
Abstract
Our immune system is based on the close collaboration of the innate and adaptive immune systems for the rapid detection of any threats to the host. Recognition of pathogen-derived molecules is entrusted to specific germline-encoded signaling receptors. The same receptors have now also emerged as efficient detectors of misplaced or altered self-molecules that signal tissue damage and cell death following, for example, disruption of the blood supply and subsequent hypoxia. Many types of endogenous molecules have been shown to provoke such sterile inflammatory states when released from dying cells. However, a group of proteins referred to as alarmins have both intracellular and extracellular functions which have been the subject of intense research. Indeed, alarmins can either exert beneficial cell housekeeping functions, leading to tissue repair, or provoke deleterious uncontrolled inflammation. This group of proteins includes the high-mobility group box 1 protein (HMGB1), interleukin (IL)-1α, IL-33 and the Ca2+-binding S100 proteins. These dual-function proteins share conserved regulatory mechanisms, such as secretory routes, post-translational modifications and enzymatic processing, that govern their extracellular functions in time and space. Release of alarmins from mesenchymal cells is a highly relevant mechanism by which immune cells can be alerted of tissue damage, and alarmins play a key role in the development of acute or chronic inflammatory diseases and in cancer development.
Similar content being viewed by others
INTRODUCTION
The immune recognition of an infection and the subsequent battle against the infecting pathogen is governed by the concerted efforts of both the innate and the adaptive immune systems. The first sensing of microbial invasion requires germline-encoded signaling receptors, also known as pattern-recognition receptors (PRRs), that evolved to acquire sensing specificity for foreign signature molecules generally termed pathogen-associated molecular patterns. This system was first theorized by Charles Janeway1 25 years ago, and has since been extended to include the more recently recognized ability of the innate immune system to also sense tissue damage by recognizing mislocalized or altered endogenous molecules termed damage-associated molecular patterns (DAMPs).2, 3
These molecules found in a foreign environment trigger sterile inflammation and, in the best case, promote tissue repair and the resolution of the inflammation. However, innate immune responses can also fuel uncontrolled or chronic inflammation. Since the introduction of the DAMP model by Polly Matzinger,2 many molecules that are released during proinflammatory cell death have been ascribed a DAMP function. These include heat-shock proteins (HSPs), adenosine triphosphate (ATP), nucleosomes, mitochondrial components and several alarmins: dual-function proteins that have distinct roles inside or outside the cells. The term alarmin describes protein functions leading to a rapid inflammatory response upon the release of these biologically active molecules. Notably, alarmins are also found in the nucleus in the resting state and likely exert biologically meaningful, yet understudied, functions. For example, failure of interleukin (IL)-33 to translocate to the nucleus results in a lethal inflammatory response, suggesting that the nuclear localization of, in this case, IL-33 has a protective function.4 In this review, we summarize recent knowledge on the most thoroughly studied dual-function proteins, the high-mobility group box 1 (HMGB1) protein, IL-1α, IL-33 and S100 proteins, with a focus on their extracellular functions.
HIGH-MOBILITY GROUP BOX 1
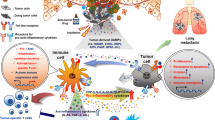
HMGB1 is one of the most abundant nonhistone nuclear proteins and is a member of the HMG protein family that contributes to chromatin architecture and modulates gene expression. Once released from stromal or immune cells, and upon interaction with a large panel of cell surface receptors, HMGB1 exerts a plethora of cell regulatory functions, from maturation, proliferation and motility to inflammation, survival and cell death (Figure 1).

Role of HMGB1 in inflammation. Under resting conditions, HMGB1 is localized in the nucleus, where it plays an important role in chromatin structure and gene expression. The translocation of HMGB1 to the cytoplasm is regulated by post-translational modifications such as acetylation, methylation and phosphorylation (1). Because of the lack of a secretion signal, HMGB1 is actively secreted through a caspase-1-dependent, noncanonical secretory vesicular pathway (2). HMGB1 can also be passively released from damaged cells either alone or in complex with RNA, DNA or nucleosomes (3). Interestingly, during apoptotic cell death, ROS production induces the terminal oxidation of HMGB1 that inhibits its proinflammatory function and switches HMGB1 function toward tolerogenicity (4). Once in the extracellular space, HMGB1 binds to several receptors in either free or complexed form (5). HMGB1 receptors, including RAGE and TLR4, bind free HMGB1 or HMGB1 in complex with DNA or LPS. Through its interaction with RAGE, the internalization of the HMGB1–DNA complex increases the activation of TLR9 localized in the endosome. However, HMGB1 complex formation with nucleic acids and potentially with other molecules can be inhibited by direct interaction with TIM-3. Other receptors, such as TLR2, IL-1R and CXCR4, recruit HMGB1 in complex with nucleosomes, IL-1β or CXCL12, respectively. Thus, sensing of HMGB1 mediates mechanisms of inflammation, cell migration, proliferation and differentiation (6). Furthermore, acting through the CXCL12/CXR4 axis, HMGB1 enhances chemotaxis (7). HMGB1, high-mobility group box 1 protein; IL, interleukin; LPS, lipopolysaccharide; RAGE, receptor for advanced glycation end-products; ROS, reactive oxygen species; TLR, Toll-like receptor.
HMGB1 expression, cellular localization and post-translational modifications
In resting cells, HMGB1 is localized in the nucleus, where it exerts an important function for chromatin structure and gene expression (Figure 1).5, 6 HMGB1 is involved in chromosomal DNA repair and contributes to nucleosome mobility by promoting histone sliding along the DNA strand. Consequently, Hmgb1 gene knockout is lethal, further arguing for the importance of HMGB1. Structurally, HMGB1 is composed of two DNA-binding HMG-box domains, namely Box A and Box B, followed by a flexible and negatively charged C-terminal tail. The C-terminal tail is believed to mediate a change in the three-dimensional structure of HMGB1 from a collapsed to a more linear conformation that most likely regulates HMGB1 binding to its ligands.7 Interestingly, post-translational modifications, such as acetylation, methylation and phosphorylation, have been shown to govern HMGB1 cellular localization (Figure 1).8, 9, 10, 11 Upon activation, monocytes and macrophages were found to hyperacetylate HMGB1 at nuclear localization sites, leading to its cytosolic relocalization. This process was recently shown to be mediated by the activation of the JAK/STAT1 (Janus kinase/signal transducer and activator of transcription 1) pathway.12 In addition, in monocytes, HMGB1 cytoplasmic localization can be regulated by the phosphorylation of its nuclear localization signal,11 and this was found to depend on protein kinase C activity.9 Furthermore, in neutrophils, HMGB1 cytosolic translocation seems to depend on the methylation of Lys42 that decreases HMGB1 binding affinity to DNA and thus enables its translocation to the cytosol.8 These studies investigated the role of each modification in a rather exclusive manner. It is therefore difficult to conclude whether these mechanisms regulating HMGB1 cellular localization function in parallel or are differentially modulated in a cell type- or cell activation-dependent manner. Because the nuclear localization of HMGB1 is likely to act as a regulatory mechanism with regard to HMGB1 extracellular function, a more integrative study of HMGB1 post-translational modification will be necessary to better comprehend the regulation of HMGB1 cytosolic translocation before its release.
Adding to the complexity of HMGB1 activity, the redox state of HMGB1 is believed to orchestrate its extracellular function.13 HMGB1 possesses three cysteines at positions 23, 45 and 106. These enable three different oxidation states of HMGB1 that license three mutually exclusive functions: alarmin, chemoattraction or tolerance.14, 15, 16 In fact, the formation of a disulfide bond between Cys23 and Cys45 was shown to confer proinflammatory properties to HMGB1.14, 15 Reduced HMGB1, with all cysteines in the thiol state, loses its alarmin function and behaves as a chemoattractant.15 In addition, upon cellular stress or apoptotic cell death, reactive oxygen species (ROS) production by mitochondria leads to the terminal oxidation of HMGB1 (sulfonate cysteines), granting HMGB1 tolerogenic properties that seem to mainly depend on Cys106 (Figure 1).16
Consequently, post-translational modifications orchestrate HMGB1 activity from cell localization to extracellular function and thus act as a crucial functional switch.
HMGB1 release and extracellular functions
HMGB1 can be released either passively by necrotic and damaged cells or by active mechanisms triggered upon immune cell activation. Once released in the extracellular space, HMGB1 mediates inflammation, cell migration, proliferation and differentiation (Figure 1).17, 18, 19 In fact, extracellular HMGB1 was shown to act as a chemoattractant for myeloid cells,20 smooth muscle cells (SMCs)21 and mesoangioblasts, thereby promoting muscle tissue repair.22
HMGB1 is considered to be one of the most mobile nuclear proteins and interacts only very transiently with chromosomal DNA. This loose nuclear DNA binding enables the leakage of HMGB1 upon cell damage or necrosis.5, 23 In contrast, during apoptosis, HMGB1 was long believed to be trapped inside the nucleus, where it binds strongly to hypoacetylated chromatin.23 However, it was since demonstrated that oxidized HMGB1 can be released from late-stage apoptotic cells, thereby promoting tolerance in a caspase-1-dependent manner.16, 24, 25 It was also proposed that macrophages secrete HMGB1 following the phagocytosis of apoptotic cells,26 thus further challenging the idea that apoptosis is a ‘silent’ death.
In addition to the cell death-dependent release of HMGB1, which likely represents its most important role, immune cells such as macrophages and monocytes are known to actively secrete HMGB1 once stimulated by cytokines (interferon-γ (IFNγ), tumor necrosis factor (TNF) and IL-1) or pathogen-derived molecules (lipopolysaccharide (LPS)).27, 28, 29, 30 Moreover, macrophages release HMGB1 following the activation of the NLRP3 or NLRC4 inflammasomes.27, 28, 31 Because HMGB1 lacks a leader sequence that would enable its transfer to the endoplasmic reticulum (ER) and the Golgi, active secretion of HMGB1 follows a nonclassical vesicular pathway.32 Indeed, several proteins, including not only HMGB1 but also IL-1β and IL-18, have been shown to depend on autophagy for their active secretion through the unconventional secretory pathway.33, 34 Interestingly, following cell starvation, HMGB1 was proposed to enhance autophagy through its interaction with Beclin1, and this was dependent on ROS production and the HMGB1 redox state.35 HMGB1 cytoplasmic translocation both promoted autophagy and limited the apoptotic pathway. Thus, HMGB1 cytoplasmic translocation not only enables release but also can actively promote its own secretion.
