are needed to define the role of B cells in the pathogenesis of autoimmune diseases such as lupus, systemic sclerosis, RA and T1D, in which epigenetic treatments, such as HDAC inhibitors, have shown therapeutic effects. Remarkably, B cells from circulation and local B cells from inflammatory sites can now be analyzed by single-cell sequencing and other advanced techniques. With the advent of the epigenomic era, new technologies will facilitate the investigation of epigenetic dysregulation in B cells and its implication in disease pathogenesis, which may lead to the identification of potential biomarkers and novel therapeutic targets.
References
Niewold TB. Advances in lupus genetics. Curr Opin Rheumatol 2015; 27: 440–447.
Ceccarelli F, Agmon-Levin N, Perricone C. Genetic factors of autoimmune diseases. J Immunol Res 2016; 2016: 3476023.
Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet 2013; 14: 661–673.
Renaudineau Y, Garaud S, Le Dantec C, Alonso-Ramirez R, Daridon C, Youinou P. Autoreactive B cells and epigenetics. Clin Rev Allergy Immunol 2010; 39: 85–94.
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA 2005; 102: 10604–10609.
Xiang Z, Yang Y, Chang C, Lu Q. The epigenetic mechanism for discordance of autoimmunity in monozygotic twins. J Autoimmun 2017; 83: 43–50.
Rodriguez-Cortez VC, Del Pino-Molina L, Rodriguez-Ubreva J, Ciudad L, Gomez-Cabrero D, Company C et al. Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naive-to-memory B-cell transition. Nat Commun 2015; 6: 7335.
Wu H, Zhao M, Tan L, Lu Q. The key culprit in the pathogenesis of systemic lupus erythematosus: aberrant DNA methylation. Autoimmun Rev 2016; 15: 684–689.
Suarez-Alvarez B, Rodriguez RM, Fraga MF, Lopez-Larrea C. DNA methylation: a promising landscape for immune system-related diseases. Trends Genet 2012; 28: 506–514.
Huang X, Wu H, Qiu H, Yang H, Deng Y, Zhao M et al. The expression of Bcl-6 in circulating follicular helper-like T cells positively correlates with the disease activity in systemic lupus erythematosus. Clin Immunol 2016; 173: 161–170.
Liu X, Lu H, Chen T, Nallaparaju KC, Yan X, Tanaka S et al. Genome-wide analysis identifies Bcl6-controlled regulatory networks during T follicular helper cell differentiation. Cell Rep 2016; 14: 1735–1747.
Xiao F, Lin X, Tian J, Wang X, Chen Q, Rui K et al. Proteasome inhibition suppresses Th17 cell generation and ameliorates autoimmune development in experimental Sjogren's syndrome. Cell Mol Immunol 2017; 14: 924–934.
Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell 2007; 128: 669–681.
Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep 2011; 12: 647–656.
Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013; 502: 472–479.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009; 324: 930–935.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011; 333: 1300–1303.
He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333: 1303–1307.
Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 2011; 286: 35334–35338.
Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta 2014; 1839: 627–643.
Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol 2011; 2011: 371832.
Renaudineau Y, Youinou P. Epigenetics and autoimmunity, with special emphasis on methylation. Keio J Med 2011; 60: 10–16.
Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 2012; 48: 491–507.
Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature 2009; 458: 422–429.
Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell 2008; 29: 653–663.
Rossetto D, Avvakumov N, Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 2012; 7: 1098–1108.
Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004; 303: 83–86.
Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 2010; 79: 351–379.
Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 2009; 11: 228–234.
Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J 2011; 278: 1598–1609.
Shaknovich R, Cerchietti L, Tsikitas L, Kormaksson M, De S, Figueroa ME et al. DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 2011; 118: 3559–3569.
Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol 2007; 8: 463–470.
Odegard VH, Kim ST, Anderson SM, Shlomchik MJ, Schatz DG. Histone modifications associated with somatic hypermutation. Immunity 2005; 23: 101–110.
Azagra A, Roman-Gonzalez L, Collazo O, Rodriguez-Ubreva J, de Yebenes VG, Barneda-Zahonero B et al. In vivo conditional deletion of HDAC7 reveals its requirement to establish proper B lymphocyte identity and development. J Exp Med 2016; 213: 2591–2601.
Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol 2012; 12: 517–531.
Chowdhury M, Forouhi O, Dayal S, McCloskey N, Gould HJ, Felsenfeld G et al. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc Natl Acad Sci USA 2008; 105: 15872–15877.
Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med 2011; 208: 1649–1660.
White CA, Pone EJ, Lam T, Tat C, Hayama KL, Li G et al. Histone deacetylase inhibitors upregulate B cell microRNAs that silence AID and Blimp-1 expression for epigenetic modulation of antibody and autoantibody responses. J Immunol 2014; 193: 5933–5950.
Frasca D, Diaz A, Romero M, Ferracci F, Blomberg BB. MicroRNAs miR-155 and miR-16 decrease AID and E47 in B cells from elderly individuals. J Immunol 2015; 195: 2134–2140.
Dominguez PM, Teater M, Chambwe N, Kormaksson M, Redmond D, Ishii J et al. DNA methylation dynamics of germinal center B cells are mediated by AID. Cell Rep 2015; 12: 2086–2098.
Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010; 463: 1101–1105.
Rajagopal D, Maul RW, Ghosh A, Chakraborty T, Khamlichi AA, Sen R et al. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med 2009; 206: 1237–1244.
Li G, White CA, Lam T, Pone EJ, Tran DC, Hayama KL et al. Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14-3-3 adaptors and AID to specify antibody class-switch DNA recombination. Cell Rep 2013; 5: 702–714.
Borchert GM, Holton NW, Edwards KA, Vogel LA, Larson ED. Histone H2A and H2B are monoubiquitinated at AID-targeted loci. PLoS One 2010; 5: e11641.
Rodriguez-Cortez VC, Del Pino-Molina L, Rodriguez-Ubreva J, Lopez-Granados E, Ballestar E. Dissecting epigenetic dysregulation of primary antibody deficiencies. J Clin Immunol 2016; 36 (Suppl 1): 48–56.
Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol 2006; 6: 573–583.
Maul RW, Gearhart PJ. AID and somatic hypermutation. Adv Immunol 2010; 105: 159–191.
Aida M, Hamad N, Stanlie A, Begum NA, Honjo T. Accumulation of the FACT complex, as well as histone H3.3, serves as a target marker for somatic hypermutation. Proc Natl Acad Sci USA 2013; 110: 7784–7789.
Loeffler M, Kreuz M, Haake A, Hasenclever D, Trautmann H, Arnold C et al. Genomic and epigenomic co-evolution in follicular lymphomas. Leukemia 2015; 29: 456–463.
Begum NA, Stanlie A, Nakata M, Akiyama H, Honjo T. The histone chaperone Spt6 is required for activation-induced cytidine deaminase target determination through H3K4me3 regulation. J Biol Chem 2012; 287: 32415–32429.
Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S et al. Multiple layers of B cell memory with different effector functions. Nat Immunol 2009; 10: 1292–1299.
Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK et al. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 2016; 17: 323–330.
Ochiai K, Muto A, Tanaka H, Takahashi S, Igarashi K. Regulation of the plasma cell transcription factor Blimp-1 gene by Bach2 and Bcl6. Int Immunol 2008; 20: 453–460.
Lemercier C, Brocard MP, Puvion-Dutilleul F, Kao HY, Albagli O, Khochbin S. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J Biol Chem 2002; 277: 22045–22052.
Lee SC, Bottaro A, Insel RA. Activation of terminal B cell differentiation by inhibition of histone deacetylation. Mol Immunol 2003; 39: 923–932.
Gururajan M, Haga CL, Das S, Leu CM, Hodson D, Josson S et al. MicroRNA 125b inhibition of B cell differentiation in germinal centers. Int Immunol 2010; 22: 583–592.
Zhang J, Jima DD, Jacobs C, Fischer R, Gottwein E, Huang G et al. Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood 2009; 113: 4586–4594.
Nie K, Gomez M, Landgraf P, Garcia JF, Liu Y, Tan LH et al. MicroRNA-mediated down-regulation of PRDM1/Blimp-1 in Hodgkin/Reed-Sternberg cells: a potential pathogenetic lesion in Hodgkin lymphomas. Am J Pathol 2008; 173: 242–252.
King JK, Ung NM, Paing MH, Contreras JR, Alberti MO, Fernando TR et al. Regulation of marginal zone B-cell differentiation by microRNA-146a. Front Immunol 2016; 7: 670.
Nie K, Zhang T, Allawi H, Gomez M, Liu Y, Chadburn A et al. Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: a potential role of the microRNA let-7. Am J Pathol 2010; 177: 1470–1479.