Several receptors have been shown to trigger downstream signaling upon binding to HMGB1 either directly or in complex with other molecules (Figure 1). In macrophages and dendritic cells, HMGB1 binds directly to Toll-like receptor 4 (TLR4) and induces the secretion of proinflammatory cytokines.36 Interestingly, binding of HMGB1 to TLR4 depends on reduced Cys106.14, 36 Moreover, the activation of TLR4 by proinflammatory HMGB1 (disulfide form) was recently shown to require myeloid differentiation factor 2 (MD-2).37 Upon binding to TLR4-MD-2, HMGB1 triggers the MyD88-dependent activation of nuclear factor (NF)-κB and the subsequent release of proinflammatory cytokines (i.e., TNFα, IL-1β and IL-6). Intriguingly, HMGB1 can also bind directly to LPS, thereby strengthening its ability to activate TLR4 through CD14.38 In contrast, binding of HMGB1 to CD24 and the further mobilization of Siglec-10 (or mouse Siglec-G) was shown to antagonize HMGB1-induced TLR4 activation in dendritic cells.39 In this study, Chen et al.39 found that the effect of CD24/Siglec-10 was restricted to stimulation with endogenous proteins (HMGB1, HSP70 and HSP90) and did not hold upon the activation of cells with pathogen-derived molecules. Hence, the authors proposed that the CD24/Siglec-10 pathway is a self-regulatory mechanism, limiting deleterious immune activation by endogenous damage-signaling molecules.
Another important cell surface receptor for HMGB1 is the receptor for advanced glycation end-products (RAGE). In fact, HMGB1 binding to RAGE was demonstrated shortly after RAGE discovery40 and has since been characterized in many settings. First, RAGE proved to mediate the previously described role of HMGB1 in neurite outgrowth during the development of the nervous system.40, 41 In dendritic cells, HMGB1 release and sensing by RAGE was shown to be critical for homing to the lymph nodes and further cross-activation of T lymphocytes.42, 43, 44 In endothelial cells, HMGB1 was further shown to promote the expression of RAGE and surface adhesion proteins (intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1)) and also to induce RAGE-dependent cytokine production.45, 46, 47
In addition to TLR4 and RAGE, HMGB1 interacts with several more receptors once in complex with other molecules. Indeed, extracellular HMGB1 can interact with IL-1β,48 CXCL12,49 and nucleosomes,50 thereby promoting the activation of IL-1 receptor (IL-1R), CXCR4 and TLR2, respectively. Furthermore, extracellular HMGB1, like other HMGB proteins, is found in complex with either DNA or RNA, promoting sensing by their putative receptors.51, 52, 53 Interestingly, yet another receptor of HMGB1, TIM-3, expressed at the surface of tumor-associated dendritic cells, was recently shown to compete with nucleic acids for binding to HMGB1, thereby dampening the efficacy of antitumor DNA vaccines or chemotherapy.54 These studies revealed the importance of controlling the purity of the recombinant HMGB1 protein used in experiments. In fact, several publications have argued that high-purity HMGB1 has a limited proinflammatory activity on its own.48, 51, 53, 55 Yet, the propensity of HMGB1 to co-purify with innate immune stimulants is not the only characteristic that can influence its biological function. One has to take into account that different HMGB1 preparations can vary in their amount of protein oxidation, thus influencing the biological activity of recombinant HMGB1. In addition, although HMGB1 redox status has now been described as a major regulatory mechanism of HMGB1 extracellular function, the role of other known post-translational modifications of HMGB1 in the extracellular space remains unknown. Hence, in the future, it would be beneficial to further analyze the extracellular functions of HMGB1, paying special attention to its purity and to the exact nature of its post-translational state.
HMGB1 in sepsis and sterile injury
HMGB1 was shown to mediate several acute effects in the context of infection or sterile tissue damage following myocardial infarction, stroke, acute lung injury or ischemia–reperfusion injury often occurring after transplantation and trauma. During sepsis, HMGB1 acts as a late mediator of inflammation that can be sustained for several days and correlates with an unfavorable prognosis.29 In fact, upon infection, the presence of large amounts of pathogen-derived molecules, such as endotoxin, induces a biphasic secretion of cytokines:56 (1) the release of conventional proinflammatory cytokines, such as TNFα and IL-1β (peak levels are found within hours) and (2) the late release of HMGB1 that often leads to lethality. Interestingly, treatment of animals with anti-HMGB1 antibodies or the HMGB1-binding antagonist thrombomodulin provides protection even when administered several hours after the peak secretion of the early cytokines.45, 57 In accordance with these findings, conditional deletion of HMGB1 in myeloid cells protected mice against LPS-induced endotoxic shock.58 Furthermore, a recent clinical study in which septic patients were treated with a combination of Polymyxin B and thrombomodulin found improved survival of the treated cohort.59 These data argue that HMGB1 may represent a pharmacological target for the treatment of septic shock. However, a study with larger groups and across different microbes and infection routes will be necessary before large-scale use in the clinic.
It is worth noting that the treatment of septic animals with recombinant HMGB1 A box, an antagonist competing with full-length HMGB1 for receptor binding, was also shown to provide protection against sepsis.60 In addition, the use of a monoclonal anti-RAGE antibody after cecal ligation and puncture (CLP) reduced lethality even when it was administered 36 h after CLP.61 This study followed the previous work by Liliensiek et al.,62 who first indicated a role for RAGE in the development of acute inflammation during sepsis. In addition to RAGE, the effects of HMGB1 in sepsis are believed to be partly mediated through TLR4.37, 38 Further reinforcing this hypothesis, treatment of CLP-elicited septic mice with a specific inhibitor of HMGB1 binding to MD-2 (P5779) decreased lethality.37 However, as mentioned earlier, binding of HMGB1 to LPS strengthens TLR4 activation and thus could potentially contribute to HMGB1-induced acute inflammation following bacterial infection.
In contrast to its role during sepsis, in the context of sterile tissue damage and cell death, HMGB1 mediates early inflammation responses that only last hours. Indeed, HMGB1 levels increase in the circulation following major events such as stroke, myocardial infarction or hemorrhagic shock. These conditions induce an ischemia–reperfusion injury in tissues where HMGB1 is passively released.63, 64, 65, 66, 67, 68, 69 In these settings, both TLR4- and RAGE-dependent inflammatory pathways mediate the early effect of HMGB1 release. Indeed, in the ischemic liver and kidney, TLR4 was shown to trigger inflammation in response to HMGB1 release.64, 65 Furthermore, RAGE expressed at the surface of microglial cells was found to mediate part of the deleterious function of extracellular HMGB1 in the ischemic brain.68 Strikingly, following brain injury, released HMGB1 was found to signal through RAGE in the lung, thereby mediating pulmonary dysfunction after lung transplantation.69, 70 In accordance with this finding, HMGB1 release from gut epithelial cells was recently shown to mediate lung injury following trauma and hemorrhagic shock.71 These studies illustrate the ability of HMGB1 to mediate its alarmin function in a systemic manner across the entire body. Here again, collaboration between RAGE and TLR4 is responsible for the deleterious effects of HMGB1 in acute sterile inflammation. Hence, direct targeting of HMGB1 with antibodies or thrombomodulin will probably be the most appropriate treatment strategy for transplanted patients or for those suffering from a traumatic injury. In fact, anti-HMGB1 antibodies have already shown some success in animal models of sterile inflammation and could therefore be promising for use in humans.68, 72, 73
Role of HMGB1 in cancer
HMGB1 was studied for many years for its role in cancer and is now considered a central modulator of cancer development.74 In fact, HMGB1 expression increases in many types of cancer,75, 76, 77, 78, 79 correlates with tumor invasion and metastasis and relates to unfavorable prognosis.76, 77, 80, 81 At the onset of tumorigenesis, rapid cell growth can overwhelm the limited blood circulation, thereby forming an ischemic environment that induces local cell necrosis and contributes to HMGB1 release.78 Moreover, during hypoxia, released HMGB1 activates the NLRP3 inflammasome and the subsequent caspase-1-dependent release of IL-1β and IL-18, fostering the inflammatory environment and promoting cell invasion.78 Ultraviolet irradiation of melanoma cells and the subsequent release of HMGB1 from damaged keratinocytes was also recently shown to induce a TLR4-dependent inflammatory environment that proved to be an important step toward perivascular invasion and metastasis in melanoma.82 Intriguingly, in pancreatic cancer cell lines, the presence of extracellular HMGB1 and its binding to RAGE seems necessary for mitochondrial ATP production and sustained cell growth.79 This study proposed that upon HMGB1 binding, RAGE translocates to the mitochondria, where it interacts with electron transport chain complexes I and II and thereby positively regulates oxidative phosphorylation. In addition, in the context of colorectal cancer, released HMGB1 can act on endothelial cells, promoting cell proliferation and neovascularization.81, 83 Indeed, in vitro extracellular treatment with recombinant HMGB1 promotes endothelial cell migration and the NF-κB-dependent expression of adhesion and angiogenic proteins. Moreover, treatment with recombinant HMGB1 leads to TLR4 and RAGE expression. However, knockdown studies showed that HMGB1-dependent neovascularization is mainly mediated through RAGE.81 Further emphasizing the role of the RAGE–HMGB1 axis in cancer progression, blockade of either HMGB1 or RAGE can reduce malignant mesothelioma and glioma tumor growth and metastasis.80, 84 Interestingly, upon anticancer treatment, the HMGB1 redox state balances the cell fate between autophagy-mediated cell survival or apoptosis.85, 86 Indeed, using pancreatic and colon cancer cell lines (Panc2.03 and HCT116, respectively), Tang et al.85, 86 demonstrated that treatment with cytotoxic anticancer agents could induce both the autophagy and apoptosis pathways and subsequent HMGB1 release. In this study, the inhibition of autophagy decreased HMGB1 release and pushed the balance toward apoptosis. Reduced extracellular HMGB1 promoted autophagy and cell survival through a RAGE- and Beclin-1-dependent pathway. In contrast, oxidized HMGB1 promoted cell death and increased the efficiency of chemotherapeutic drugs. In accordance with these data, studies using different cancer cell lines (leukemia, colorectal carcinoma and pancreatic cancer cells) demonstrated a role for both RAGE and HMGB1 in the balance between autophagy and apoptosis through the modulation of p53-dependent apoptosis and the activation of ATG5-dependent autophagy pathways.87, 88, 89 In contrast, HMGB1 has been implicated in the antitumor immune response induced by radiation therapy or chemotherapy.90, 91 In this case, HMGB1 enables the TLR4-dependent activation of dendritic cells and promotes antigen cross-presentation to cytotoxic T cells. However, these experiments did not consider the HMGB1 redox state. Hence, further analysis of the HMGB1 redox state in this setting would be of interest.