Barnes NA, Stephenson S, Cocco M, Tooze RM, Doody GM. BLIMP-1 and STAT3 counterregulate microRNA-21 during plasma cell differentiation. J Immunol 2012; 189: 253–260.
Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y et al. Regulation of the germinal center response by microRNA-155. Science 2007; 316: 604–608.
Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007; 316: 608–611.
Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity 2007; 27: 847–859.
Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol 2005; 5: 230–242.
Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol 2000; 20: 2592–2603.
Gyory I, Wu J, Fejer G, Seto E, Wright KL. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol 2004; 5: 299–308.
Zan H, Casali P. Epigenetics of peripheral B-cell differentiation and the antibody response. Front Immunol 2015; 6: 631.
Li G, Zan H, Xu Z, Casali P. Epigenetics of the antibody response. Trends Immunol 2013; 34: 460–470.
Baxter J, Sauer S, Peters A, John R, Williams R, Caparros ML et al. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J 2004; 23: 4462–4472.
Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest 2013; 123: 5009–5022.
Good-Jacobson KL. Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front Immunol 2014; 5: 596.
Lai AY, Mav D, Shah R, Grimm SA, Phadke D, Hatzi K et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res 2013; 23: 2030–2041.
Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J Jr., Miljkovic V et al. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci USA 2003; 100: 2639–2644.
Calame K. MicroRNA-155 function in B cells. Immunity 2007; 27: 825–827.
D'Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythematosus. Lancet 2007; 369: 587–596.
Wu H, Fu S, Zhao M, Lu L, Lu Q. Dysregulation of cell death and its epigenetic mechanisms in systemic lupus erythematosus. Molecules 2016; 22: pii: E30.
Wu H, Zhao M, Yoshimura A, Chang C, Lu Q. Critical link between epigenetics and transcription factors in the induction of autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 2016; 50: 333–344.
Wu H, Zhao M, Chang C, Lu Q. The real culprit in systemic lupus erythematosus: abnormal epigenetic regulation. Int J Mol Sci 2015; 16: 11013–11033.
Long H, Yin H, Wang L, Gershwin ME, Lu Q. The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J Autoimmun 2016; 74: 118–138.
Richardson BC, Patel DR. Epigenetics in 2013. DNA methylation and miRNA: key roles in systemic autoimmunity. Nat Rev Rheumatol 2014; 10: 72–74.
Garaud S, Le Dantec C, Jousse-Joulin S, Hanrotel-Saliou C, Saraux A, Mageed RA et al. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J Immunol 2009; 182: 5623–5632.
Fali T, Le Dantec C, Thabet Y, Jousse S, Hanrotel C, Youinou P et al. DNA methylation modulates HRES1/p28 expression in B cells from patients with lupus. Autoimmunity 2014; 47: 265–271.
Nakkuntod J, Avihingsanon Y, Mutirangura A, Hirankarn N. Hypomethylation of LINE-1 but not Alu in lymphocyte subsets of systemic lupus erythematosus patients. Clin Chim Acta 2011; 412: 1457–1461.
Mazari L, Ouarzane M, Zouali M. Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc Natl Acad Sci USA 2007; 104: 6317–6322.
Giambra V, Volpi S, Emelyanov AV, Pflugh D, Bothwell AL, Norio P et al. Pax5 and linker histone H1 coordinate DNA methylation and histone modifications in the 3' regulatory region of the immunoglobulin heavy chain locus. Mol Cell Biol 2008; 28: 6123–6133.
Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol 2010; 11: 607–620.
Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y et al. MicroRNA-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with Lyn. Arthritis Rheum 2013; 65: 1603–1611.
Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity 2008; 28: 630–638.
de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med 2008; 205: 2199–2206.
Yuan Y, Kasar S, Underbayev C, Vollenweider D, Salerno E, Kotenko SV et al. Role of microRNA-15a in autoantibody production in interferon-augmented murine model of lupus. Mol Immunol 2012; 52: 61–70.
Luo S, Liu Y, Liang G, Zhao M, Wu H, Liang Y et al. The role of microRNA-1246 in the regulation of B cell activation and the pathogenesis of systemic lupus erythematosus. Clin Epigenetics 2015; 7: 24.
Garchow BG, Bartulos Encinas O, Leung YT, Tsao PY, Eisenberg RA, Caricchio R et al. Silencing of microRNA-21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med 2011; 3: 605–615.
Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 2008; 9: 405–414.
Duroux-Richard I, Cuenca J, Ponsolles C, Pineiro AB, Gonzalez F, Roubert C et al. MicroRNA profiling of B cell subsets from systemic lupus erythematosus patients reveals promising novel biomarkers. Int J Mol Sci 2015; 16: 16953–16965.
Forster N, Gallinat S, Jablonska J, Weiss S, Elsasser HP, Lutz W. p300 protein acetyltransferase activity suppresses systemic lupus erythematosus-like autoimmune disease in mice. J Immunol 2007; 178: 6941–6948.
Kourilovitch M, Galarza-Maldonado C, Ortiz-Prado E. Diagnosis and classification of rheumatoid arthritis. J Autoimmun 2014; 48-49: 26–30.
Zhu X, Song Y, Huo R, Zhang J, Sun S, He Y et al. Cyr61 participates in the pathogenesis of rheumatoid arthritis by promoting proIL-1beta production by fibroblast-like synoviocytes through an AKT-dependent NF-kappaB signaling pathway. Clin Immunol 2015; 157: 187–197.
Rui K, Zhang Z, Tian J, Lin X, Wang X, Ma J et al. Olfactory ecto-mesenchymal stem cells possess immunoregulatory function and suppress autoimmune arthritis. Cell Mol Immunol 2016; 13: 401–408.
Meda F, Folci M, Baccarelli A, Selmi C. The epigenetics of autoimmunity. Cell Mol Immunol 2011; 8: 226–236.
Ngalamika O, Zhang Y, Yin H, Zhao M, Gershwin ME, Lu Q. Epigenetics, autoimmunity and hematologic malignancies: a comprehensive review. J Autoimmun 2012; 39: 451–465.
Grabiec AM, Reedquist KA. The ascent of acetylation in the epigenetics of rheumatoid arthritis. Nat Rev Rheumatol 2013; 9: 311–318.
Maciejewska-Rodrigues H, Karouzakis E, Strietholt S, Hemmatazad H, Neidhart M, Ospelt C et al. Epigenetics and rheumatoid arthritis: the role of SENP1 in the regulation of MMP-1 expression. J Autoimmun 2010; 35: 15–22.
Klein K, Gay S. Epigenetics in rheumatoid arthritis. Curr Opin Rheumatol 2015; 27: 76–82.
Viatte S, Plant D, Raychaudhuri S. Genetics and epigenetics of rheumatoid arthritis. Nat Rev Rheumatol 2013; 9: 141–153.
Ospelt C, Reedquist KA, Gay S, Tak PP. Inflammatory memories: is epigenetics the missing link to persistent stromal cell activation in rheumatoid arthritis? Autoimmun Rev 2011; 10: 519–524.
Kuchen S, Seemayer CA, Rethage J, von Knoch R, Kuenzler P, Beat AM et al. The L1 retroelement-related p40 protein induces p38delta MAP kinase. Autoimmunity 2004; 37: 57–65.
Grabiec AM, Tak PP, Reedquist KA. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res Ther 2008; 10: 226.
de la Rica L, Urquiza JM, Gomez-Cabrero D, Islam AB, Lopez-Bigas N, Tegner J et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun 2013; 41: 6–16.
Hampe CS. B cell in autoimmune diseases. Scientifica (Cairo) 2012; 2012, Article ID: 215308, p18.
Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci USA 2011; 108: 11193–11198.
Alivernini S, Kurowska-Stolarska M, Tolusso B, Benvenuto R, Elmesmari A, Canestri S et al. MicroRNA-155 influences B-cell function through PU.1 in rheumatoid arthritis. Nat Commun 2016; 7: 12970.
van Nieuwenhuijze A, Dooley J, Humblet-Baron S, Sreenivasan J, Koenders M, Schlenner SM et al. Defective germinal center B-cell response and reduced arthritic pathology in microRNA-29a-deficient mice. Cell Mol Life Sci 2017; 74: 2095–2106.
Kong S, Yeung P, Fang D. The class III histone deacetylase sirtuin 1 in immune suppression and its therapeutic potential in rheumatoid arthritis. J Genet Genomics 2013; 40: 347–354.
Hawtree S, Muthana M, Wilkinson JM, Akil M, Wilson AG. Histone deacetylase 1 regulates tissue destruction in rheumatoid arthritis. Hum Mol Genet 2015; 24: 5367–5377.
Angiolilli C, Kabala PA, Grabiec AM, Van Baarsen IM, Ferguson BS, Garcia S et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann Rheum Dis 2017; 76: 277–285.