HMGB1 mediates chronic inflammation
There are many examples of the central role of HMGB1 in the development and aggravation of chronic inflammation. As a first example, extracellular HMGB1 has been associated with the development of arthritis, particularly with the inflammation of the synovial space. Indeed, HMGB1 levels are increased in the serum and synovial fluid of patients with rheumatoid arthritis (RA).92, 93 The use of recombinant HMGB1 demonstrated that extracellular HMGB1 promotes the macrophage- and neutrophil-dependent development of arthritis.93, 94 HMGB1 is actively secreted by synovial macrophages and fibroblasts in collagen-induced arthritis and hypoxic conditions.95 In this system, the effect of HMGB1 was shown to be partly mediated by TLR2 and TLR4.96 Consequently, the use of anti-HMGB1 monoclonal antibodies, thrombomodulin or A box peptide efficiently diminished inflammation and arthritis.95, 97, 98 Similarly, blocking HMGB1 release by nuclear sequestration using oxaliplatin had protective effects.99 In accordance with these data, gold sodium thiomalate, a molecule often used to treat RA, was shown to block HMGB1 cytosolic translocation and thereby inhibit its secretion from macrophage cell lines.100
In addition, HMGB1 has been studied for its role in the development of systemic lupus erythematosus. Indeed, several studies have found the level of HMGB1 in the serum and skin lesions to be increased in patients with lupus, and this correlated with disease activity.101, 102, 103, 104, 105 Extracellular nucleosomes in complex with HMGB1 are believed to disrupt self-tolerance and promote the production of anti-double-stranded DNA antibodies.50, 106 HMGB1 binding to DNA contained in immune complexes was also shown to enhance RAGE-dependent DNA intake and TLR9 activation, further increasing cytokine secretion and inflammation in lupus patients.51, 53
Finally, in atherosclerotic plaques, SMCs are activated to secrete HMGB1. Interestingly, HMGB1 expression can be induced upon treatment with cholesterol, further linking HMGB1 to atherosclerosis.107, 108 Upon sensing the Box B domain of HMGB1, SMCs proliferate and migrate. However, SMCs are not the only cell type secreting HMGB1 in atherosclerotic plaques. In fact, endothelial cells, macrophages, foam cells and activated platelets are also able to secrete HMGB1.109 As the NLRP3 inflammasome contributes to the formation of atherosclerotic plaques upon activation with cholesterol crystals,110 and its activation in macrophages leads to the active release of HMGB1,27, 28 it is likely that the secretion of HMGB1 by plaque-infiltrating macrophages is NLRP3 dependent and contributes to the inflammation induced by cholesterol crystals present in the plaques. In this context, in vitro experiments showed that treatment with extracellular HMGB1 increases the expression of adhesion molecules (ICAM-1 and VCAM-1) and promotes the secretion of proinflammatory cytokines (i.e., TNFα) and chemokines (i.e., CXCL8 and CCL2).46, 47 In addition, RAGE expressed in endothelial cells is an important mediator of plaque formation. In fact, RAGE has long been known to promote the expression of adhesion proteins such as VCAM-1 in endothelial cells.111 During atherosclerosis, RAGE increases HMGB1 expression and release,112, 113 and in vitro treatment of endothelial cells (human umbilical vein endothelial cells) with extracellular HMGB1 induces RAGE-dependent ER stress.114 This implication of the RAGE–HMGB1 axis in the induction of ER stress could contribute to the previously mentioned induction of autophagy by HMGB1 through RAGE.89, 115 Furthermore, during atherosclerosis, the HMGB1–RAGE axis has recently been implicated in platelet activation.116 Hence, in atherosclerotic plaques, HMGB1 sensing by platelets, endothelial cells and SMCs promotes the migration and adhesion of immune cells, thereby fostering plaque formation and growth.
For the past two decades, HMGB1 commanded the attention of many groups because of its central role in signaling infection and cellular damage. It has become clear that even if HMGB1 is an important mediator of necessary antimicrobial and tissue repair mechanisms, it also often acts as powerful deleterious ‘double-agent’ in the development of multifactorial diseases such as cancer and acute or chronic inflammation. Hence, the development of clinical tools targeting HMGB1 to moderate its negative effects will be crucial for the efficient treatment of many patients.
INTERLEUKIN-1α
The IL-1 family of proteins contains 11 members. The best-studied family members are IL-1α and IL-1β. Both are highly similar in structure and bind to the same cell membrane receptor, IL-1R. Interestingly, IL-1 receptor antagonist (IL-1Ra) is a naturally expressed member of the IL-1 family that is nonimmune stimulatory and inhibits both IL-1α and IL-β function by competing for binding to their receptor. Members of the IL-1 family are expressed as pro-forms (pIL-1) that are usually matured through enzymatic cleavage. Unlike IL-1β but similarly to all alarmins described herein, IL-1α is a dual-function cytokine that presents both nuclear and extracellular functions. Extracellular IL-1α is now recognized to be an important player in sterile inflammatory diseases and cancer.117, 118, 119
IL-1α expression and intracellular function
IL-1α precursor (pIL-1α) is constitutively expressed in most resting nonhematopoietic cells, such as epithelial cells lining the gastrointestinal tract, liver, kidney and skin.120, 121 Moreover, pIL-1α expression can be increased in conditions of stress and inflammation.122 In resting cells, pIL-1α is found in the nucleus, where it promotes gene expression by acting as a transcription factor (Figure 2),123 regulating cell growth and differentiation. This relies on the N-terminal domain containing the nuclear localization signal,124 which is absent in mature IL-1α.124, 125 Interestingly, following cleavage, the N-terminal domain of IL-1α was shown to independently translocate to the nucleus, where it interacts with certain members of the RNA splicing and processing machinery.126 In the nucleus, pIL-1α (but not mature IL-1α) also interacts with histone acetyltransferases and thus acts as a transcriptional regulator.123, 127 In addition, upon stimulation with LPS or TNFα, pIL-1α translocates to the nucleus, where it was shown to promote the expression of inflammatory genes such as IL-6 and IL-8.125, 128 In accordance with these findings, in systemic sclerosis fibroblasts, pIL-1α translocation was shown to depend on binding to HS1-associated protein X-1 (HAX-1) and induce the expression of IL-6 and pro-collagen (Figure 2).129 Interestingly, HAX-1 was also found to interact with the cleaved IL-1α N-terminal domain.130 Together, these studies suggest that the nuclear function of pIL-1α is predominantly dependent on its N-terminal domain. Upon cell activation, pIL-1α cleavage acts as a regulatory mechanism, keeping IL-1α outside the nucleus and promoting its secretion. Furthermore, pIL-1α cleavage could support a distinct transcriptional function for the ‘freed’ nuclear N-terminal peptide. However, this hypothesis remains to be proven.

Proinflammatory role of extracellular interleukin (IL)-1α. IL-1α precursor (pIL-1α) is constitutively expressed in most resting nonhematopoietic cells. In these cells, IL-1α is primarily localized in the nucleus, where it promotes gene expression by acting as a transcription factor (1). IL-1α nuclear transport depends on its interaction with HS1-associated protein X-1 (HAX-1) (2). In stimulated cells, pIL-1α is processed by the membrane-bound protease calpain, a calcium-dependent cysteine protease, before active release into the extracellular space through a noncanonical vesicular pathway (3). Interestingly, HAX-1 was also found to interact with the cleaved IL-1α N-terminal domain, suggesting its independent nuclear function (2). pIL-1α processing is inhibited by the cytosolic expression of IL-1 receptor-2 (IL-1R2) that binds to pIL-1α and thereby prevents its interaction with calpain and its subsequent secretion (4). However, active caspase-1 cleaves IL-1R2 and thereby enables pIL-1α processing. pIL-1α can also be passively released from damaged cells and, similar to mature IL-1α, interacts with IL-1 receptor-1 (IL-1R1) (5). In addition to being released in the extracellular space, IL-1α can be displayed at the plasma membrane, where it activates IL-1R1-expressing juxtaposing cells (6). Once sensed by IL-1R1, soluble or membrane-bound IL-1α induces the recruitment of the accessory receptor IL-1R3 and triggers proinflammatory signaling pathways (7).
IL-1α secretion and extracellular activity
In stimulated cells, IL-1α is processed by the membrane-bound protease calpain, a calcium-dependent cysteine protease, and then released into the extracellular space.131, 132, 133, 134 Similar to IL-1β and HMGB1, IL-1α does not contain any secretion signal and thus relies on the noncanonical vesicular secretion pathway (Figure 2). As for IL-1β, IL-1α secretion is believed to partially depend on caspase-1 activity.135, 136 In fact, it was recently demonstrated that IL-1α secretion is antagonized by the intracellular presence of IL-1R2 which, through binding to pIL-1α, prevents its processing by calpain and further secretion.134 Active caspase-1 cleaves IL-1R2, thereby enabling pIL-1α processing by calpain and the release of mature IL-1α. However, Gross et al.137 showed that caspase-1 dependency for IL-1α secretion can differ depending on the stimulus and the targeted inflammasomes and does not seem to require the protease activity of caspase-1. IL-1α secretion may therefore rely on redundant parallel pathways. In fact, elevated intracellular Ca2+ triggers IL-1α secretion from bone marrow-derived cells.132, 137, 138 An increase in the Ca2+ concentration was shown to be sufficient for inflammasome-independent IL-1α processing by calpain and its subsequent secretion.137 Interestingly, increased levels of Cu2+ were also shown to promote IL-1α secretion through an S100A13-dependent secretion mechanism.139, 140 Further differentiating itself from IL-1β and HMGB1, IL-1α activation and secretion is inhibited by autophagy. Indeed, in the context of Mycobacterium tuberculosis infection, ATG5-dependent autophagy was shown to block both the calpain-dependent activation of IL-1α and its secretion, thereby limiting lung inflammation and tissue damage.141
Another regulatory mechanism seems to reside in the close proximity of IL-1α to the nucleus. Indeed, like HMGB1, cells undergoing apoptosis retain IL-1α in the chromatin fraction, whereas necrotic cells can release pIL-1α, resulting in myeloid cell chemotaxis and inflammation.142 In fact, the IL-1α pro-form is known to bind to IL-1R1 and to induce inflammation. To date, however, it is not entirely clear which of the two forms is the most relevant following cell damage. Notably, IL-1α release by stressed endothelial cells was also proposed to be mediated by the formation of pIL-1α-containing apoptotic bodies that possessed chemotactic and proinflammatory properties.143
In addition to calpain- and caspase-1-dependent pIL-1α processing, other proteases, such as granzyme B, elastase or chymase, have been proposed to cleave pIL-1α, producing the mature form of IL-1α and potentiating its proinflammatory activity.144 Interestingly, IL-1α may also be displayed at the cell surface, where it can activate juxtaposing target cells expressing its receptor, such as T cells or endothelial cells (Figure 2).145, 146, 147, 148
As for IL-1β, once it is released into the extracellular space, soluble or membrane-bound IL-1α binds to IL-1R1 and further triggers the recruitment of the accessory receptor IL-1R3 (also known as IL-1RAcP) for signal transduction. Downstream signaling is triggered by the recruitment of the MyD88 adaptor protein to the Toll–interleukin receptor domain of IL-1R3. As for other MyD88-dependent TLR signaling, MyD88 recruitment activates IRAK4/IRAK1 and subsequent downstream mitogen-activated protein kinase (MAPK) signaling and NF-κB activation (Figure 2).149, 150 Thus, IL-1α activates neighboring fibroblasts or epithelial cells, further triggering the release of chemokines that leads immune cells, preferentially neutrophils,151 to infiltrate and enhance local inflammation.
IL-1α in sterile inflammation: from acute to chronic disease
As previously mentioned, pIL-1α is constitutively expressed in most resting cells. It is therefore not surprising for pIL-1α to play a key role in inflammation induced by necrosis or tissue damage following ischemia. Indeed, liver lysates containing IL-1α promote neutrophilic migration in matrigel.152 Moreover, in peritonitis, IL-1α is released from necrotic cells and subsequently activates IL-1R1 present at the surface of mesothelial cells. This was proposed to be the prime event inducing neutrophil infiltration and peritoneal inflammation rather than the HMGB1 pathway.152, 153 This proinflammatory role of IL-1α was recently confirmed in a mouse model of acute colon inflammation (dextran sodium sulfate-induced colitis), where epithelial IL-1α played a key role in the establishment of inflammation.121 In contrast, IL-1β originating from myeloid cells promoted healing and tissue repair.
One of the main triggers of cell necrosis is the hypoxia that follows poor oxygen supply associated with, for example, early-stage tumor growth or blood vessel clogging. Interestingly, pIL-1α expression was shown to be increased before epithelial cell necrosis in a hypoxia-induced factor (HIF)-1a- and HIF-2a-dependent manner.154 The release of IL-1α promotes chemokine secretion and neutrophil infiltration. Moreover, extracellular pIL-1α itself can trigger the increased expression of pIL-1α (and pIL-1β) in surrounding cells, thereby fueling local inflammation.151, 155 Following this early IL-1α-dependent inflammation and recruitment of neutrophils, inflammasomes and caspase-1 activation enable the cleavage of pIL-1β, promoting inflammation through the recruitment of macrophages.151, 156 This mechanism is confirmed by the IL-1α dependency of IL-1β secretion following inflammasome activation.137
During brain ischemia, activated platelets present in the brain vasculature were shown to release pIL-1α and thereby stimulate endothelial cells to secrete the chemokine CXCL1 and express the endothelial surface adhesion proteins VCAM-1 and ICAM-1. Hence, upon ischemic injury, pIL-1α fosters inflammation by enabling neutrophils to migrate through the endothelial barrier.157 Interestingly, platelets are known to be one of the first cell types to reach the brain and to be activated in response to stroke or in patients with multiple sclerosis.158, 159, 160 Thus, platelet-originating IL-1α would be an early signal playing a central role in the establishment of cerebrovascular inflammation and subsequently devastating brain injury.
Notably, similar mechanisms are involved in the ischemic heart following myocardial infarction. Indeed, IL-1α release from necrotic myocytes was shown to trigger acute inflammation by activating surrounding myofibroblasts.161
The importance of IL-1α and IL-1β in sterile inflammation is further highlighted by the case of patients born with a deficiency in the IL-1R antagonist, IL-1Ra.162, 163 Indeed, deficiency in IL-1Ra triggers IL-1-dependent inflammation and the infiltration of neutrophils to the skin, joints and bones. Systemic treatment with recombinant IL-1Ra (anakinra) has been an efficient way to decrease inflammation and prevent death.156 However, because anakinra can inhibit both IL-1α and IL-1β by binding to their common receptor, it is unclear which is the main cytokine at the root of the sterile inflammation affecting these patients. Nonetheless, anakinra is currently also used to treat RA as well as neonatal-onset multisystem inflammatory disease.164 Other drugs, such as monoclonal antibodies (canakinumab) and decoy receptors (rilonacept), are also used to treat patients presenting with, for example, juvenile idiopathic arthritis or cryopyrin-associated autoinflammatory syndrome, this time with a clear aim at blocking IL-1β-derived inflammation.156
Yet, some evidence suggests a role for IL-1α in the development of these chronic inflammatory diseases. In fact, early IL-1α release is recognized as an important step that initiates inflammation, and IL-1α is thus considered a promising target for the treatment of rheumatologic disease.156, 164 Interestingly, RA patients producing IL-1α-specific autoantibodies in the joint synovial fluid show reduced joint erosion.165 This supports the idea that IL-1α is a major player in the establishment of long-term inflammation. Furthermore, in a recent clinical trial, Coleman et al.166 used a monoclonal antibody (MABp1) targeting IL-1α for the treatment of psoriasis.
The role of IL-1α in cardiovascular diseases is not restricted to its proinflammatory function following myocardial infarction. Indeed, mounting evidence now suggests an important role for IL-1α in the development of atherosclerosis.167, 168 This might come as a surprise because IL-1β has long been predicted to be the most important IL-1 cytokine responsible for vascular inflammation and the development of atherosclerotic plaques. Indeed, mice lacking either IL-1R or IL-1β or those treated with IL-1β-blocking antibodies presented decreased plaque formation.169, 170 However, others have since shown that IL-1α deficiency in hematopoietic cells has an even more important protective effect.171, 172 Together, these results therefore indicate a potential role for both IL-1α and IL-1β in atherosclerosis.
Perhaps, the greater effect of IL-1α in the development of aortic plaques might be in part attributed to the fact that IL-1α deficiency induces the decreased expression of IL-1β,172 thereby not only acting on IL-1α-dependent cell signaling but also acting in parallel on IL-1β function. Nevertheless, another study recently confirmed the results of Kamari et al.172 and further showed that IL-1α (but not IL-1β) secretion from LPS-primed macrophages is induced by unsaturated fatty acids (i.e., oleic acid) in an inflammasome-independent manner.173. In this study, unsaturated fatty acids were shown to act on the mitochondrial respiratory chain and induce its uncoupling, thereby promoting calcium fluxes and subsequent calpain-dependent IL-1α release. Moreover, by inducing mitochondrial uncoupling, oleic acid not only promoted IL-1α secretion but was also shown to inhibit IL-1β release. Hence, unsaturated fatty acids accumulating in atherosclerotic plaques would shift the IL-1 secretion by macrophages from IL-1β to IL-1α. Strikingly, analysis of IL-1α and IL-1β expression in either IL-1α/β-deficient macrophages indicated that not only IL-1α was upstream of IL-1β secretion but also that the reverse was potentially true. Indeed, IL-1β-deficient but not IL-1R-deficient macrophages showed markedly decreased IL-1α secretion and LPS-induced expression. This may indicate the existence of an IL-1β-dependent intracellular transcriptional control of IL-1α expression. However, this effect was only found in macrophages derived from the Il1btm1Yiw background. Macrophages derived from the Il1btm1Dch background had no effect on IL-1α secretion. These findings reveal the potential misconception regarding the role of IL-1β in atherosclerosis that might have been the result of underlying partial IL-1α downregulation. Caution will therefore be needed in the future when studying the role of IL-1β in atherosclerosis development.
IL-1α in cancer development
The existence of a link between chronic inflammation and the onset of tumorigenesis has long been postulated and is supported by many clinical conditions as well as cancer animal models.174, 175, 176 Yet, inflammation is also necessary for the organism to fight against the tumor.177 In the middle of this tight equilibrium, the presence of proinflammatory signals, such as the previously mentioned HMGB1, can lead to tumor growth and invasion either way.74 Interestingly, IL-1R autocrine stimulation by IL-1α and downstream MyD88 activation were shown to be crucial for the establishment of the inflammation involved in RAS-dependent carcinogenesis.178 In these settings, IL-1α and MyD88 (but not IL-1β) activation trigger the loss of keratinocyte differentiation and promote invasiveness. Similar results have previously been found in melanoma patients, where IL-1α induced the MyD88-dependent activation of the NF-κB and MAPK pathways as well as an increase in ROS production, thereby promoting tumor progression.179 Furthermore, in pancreatic ductal adenocarcinoma, tumor-associated IL-1α release, probably through cell damage, can also induce tumor growth via immune activation of the surrounding tumor fibroblasts.180 IL-1α is furthermore implicated in tumor vascularization by promoting the expression of vascular endothelial growth factor (VEGF) in endothelial cells.181 Interestingly, IL-1α seemed to have a more dramatic effect in hypoxic conditions, which fits to the proposed role of IL-1α in the often hypoxic tumor microenvironment. Following these first steps of tumor development, IL-1β takes over and is thus believed to be of greater importance.182 Yet, clinical trials are underway using MABp1 antibody to target IL-1α in the treatment of refractory cancers with metastasis.183
In contrast to the role of secreted IL-1α, intracellular and membrane-bound IL-1α activates immune mechanisms that lead to tumor destruction. Indeed, in a large panel of cancer cell lines, pIL-1α modulates cell cycle and induces apoptosis.126, 184 In addition, overexpression of membrane-bound IL-1α at the surface of fibrosarcoma or lymphoma cell lines induced loss of tumorigenicity. Once injected into mice, such cells form tumors that rapidly regress or cannot grow.185, 186, 187 Furthermore, after injection into wild-type (WT) mice, these cells induced the appearance of a long-term memory against tumor cells that relied on the infiltration of CD8+ T cells, natural killer (NK) cells and macrophages.
Together, these data show that IL-1α could, in certain settings, represent a key initiator of proinflammatory mechanisms that drive chronic inflammatory diseases. Drugs in clinical use, such as anakinra, give hope for the targeting of IL-1α in these pathologies. However, the complex multifaceted functions of IL-1α, sometimes beneficial and at other times deleterious, make IL-1α a difficult clinical target.
INTERLEUKIN-33
IL-33 is a member of the IL-1 family that is mainly expressed in the epithelium lining the surfaces in contact with the environment. IL-33 has long been considered to exclusively mediate type-2 immune responses. However, recent findings extended its influence to a broader panel of functions at the junction between innate and adaptive immune responses, from the maintenance of homeostasis to the mediation of deleterious proinflammatory reactions.