Asadipour M, Hassan-Zadeh V, Aryaeian N, Shahram F, Mahmoudi M. Histone variants expression in peripheral blood mononuclear cells of patients with rheumatoid arthritis. Int J Rheum Dis 2017 e-pub ahead of print 21 July 2017 doi:10.1111/1756-185X.13126.
Cantley MD, Fairlie DP, Bartold PM, Marino V, Gupta PK, Haynes DR. Inhibiting histone deacetylase 1 suppresses both inflammation and bone loss in arthritis. Rheumatology (Oxford) 2015; 54: 1713–1723.
Zhang Y, Zhang B. Trichostatin A, an inhibitor of histone deacetylase, inhibits the viability and invasiveness of hypoxic rheumatoid arthritis fibroblast-like synoviocytes via PI3K/Akt signaling. J Biochem Mol Toxicol 2016; 30: 163–169.
O'Neill SK, Liu E, Cambier JC. Change you can B(cell)eive in: recent progress confirms a critical role for B cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 2009; 16: 293–298.
Hinman RM, Smith MJ, Cambier JC. B cells and type 1 diabetes in mice and men. Immunol Lett 2014; 160: 128–132.
Lu Q. The critical importance of epigenetics in autoimmunity. J Autoimmun 2013; 41: 1–5.
Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes 1997; 46: 941–946.
Burgio E, Lopomo A, Migliore L. Obesity and diabetes: from genetics to epigenetics. Mol Biol Rep 2015; 42: 799–818.
Dang MN, Buzzetti R, Pozzilli P. Epigenetics in autoimmune diseases with focus on type 1 diabetes. Diabetes Metab Res Rev 2013; 29: 8–18.
Paul DS, Teschendorff AE, Dang MA, Lowe R, Hawa MI, Ecker S et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat Commun 2016; 7: 13555.
Zheng Q, Xu Y, Liu Y, Zhang B, Li X, Guo F et al. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J Mol Med (Berl) 2009; 87: 1191–1205.
Bacha F, Klinepeter Bartz S. Insulin resistance, role of metformin and other non-insulin therapies in pediatric type 1 diabetes. Pediatr Diabetes 2016; 17: 545–558.
Al Khalifah RA, Alnhdi A, Alghar H, Alanazi M, Florez ID. The effect of adding metformin to insulin therapy for type 1 diabetes mellitus children: a systematic review and meta-analysis. Pediatr Diabetes 2017; 18: 664–673.
Khan S, Jena G. Valproic acid improves glucose homeostasis by increasing beta-cell proliferation, function, and reducing its apoptosis through HDAC inhibition in juvenile diabetic rat. J Biochem Mol Toxicol 2016; 30: 438–446.
Khan S, Jena GB. Protective role of sodium butyrate, a HDAC inhibitor on beta-cell proliferation, function and glucose homeostasis through modulation of p38/ERK MAPK and apoptotic pathways: study in juvenile diabetic rat. Chem Biol Interact 2014; 213: 1–12.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81220108017, 81522038, 81602767, 81430074, 91442116, 81373195 and 81771761), National Basic Research Program of China (No. 2014CB541904), the Programs of Science-Technology Commission of Hunan Province (2013F J4202), the Natural Science Foundation of Hunan Province (2017JJ3453) and the Natural Key Clinical Speciality Construction Project of National Health and Family Planning Commission of the People’s Republic of China.
Author information
Authors and Affiliations
Contributions
HW wrote the manuscript. YD, YF, DL, KM, XW and MZ conducted editing and LL and QL revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Wu, H., Deng, Y., Feng, Y. et al. Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell Mol Immunol 15, 676–684 (2018). https://doi.org/10.1038/cmi.2017.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2017.133
Keywords
This article is cited by
-
T cell expressions of aberrant gene signatures and Co-inhibitory receptors (Co-IRs) as predictors of renal damage and lupus disease activity
Journal of Biomedical Science (2024)
-
Plasma Levels of mir-34a-5p Correlate with Systemic Inflammation and Low Naïve CD4 T Cells in Common Variable Immunodeficiency
Journal of Clinical Immunology (2024)
-
The critical importance of epigenetics in autoimmune-related skin diseases
Frontiers of Medicine (2023)
-
Epigenetic regulation of B cells and its role in autoimmune pathogenesis
Cellular & Molecular Immunology (2022)
-
B Cell Aberrance in Lupus: the Ringleader and the Solution
Clinical Reviews in Allergy & Immunology (2022)