IL-33 protein expression and post-translational modification
Similar to IL-1α, IL-33 is an alarmin mostly expressed in stromal cells, such as endothelial cells, fibroblasts and the epithelial cells of tissues in contact with the environment. IL-33 (before known as NF-HEV) localizes to the nucleus thanks to a signal present at its N-terminal end (Figure 3).188, 189 Because of its nuclear location and its release upon the loss of cell integrity, IL-33 was proposed to be another dual-function alarmin in line with HMGB1 and IL-1α.190 However, unlike HMGB1 and IL-1α, nuclear IL-33 represses gene expression by facilitating chromatin compaction.191 Furthermore, in addition to repressing gene expression, IL-33 nuclear location was recently shown to act as a strong regulator of IL-33 extracellular function.4 Indeed, in chimeric mice expressing a N-terminally truncated IL-33, the exclusion of IL-33 from the nucleus induced lethal inflammation, suggesting that its nuclear location acts as a sink, preventing its uncontrolled extracellular release. This IL-33 nuclear ‘docking’ is even more important since its release is independent of prior processing either by caspase-1, caspase-8 (required for IL-1β or IL-18) or calpain (required for IL-1α).192, 193 In fact, processing by capsase-1 or apoptotic caspases (caspase-3 and -7) occurs inside the IL-1-like domain, after asparagine 178, and was reported to inactivate the alarmin function of IL-33.194, 195 Inactivation of IL-33 by caspases led at first to the consensus that full-length IL-33 is the active form (Figure 3). Yet, it was since proposed that IL-33 is cleaved to a mature and more proinflammatory form.196 In fact, in vitro studies demonstrated the cleavage of IL-33 by neutrophil elastase and cathepsin G. The resulting C-terminal cleavage products, containing the entire IL-1-like domain, had stronger proinflammatory activity (Figure 3). However, it remains unclear whether IL-33 processing is physiologically important, and deeper study of the nature of released IL-33 in a pathological environment is required. Once released, IL-33 binds to a member of the IL-1R family, ST2 (also known as IL-1RL1),197 through which IL-33 can activate both innate and adaptive immune cells. Indeed, ST2 is particularly expressed in lymphocytes, such as T helper 2 (Th2) cells and mast cells.198, 199

Extracellular role of interleukin (IL)-33. IL-33 is present in the nucleus of most stromal cells of tissues in contact with the environment. In the nucleus, IL-33 represses gene expression by facilitating chromatin compaction. Little is known concerning IL-33 translocation to the cytoplasm (1). IL-33 lacks a secretion signal and, therefore, active release may require packaging in noncanonical secretory vesicles (2). Processing by caspases inhibits the proinflammatory function of IL-33 (3). It is therefore likely that IL-33 is released in its full-length form either actively or passively following cell damage (4). However, in the extracellular space, cleavage of IL-33 by neutrophil cathepsin G or elastase promotes its proinflammatory activity (5). Both full-length and cleaved IL-33 interact with the ST2 receptor and further recruit the accessory receptor, IL-1 receptor-3 (IL-1R3), to trigger proinflammatory signals or T helper 2 (Th2)-type cell maturation and response (6). Interestingly, IL-33 extracellular function is regulated in time and space by rapidly occurring oxidation that inhibits its binding to ST2 (7).
Further amplifying the similarity of IL-33 with other alarmins, a recent study demonstrated that post-translational modification of IL-33 is an important regulator of IL-33 inflammatory function.200 However, unlike HMGB1, the oxidation of IL-33, occurring rapidly in the extracellular space, proved to be a crucial downregulator of IL-33 function (Figure 3). Such a mechanism would limit IL-33 in time and space, restricting its activity to the local environment. Interestingly, this study also showed that all IL-1 family members (except IL-1α and IL-36β) are subject to oxidation, thus revealing a potential common regulatory mechanism.
IL-33 downstream signaling
Similar to IL-1/IL-1R1 signaling, upon binding of IL-33, ST2 activation requires the accessory receptor IL-1R3 to function.198, 199 Upon activation, ST2/IL-1R3 trigger signal transduction through a MyD88-IRAK-dependent pathway, leading to NF-κB, c-Jun N-terminal kinase (JNK) and MAPK activation.197 Thus, in mast cells, activation of NF-κB leads to the production of proinflammatory cytokines, such as IL-1β, IL-6 and TNFα.193, 201, 202 However, in mast cells or T cells, production of IL-5, IL-13 and the chemokines CCL5, CCL17 and CCL24 depends on the activation of MAPK signaling rather than NF-κB. Importantly, IL-33 activates naive T cells and promotes their maturation toward a Th2 phenotype, leading to the release of Th2-type cytokines and chemokines.203, 204
Interestingly, similar to RAGE, several ST2 splicing variants exist, producing either full-length, membrane-bound ST2 or a soluble protein thought to act as decoy receptor (sST2).205 For example, the use of recombinant soluble ST2 antagonizes the effect of IL-33 in allergic airway inflammation.206 In fact, soluble ST2 can bind to extracellular IL-33, thereby inhibiting its interaction with membrane-bound and signaling-competent ST2 (Figure 3).
IL-33 in infection and sepsis
Depending on the context, IL-33 was found to exert either deleterious or protective effects upon parasitic or viral infection by driving Th2-type responses. The role for IL-33 in parasitic infection was first suggested by the finding that upon infection of mice with Leishmania major, treatment with ST2-specific blocking antibodies ameliorated the disease, decreased parasite replication and switched the T cells toward a protective Th1 response.207 In a similar manner, following infection with respiratory syncytial virus, treatment with ST2-blocking antibodies reduced lung inflammation and the disease severity in mice primed for Th2-type but not Th1-type responses.208 In contrast, in the context of Trichuris muris infection, IL-33 expression was enhanced in resistant mice. After infection with T. muris, treatment with recombinant IL-33 conferred resistance against the infection and promoted a Th2 response (IL-4, IL-13 and IL-9) whereas it suppressed Th1- and Th17-type cytokine expression (IFNγ, TNFα and IL-6).209
In the context of systemic inflammation, such as that occurring during sepsis, IL-33 was found to be beneficial.210 Indeed, in the murine CLP model, extracellular IL-33 inhibited the expression of G protein-coupled receptor kinase-2 (GRK2) and thereby blocked the TLR4-dependent downregulation of the chemokine receptor CXCR2 in neutrophils. This fostered neutrophil recruitment and microbial clearance at the local site of infection. Interestingly, although IL-33 did not affect the local release of proinflammatory cytokines, systemic IL-33 treatment reduced the concentration of blood and lung cytokines, thereby limiting the systemic inflammation induced by sepsis.
IL-33 and chronic inflammatory diseases
In the lung, epithelial cells are the main source of IL-33. Indeed, upon inflammatory stress, epithelial cells can be damaged and release IL-33 into the extracellular space. For example, IL-33 was shown to be deleterious during the development of asthma. In fact, the levels of IL-33 are higher in asthma patients than in normal individuals,211, 212 and the same is true in a mouse model of ovalbumin (OVA)-elicited inflammation.213 Interestingly, when treated with blocking antibodies specific for ST2 or IL-33 at the time of OVA stimulation, mice presented diminished eosinophil infiltration, less inflammation and reduced airway hyperresponsiveness (AHR).214, 215
The role for IL-33 in the development of exacerbated lung inflammation was recently further characterized in the context of chronic obstructive pulmonary disease (COPD) and viral infection-induced development of AHR.216 Cigarette smoke is considered one of the most important causes of COPD development. Moreover, in COPD patients, viral infection is often associated with acute exacerbation and AHR.217, 218, 219 Kearley et al showed that cigarette smoke induces the increased expression of IL-33 in epithelial cells.216 Upon tissue damage induced by viral infection, this increased supply of IL-33 is released and exacerbates inflammation. Interestingly, the authors also demonstrated that cigarette smoke induced a shift in the type of immune response by controlling the expression of ST2 and IL-1R3. Indeed, cigarette smoke induced a dramatic decrease in ST2/IL-1R3 expression in Th2 T cells and type 2 innate lymphoid cells, thereby reducing the release of Th2-related cytokines (i.e., IL-5 and IL-13) by these cells. In parallel, cigarette smoke induced an increase in ST2/IL-1R3 expression in macrophages and NK cells that led to an increase in the secretion of TNFα, IL-12 and IFNγ. This could therefore explain the exacerbated inflammation found in COPD patients.
Like HMGB1 and IL-1α, IL-33 was shown to be involved in other chronic inflammatory settings, such as atherosclerosis and arthritic joint inflammation. Indeed, recombinant IL-33 treatment of high-fat-diet ApoE−/− mice, an animal model of atherosclerosis, induced increased blood concentrations of IL-4, IL-5 and IL-13, whereas it decreased the level of IFNγ.220 Hence, in these settings, IL-33 switched the immune response from a Th1-type to a protective Th2-type response. In a similar manner, IL-33 and ST2 proved to have protective effects in the context of obesity.221 In fact, treatment of obese mice (ob/ob) with IL-33 increased the ST2-dependent levels of Th2 cytokines in the blood and adipose tissue and improved the metabolic parameters of these mice.
In opposition with its role in atherosclerosis, IL-33 was previously found to be deleterious in the context of arthritic joint inflammation.222, 223 In fact, a dramatic increase in the synovial and serum levels of IL-33 was detected in patients with active RA.224 Moreover, when immunized with collagen, ST2-deficient mice developed strongly reduced arthritis when compared with WT mice. Treatment with recombinant IL-33 increased synovial inflammation and the secretion of proinflammatory cytokines by mast cells.222 As expected, treatment of collagen-immunized mice with ST2-specific blocking antibodies reduced the development of arthritic joint inflammation.223
Together, the central position of IL-33 at the junction between the innate and adaptive immune responses makes it an important modulator of inflammation in a broad array of both acute and chronic inflammatory diseases. However, similarly to HMGB1 and IL-1α, IL-33 may prove to be a challenging drug target because of its variable effects in distinct inflammatory contexts.
S100
The S100 family of proteins is composed of 25 members that exert a large variety of intracellular and/or extracellular functions. Inside cells, S100 proteins regulate cell proliferation, differentiation, migration, energy metabolism, Ca2+ homeostasis, inflammation and cell death. Once released into the extracellular space, particular S100 proteins act as alarmins through their interaction with different receptors, orchestrating both innate and adaptive immune responses, cell migration and chemotaxis as well as tissue development and repair.225, 226, 227
S100 protein expression and intracellular functions
S100 proteins are part of the calcium homeostasis machinery that has a crucial role in maintaining sufficient concentrations of intracellular Ca2+ for cell metabolism yet restricting excessive storage that could lead to Ca2+ precipitation or overwhelm the cell metabolism (Figure 4). The expression of the different S100 proteins is cell dependent and is regulated by specific growth factors, cytokines or activation of cell surface PRRs.225, 227 Most S100 proteins are of rather small size (⩽14 kDa) and are encoded by genes clustered on chromosome 1q21.228 One interesting exception to this rule is S100B, the coding gene of which is located on chromosome 21q22.3 and is found to be overexpressed in patients with Down syndrome.229, 230 S100B is also unique in the fact that its expression has been associated with cells of neural crest origin, and it is now considered one of the best biomarkers of melanoma.231, 232

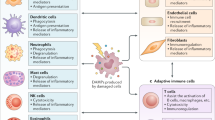
Role of S100 protein in cell housekeeping and inflammation. S100 proteins are ubiquitously expressed in all cells and are crucial regulators of the calcium homeostasis machinery (1). The intracellular functions of S100 proteins also extend to specific cell functions such as transcriptional regulation and DNA repair (2), membrane protein recruitment and trafficking (3) and cytoskeleton assembly (4). The exact mechanisms that regulate the release of S100 proteins remain unclear thus far. Nevertheless, the secretion of S100 proteins occurs either passively upon cell damage or actively following cell activation (5). Once released into the extracellular space, S100 proteins interact with several receptors, most importantly RAGE and TLR4 (6). Upon binding to their receptors, S100 proteins trigger proinflammatory pathways promoting cell migration, proliferation and differentiation (7). S100 protein-induced signaling pathways also lead to the expression of MMPs and CAMs, thereby promoting chemotaxis and tissue remodeling (8). Certain S100 proteins, such as S100A8/A9, are extremely sensitive to oxidation (9). Their redox state acts as a molecular switch from proinflammatory function to protective wound-healing and antioxidant function. In return, oxidation-sensitive S100 proteins are believed to act as scavengers of ROS and NO and thereby prevent oxidative stress. CAM, cell adhesion molecule; MMP, matrix metalloproteinase; NO, nitric oxide; RAGE, receptor for advanced glycation end-products; ROS, reactive oxygen species; TLR4, Toll-like receptor 4.
S100 proteins exert their intracellular functions in the nucleus or cytoplasm by interacting with other target molecules, such as enzymes, cytoskeletal proteins, receptors, transcription factors and nucleic acids.227 These interactions are often a critical step for Ca2+ binding and are therefore integral parts of calcium homeostasis. Another critical step for S100 protein activation is the formation of homo- or heterodimers. Binding of ions (Ca2+, Zn2+ or Cu2+) occurs via interactions with the S100 EF-hand domain, a conserved helix-loop-helix motif found in all calcium-binding proteins. Each S100 protein present in the dimer contributes to ion binding as well as to the interaction with the target molecules.233
In healthy conditions, the functions of S100 proteins are extremely diverse and not only relate to the management of calcium storage and shuffling but also extend to specific cell functions, such as scavenging of ROS and nitric oxide (i.e., S100A8/A9), cytoskeleton assembly (i.e., S100A1, S100A4, S100A6, S100A9), membrane protein recruitment and trafficking (i.e., S100A10, S100A12), transcriptional regulation and DNA repair (i.e., S100A4, S100A11, S100A14, S100B), cell differentiation (i.e., S100A6, S100A8-A9, S100B), release of cytokines and antimicrobial agents (degranulation) (i.e., S100A8-A9, S100A12, S100A13), muscle cell contractility (i.e., S100A1), cell growth and migration (i.e., S100A4, S100A8-A9, S100B, S100P) and apoptosis (i.e., S100A6, S100A9, S100B) (Figure 4). Because of space constraints, we will not discuss the physiological roles of S100 proteins in additional detail. However, others have described the biological mechanisms involving S100 proteins.225, 226, 228, 234, 235, 236
S100 protein secretion, receptor interaction and post-translational modifications
S100 proteins have been detected in the extracellular space and certain body fluids, such as serum, urine, sputum, cerebrospinal fluid and feces, where they are generally associated with a state of disease.237, 238, 239, 240 S100A8/A9, S100A12 and S100B are even considered biomarkers of specific diseases, such as cancer, atherosclerosis and stroke.231, 241, 242, 243, 244, 245
Like all alarmins previously described, S100 proteins lack a secretion leader sequence and therefore cannot be secreted through the classical ER–Golgi secretion pathway. The exact mechanisms that regulate the release of S100 proteins remain unclear thus far. Nevertheless, the secretion of S100 proteins occurs either passively upon cell necrosis or actively following cell activation (Figure 4). In fact, S100A8/A9 and S100A12 (also called calgranulins) were found to be released actively upon myeloid cell activation and subsequent tubulin-dependent translocation to the plasma membrane.245, 246, 247, 248 S100A8/A9 were also found to be part of neutrophil extracellular traps, a mesh-like agglomerate composed of proteins and nucleic acids that are released as antimicrobial weapons by activated neutrophils.249 Similarly, S100B is released upon brain, lung or muscle tissue damage.250, 251, 252, 253
Once released to the extracellular space, S100 proteins trigger immune cell activation through binding to different cell surface receptors, thereby mediating different effects depending on the S100 protein and the target cell. Similar to HMGB1, RAGE is considered the most important cell surface receptor for S100 proteins (Figure 4). S100A12 was the first S100 protein for which binding to RAGE was characterized,254 and this interaction is greatly increased by S100A12 capture of Ca2+ or Zn2+ ions.255, 256 RAGE N-glycosylation also appears to play an important role for the recruitment of several S100 proteins and is believed to promote receptor clustering.257, 258 Like S100A12, S100B binds to the RAGE V-domain and promotes RAGE multimerization and subsequent activation, thus promoting cell survival.259 In contrast, S100A6 is one of the rare RAGE ligands shown to interact with the RAGE C2-domain and was shown to have more of a proapoptotic effect.227, 260 Interestingly, at the surface of myoblasts, S100B was shown to interact with the basic fibroblast growth factor and its receptor, fibroblast growth factor receptor 1, leading to the recruitment and subsequent inactivation of RAGE.250 The S100A8/A9 heterodimer has also been proposed to bind to RAGE, triggering proinflammatory signaling and cell migration.261, 262 Yet, the binding is rather weak263 and seems to rely on S100A9 and the presence of Ca2+ or Zn2+ ions. In fact, an extracellular function of S100A8/A9 has been associated with the activation of other receptors, and this has been the subject of controversy. Indeed, the S100A8/A9 heterocomplex was first believed not to bind to TLR4, and it was rather the S100A8 homodimer that was thought to bind to TLR4.264 More contradictory results came from another study that concluded that S100A9 bound better to TLR4-MD2 and that this was enhanced by the capture of Ca2+ and Zn2+.263 Recent data reinforced the potential role of S100A8 in the activation of TLR4 in monocytes.265 Treatment of mouse and human monocytes with S100A8 closely resembled LPS stimulation on the transcript level. TLR4 gene knockdown or steric inhibition studies demonstrated the proinflammatory effect of S100A8 stimulation to be TLR4 dependent.265 Furthermore, other receptors have been implicated in sensing S100 proteins, and these include G protein-coupled receptors,266, 267 scavenger receptors 268, 269 and cell surface heparan sulfate proteoglycans.270
It is important to note that similar to HMGB1 and IL-33, some S100 protein functions can be modulated by post-translational modifications, such as oxidation.271, 272, 273 For example, S100A8/A9 are very sensitive to oxidation, and their redox state acts as a molecular switch from a proinflammatory function (reduced) to a protective wound-healing and antioxidant function (oxidized).271 In contrast, oxidation of S100B was shown to be necessary for binding to RAGE and the subsequent increase in the expression of the angiogenic factor VEGF, an important player in the development of macular degeneration.273
S100 proteins at the onset of acute inflammation
Upon release following cell death and tissue damage, several S100 proteins are passively released and act as DAMPs, signaling mainly through RAGE and TLR4, and thus contribute to the regulation of post-traumatic injury and inflammation following myocardial infarction, stroke or brain trauma.
For example, S100A1 is the most highly expressed S100 protein in cardiomyocytes and was shown to be released upon cell damage following ischemia–reperfusion injury associated with myocardial infarction.274, 275 Once released, S100A1 activates the surrounding cardiac fibroblasts through TLR4 and triggers the MAPK and NF-κB proinflammatory pathways.274 However, S100A1 not only promotes proinflammatory signals but also regulates a complex balance between pro- and anti-inflammatory pathways associated with the antifibrotic upregulation of matrix metalloproteinase 9 (MMP9) and the downregulation of type I collagen and connective tissue growth factor. Moreover, S100A1-blocking antibodies increased fibrosis and infarction size, thus resulting in greater myocardial dysfunction.274 Hence, S100A1 plays a beneficial role following heart injury, promoting muscle tissue repair and maintaining contractility. Similarly, S100A4 release following myocardial infarction was shown to be protective by promoting SMC motility, proliferation and differentiation.276 In stark contrast with S100A1 and S100A4, in a mouse model of angiotensin-induced cardiac damage, the S100A8/A9 heterodimer was shown to be released by granulocytes that infiltrated the muscle tissue.277 Once released, S100A8/A9 signaled through RAGE expressed at the surface of cardiac fibroblasts and thereby upregulated proinflammatory gene expression, inducing the critical release of cytokines and chemokines. In this model, the use of blocking S100A9 antibodies reduced the accumulation of fibroblasts, thus decreasing fibrosis while diminishing local inflammation. These data therefore indicate that the damage-associated release of S100A8/A9 has the opposite effect of S100A1 or S100A4 in that it promotes myocardial tissue inflammation and fibrotic scar formation. Similarly, following myocardial infarction, S100B can be released in high dose from injured cardiomyocytes and, through RAGE, further promote cell apoptosis,278 whereas in other contexts, RAGE promotes autophagy rather than apoptosis.279 S100B also promotes the RAGE- and NF-κB-dependent secretion of VEGF from cardiomyocytes, thereby engaging myofibroblast proliferation and scar formation.280
Much attention has also been paid to the role of S100B in the central nervous system (CNS). In the brain environment, S100B function seems to be regulated by its concentration. Indeed, at low doses, S100B promotes neuron survival. However, in higher doses, S100B triggers apoptosis and is therefore associated with brain damage and neurodegeneration. In fact, S100B is released after stroke or following surgery and has been used as a marker to measure the extent of CNS damage and predict clinical outcome following brain injury.242
S100 proteins and chronic inflammation
One example of the involvement of S100 proteins in the establishment of long-term chronic inflammatory disease is found in atherosclerosis. Indeed, while studying the influence of RAGE in the development of atherosclerosis in the ApoE−/− mouse model, Harja et al.112 found the critical contribution of the S100B–RAGE axis in vascular inflammation and endothelial cell dysfunction during atherosclerotic plaque development. RAGE expression by endothelial cells was found to increase the expression of adhesion molecules (VCAM-1), cytokines and chemokines as well as MMP2 in the aortic tissue, thereby promoting the recruitment of immune cells to the forming plaque. The in vitro stimulation of primary endothelial cells with S100B was shown to replicate these findings and to trigger the activation of the extracellular signal-regulated protein kinase-1/2 (ERK1/2) and JNK proinflammatory pathway. In addition to S100B, serum S100A12 levels are increased in patients with coronary artery disease after plaque rupture.281 Thus, S100A12 is now being considered a biomarker of coronary artery disease.244 In this circumstance, S100A12 inhibits MMP2, 3 and 9 in atherosclerotic plaques.281 Moreover, the presence of S100A12 promotes remodeling and calcification of atherosclerotic plaques in a RAGE-dependent manner in transgenic ApoE−/− mice expressing human S100A12 in SMCs.282, 283 Plaque calcification is believed to be partly mediated by SMCs that, under oxidative conditions, undergo an S100A12/RAGE-dependent procalcific phenotypic switch to osteoblast-like cells.283, 284 Similarly, S100A8/A9 are found in abundance in macrophages and foam cells present in human atherosclerotic plaques, where they were shown to promote dystrophic calcification.285, 286 However, the oxidation-sensitive nature of S100A8/A9 present in high amounts in atherosclerotic plaques might mean that the complex acts as an oxidant scavenger and thus promotes wound healing and protects other cell components against damaging oxidation.271
In correlation with their role in atherosclerosis development, S100A8/A9 were shown to drive the RAGE-dependent and hyperglycemia-induced increase of myelopoiesis occurring in diabetic mice.287 Indeed, in diabetic mice (streptozotocin-elicited or from an Ins2Akita background), the release of S100A8/A9 by activated neutrophils induced the RAGE-dependent proliferation of granulocyte and macrophage progenitor cells in the bone marrow. This increased the total quantity of monocytes and neutrophils infiltrating atherosclerotic lesions and thus impaired plaque regression. Interestingly, antidiabetic treatment normalizing the glycemia of Ldlr−/− atherosclerotic mice was sufficient to decrease the number of circulating monocytes and neutrophils and thereby reduced the atherosclerotic lesions. Most importantly, an increase in the serum white blood cell count correlated with a higher S100A8/A9 concentration in patients with coronary artery disease.287 In accordance with these data, increased serum levels of S100A8/A9 were found in obese patients and were linked to the increased expression of the macrophage marker CD68 in the visceral adipose tissue.288, 289 However, S100A8/A9 serum levels were found to be reduced to normal levels following weight loss. These results, together with the previously mentioned studies linking S100A8/A9 to atherosclerosis development, may partly explain the prevalence of atherosclerosis associated with obesity and diabetes. In addition, blood levels of S100A12 have been found to be increased in diabetic patients and to correlate with a higher risk of cardiovascular disease development.244
Further implicating S100 proteins in the development of chronic inflammatory diseases, calgranulins (S100A8/A9 and S100A12) were shown to be involved in the joint inflammation associated with RA.290 S100A8/A9 are highly upregulated in macrophages and neutrophils from patients with juvenile rheumatoid arthritis, and S100A8/A9 titers in the serum and synovial fluid correlates with disease severity.247, 291 The S100A8/A9 and S100A12 synovial fluid protein levels were found to specifically correlate with RA and were proposed as specific markers to differentiate RA from other forms of inflammatory arthritis.292, 293 In this context, S100A8/A9 were found to originate from monocytes, which release S100A8/A9 upon interaction with activated endothelial cells, thereby enhancing the inflammatory environment.247 Extracellular stimulation of human monocytes with S100A8/A9 was later shown to trigger NF-κB activation with subsequent release of proinflammatory cytokines.294 In addition to their monocytic origin, S100A8/A9 expression was found to be increased in chondrocytes present in the inflamed joints of IL-1Ra-deficient mice that rapidly develop arthritis-like symptoms. In this context, stimulation of chondrocytes with extracellular S100A8 activated the NF-κB pathway and the subsequent release of cytokines and several MMPs,295 and this stimulatory activity was subsequently attributed to signaling through TLR4.296 It is also interesting to note that RAGE expression increases in the joints of arthritic mice.297 Indeed, an Ager gene polymorphism coding for the G82S mutation was shown to increase the binding of S100A12 in vitro and promoted S100A12-induced inflammation.297 This is even more interesting because this Ager G82S polymorphism has a higher prevalence in RA patients.297 Local intra-articular corticosteroid treatment of RA patients induced reduced concentrations of serum and synovial S100A12, further linking calgranulins to the arthritic joint inflammatory environment.293 Together, these studies demonstrate the central role of S100A8/A9 and S100A12 in the establishment of the inflammatory microenvironment present in the arthritic joints and the metalloproteinase-dependent destruction of cartilage. It is also worth noting that the synovial fluid level of calgranulins found in active arthritic joints is higher than the serum levels, further supporting the idea of an intra-articular origin of calgranulins (infiltrating macrophages and granulocytes or chondrocytes).247, 294, 298 However, the exact role of TLR4 or RAGE in this context is not yet entirely clear.
In addition to calgranulins, both S100A4 and S100B were found to activate the ERK1/2 and NF-κB proinflammatory pathways through RAGE expressed at the surface of chondrocytes, thereby triggering an increase in MMP13 expression that could potentially contribute to arthritic cartilage destruction.299, 300
Notably, several S100 proteins have been linked to the development of skin lesions in psoriasis. Indeed, S100A8/A9 levels were shown to be elevated in psoriatic skin lesions.301, 302 Stimulation of normal keratinocytes with S100A8/A9 induced the secretion of proinflammatory cytokines and chemokines that in turn promoted S100A8/A9 expression in an autocrine-positive feedback loop,303 thus mimicking the proinflammatory environment found in psoriatic lesions. Furthermore, S100A7 and S100A15 were shown to promote the chemoattraction of monocytes, neutrophils and CD4+ T cells and trigger the production of proinflammatory cytokines in the context of psoriatic skin lesions.304, 305, 306 However, only S100A7 required RAGE for its chemotactic activity,304 underlining potential differences in the role of S100A7 and S100A15 in psoriasis.
S100 proteins and cancer
Like all of the alarmins described above, several S100 proteins have been identified as central mediators for the development of a multitude of cancers and have been shown to contribute to tumor cell proliferation, metastasis, angiogenesis and immune evasion.307, 308, 309 The role of S100 proteins in cancer is considered to be type and stage specific, and each type of cancer seems to exhibit a different S100 protein profile.307 Several groups recently reviewed the intricate role of S100 proteins in cancer in great detail.307, 308, 309
CONCLUDING REMARKS
A large body of evidence suggests that several proteins hidden inside cells have both intracellular roles and extracellular functions that contribute to the rapid recruitment and response of the immune system to infection and damage. During chronic damage and metabolic perturbations, these proteins are integral to complex proinflammatory events with shared regulatory mechanisms that result in either tissue repair or tissue damage and organ dysfunction. The pharmacological targeting of such DAMPs could therefore be of benefit. However, alarmins have intricate local and systemic roles which depend on their post-translational modification status, differential receptor engagement and the cell-type affected. Thus, targeting alarmins remains challenging.
References
Janeway CA . Pillars article: approaching the asymptote? Evolution and revolution in immunology. Cold spring harb symp quant boil. 1989. 54: 1–13. J Immunol 2013; 191: 4475–4487.
Matzinger P . Tolerance, danger, and the extended family. Annu Rev Immunol 1994; 12: 991–1045.
Seong S-Y, Matzinger P . Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 2004; 4: 469–478.
Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun 2014; 55: 33–41.
Agresti A, Bianchi ME . HMGB proteins and gene expression. Curr Opin Genet Dev 2003; 13: 170–178.
Stros M . HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta 2010; 1799: 101–113.
Stott K, Watson M, Howe FS, Grossmann JG, Thomas JO . Tail-mediated collapse of HMGB1 is dynamic and occurs via differential binding of the acidic tail to the A and B domains. J Mol Biol 2010; 403: 706–722.
Ito I, Fukazawa J, Yoshida M . Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem 2007; 282: 16336–16344.
Oh YJ, Youn JH, Ji Y, Lee SE, Lim KJ, Choi JE et al. HMGB1 is phosphorylated by classical protein kinase C and is secreted by a calcium-dependent mechanism. J Immunol 2009; 182: 5800–5809.
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003; 22: 5551–5560.
Youn JH, Shin J-S . Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol 2006; 177: 7889–7897.
Lu B, Antoine DJ, Kwan K, Lundbäck P, Wähämaa H, Schierbeck H et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci USA 2014; 111: 3068–3073.
Tang D, Kang R, Zeh HJ, Lotze MT . High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal 2011; 14: 1315–1335.
Yang H, Lundbäck P, Ottosson L, Erlandsson-Harris H, Vénéreau E, Bianchi ME et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 2012; 18: 250–259.
Vénéreau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 2012; 209: 1519–1528.
Kazama H, Ricci J-E, Herndon JM, Hoppe G, Green DR, Ferguson TA . Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008; 29: 21–32.
Lotze MT, Tracey KJ . High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005; 5: 331–342.
Andersson U, Tracey KJ 2011 HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29: 139–162.
Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A . HMGB1: endogenous danger signaling. Mol Med 2008; 14: 476–484.
Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ . HMG-1 as a mediator of acute lung inflammation. J Immunol 2000; 165: 2950–2954.
Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell Biol 2001; 152: 1197–1206.
Palumbo R, Sampaolesi M, De Marchis F, Tonlorenzi R, Colombetti S, Mondino A et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol 2004; 164: 441–449.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195.
Bell CW, Jiang W, Reich CF, Pisetsky DS . The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 2006; 291: C1318–C1325.
Jiang W, Bell CW, Pisetsky DS . The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J Immunol 2007; 178: 6495–6503.
Qin S, Wang H, Yuan R, Li H, Ochani M, Ochani K et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med 2006; 203: 1637–1642.
Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT-H, Taxman DJ et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 2009; 183: 2008–2015.
Lu B, Wang H, Andersson U, Tracey KJ . Regulation of HMGB1 release by inflammasomes. Protein Cell 2013; 4: 163–167.
Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999; 285: 248–251.
Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol 2003; 170: 3890–3897.
Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD et al2010 Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185: 4385–4392.
Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 2002; 3: 995–1001.
Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V . Secretory autophagy. Curr Opin Cell Biol 2015; 35: 106–116.
Deretic V, Saitoh T, Akira S Autophagy in infection, inflammation and immunity Nat Rev Immunol 2013; 13: 722–737.
Tang D, Kang R, Livesey KM, Cheh C-W, Farkas A, Loughran P et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190: 881–892.
Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA 2010; 107: 11942–11947.
Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med 2015; 212: 5–14.
Youn JH, Oh YJ, Kim ES, Choi JE, Shin J-S . High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J Immunol 2008; 180: 5067–5074.
Chen G-Y, Tang J, Zheng P, Liu Y . CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 2009; 323: 1722–1725.
Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem 1995; 270: 25752–25761.
Rauvala H, Merenmies J, Pihlaskari R, Korkolainen M, Huhtala ML, Panula P . The adhesive and neurite-promoting molecule p30: analysis of the amino-terminal sequence and production of antipeptide antibodies that detect p30 at the surface of neuroblastoma cells and of brain neurons. J Cell Biol 1988; 107: 2293–2305.
Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P . The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol 2007; 81: 84–91.
Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol 2005; 174: 7506–7515.
Manfredi AA, Capobianco A, Esposito A, De Cobelli F, Canu T, Monno A et al. Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J Immunol 2008; 180: 2270–2275.
Abeyama K, Stern DM, Ito Y, Kawahara K-I, Yoshimoto Y, Tanaka M et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest 2005; 115: 1267–1274.
Treutiger CJ, Mullins GE, Johansson A-SM, Rouhiainen A, Rauvala HME, Erlandsson-Harris H et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med 2003; 254: 375–385.
Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003; 101: 2652–2660.
Sha Y, Zmijewski J, Xu Z, Abraham E . HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol 2008; 180: 2531–2537.
Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Vénéreau E et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med 2012; 209: 551–563.
Urbonaviciute V, Fürnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 2008; 205: 3007–3018.
Ivanov S, Dragoi A-M, Wang X, Dallacosta C, Louten J, Musco G et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007; 110: 1970–1981.
Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009; 462: 99–103.
Tian J, Avalos AM, Mao S-Y, Chen B, Senthil K, Wu H et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 2007; 8: 487–496.
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 2012; 13: 832–842.
Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H . Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin). J Leukoc Biol 2007; 81: 49–58.
Andersson U, Tracey KJ . HMGB1 in sepsis. Scand J Infect Dis 2003; 35: 577–584.
Nagato M, Okamoto K, Abe Y, Higure A, Yamaguchi K . Recombinant human soluble thrombomodulin decreases the plasma high-mobility group box-1 protein levels, whereas improving the acute liver injury and survival rates in experimental endotoxemia. Crit Care Med 2009; 37: 2181–2186.
Yanai H, Matsuda A, An J, Koshiba R, Nishio J, Negishi H et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci USA 2013; 110: 20699–20704.
Yamato M, Minematsu Y, Fujii J, Mori K, Minato T, Miyagawa S et al. Effective combination therapy of polymyxin-B direct hemoperfusion and recombinant thrombomodulin for septic shock accompanied by disseminated intravascular coagulation: a historical controlled trial. Ther Apher Dial 2013; 17: 472–476.
Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA 2004; 101: 296–301.
Lutterloh EC, Opal SM, Pittman DD, Keith JC, Tan X-Y, Clancy BM et al. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit Care 2007; 11: R122.
Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 2004; 113: 1641–1650.
Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 2005; 201: 1135–1143.
Tsung A, Zheng N, Jeyabalan G, Izuishi K, Klune JR, Geller DA et al. Increasing numbers of hepatic dendritic cells promote HMGB1-mediated ischemia-reperfusion injury. J Leukoc Biol 2007; 81: 119–128.
Krüger B, Krick S, Dhillon N, Lerner SM, Ames S, Bromberg JS et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci USA 2009; 106: 3390–3395.
Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 2008; 117: 3216–3226.
Goldstein RS, Gallowitsch-Puerta M, Yang L, Rosas-Ballina M, Huston JM, Czura CJ et al. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock 2006; 25: 571–574.
Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ et al. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci 2008; 28: 12023–12031.
Weber DJ, Gracon ASA, Ripsch MS, Fisher AJ, Cheon BM, Pandya PH et al. The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation. Sci Transl Med 2014; 6: 252ra124.
Nicolls MR, Laubach VE . Traumatic brain injury: lungs in a RAGE. Sci Transl Med 2014; 6: 252fs34.
Sodhi CP, Jia H, Yamaguchi Y, Lu P, Good M, Egan C et al. Intestinal epithelial TLR-4 activation is required for the development of acute lung injury after trauma/hemorrhagic shock via the release of HMGB1 from the gut. J Immunol 2015; 194: 4931–4939.
Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 2007; 21: 3904–3916.
Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med 2006; 12: 105–114.
Kang R, Zhang Q, Zeh HJ, Lotze MT, Tang D . HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013; 19: 4046–4057.
Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A et al. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res 2007; 13: 2836–2848.
Chung HW, Lee S-G, Kim H, Hong DJ, Chung JB, Stroncek D et al. Serum high mobility group box-1 (HMGB1) is closely associated with the clinical and pathologic features of gastric cancer. J Transl Med 2009; 7: 38.
Poser I, Golob M, Buettner R, Bosserhoff AK . Upregulation of HMG1 leads to melanoma inhibitory activity expression in malignant melanoma cells and contributes to their malignancy phenotype. Mol Cell Biol 2003; 23: 2991–2998.
Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH et al. High mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 2012; 55: 1863–1875.
Kang R, Tang D, Schapiro NE, Loux T, Livesey KM, Billiar TR et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014; 33: 567–577.
Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I et al. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res 2012; 72: 3290–3301.
van Beijnum JR, Nowak-Sliwinska P, van den Boezem E, Hautvast P, Buurman WA, Griffioen AW . Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene 2013; 32: 363–374.
Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014; 507: 109–113.
van Beijnum JR, Dings RP, van der Linden E, Zwaans BMM, Ramaekers FCS, Mayo KH et al. Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood 2006; 108: 2339–2348.
Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000; 405: 354–360.
Tang D, Loze MT, Zeh HJ, Kang R . The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy 2010; 6: 1181–1183.
Tang D, Kang R, Cheh C-W, Livesey KM, Liang X, Schapiro NE et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010; 29: 5299–5310.
Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011; 25: 23–31.
Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res 2012; 72: 1996–2005.
Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 2010; 17: 666–676.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007; 13: 1050–1059.
Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev 2007; 220: 47–59.
Kokkola R, Sundberg E, Ulfgren AK, Palmblad K, Li J, Wang H et al. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum 2002; 46: 2598–2603.
Taniguchi N, Kawahara K-I, Yone K, Hashiguchi T, Yamakuchi M, Goto M et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum 2003; 48: 971–981.
Pullerits R, Jonsson I-M, Verdrengh M, Bokarewa M, Andersson U, Erlandsson-Harris H et al. High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum 2003; 48: 1693–1700.
Hamada T, Torikai M, Kuwazuru A, Tanaka M, Horai N, Fukuda T et al. Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Rheum 2008; 58: 2675–2685.
Liu-Bryan R, Terkeltaub R . Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum 2010; 62: 2004–2012.
Kokkola R, Li J, Sundberg E, Aveberger A-C, Palmblad K, Yang H et al. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum 2003; 48: 2052–2058.
van de Wouwer M, Plaisance S, De Vriese A, Waelkens E, Collen D, Persson J et al. The lectin-like domain of thrombomodulin interferes with complement activation and protects against arthritis. J Thromb Haemost 2006; 4: 1813–1824.
Ostberg T, Wähämaa H, Palmblad K, Ito N, Stridh P, Shoshan M et al. Oxaliplatin retains HMGB1 intranuclearly and ameliorates collagen type II-induced arthritis. Arthritis Res Ther 2008; 10: R1.
Zetterström CK, Jiang W, Wähämaa H, Ostberg T, Aveberger A-C, Schierbeck H et al. Pivotal advance: inhibition of HMGB1 nuclear translocation as a mechanism for the anti-rheumatic effects of gold sodium thiomalate. J Leukoc Biol 2008; 83: 31–38.
Popovic K, Ek M, Espinosa A, Padyukov L, Harris HE, Wahren-Herlenius M et al. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum 2005; 52: 3639–3645.
Barkauskaite V, Ek M, Popovic K, Harris HE, Wahren-Herlenius M, Nyberg F . Translocation of the novel cytokine HMGB1 to the cytoplasm and extracellular space coincides with the peak of clinical activity in experimentally UV-induced lesions of cutaneous lupus erythematosus. Lupus 2007; 16: 794–802.
Li J, Xie H, Wen T, Liu H, Zhu W, Chen X . Expression of high mobility group box chromosomal protein 1 and its modulating effects on downstream cytokines in systemic lupus erythematosus. J Rheumatol 2010; 37: 766–775.
Ma C-Y, Jiao Y-L, Zhang J, Yang Q-R, Zhang Z-F, Shen Y-J et al. Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumat Int 2010; 32: 395–402.
Abdulahad DA, Westra J, Bijzet J, Limburg PC, Kallenberg CGM, Bijl M . High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Arthritis Res Ther 2011; 13: R71.
Urbonaviciute V, Voll RE . High-mobility group box 1 represents a potential marker of disease activity and novel therapeutic target in systemic lupus erythematosus. J Intern Med 2011; 270: 309–318.
Porto A, Palumbo R, Pieroni M, Aprigliano G, Chiesa R, Sanvito F et al. Smooth muscle cells in human atherosclerotic plaques secrete and proliferate in response to high mobility group box 1 protein. FASEB J 2006; 20: 2565–2566.
Inoue K, Kawahara K-I, Biswas KK, Ando K, Mitsudo K, Nobuyoshi M et al. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovasc Pathol 2007; 16: 136–143.
Kalinina N, Agrotis A, Antropova Y, DiVitto G, Kanellakis P, Kostolias G et al. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterioscler Thromb Vasc Biol 2004; 24: 2320–2325.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464: 1357–1361.
Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 1995; 96: 1395–1403.
Harja E, Bu D-X, Hudson BI, Chang JS, Shen X, Hallam K et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest 2008; 118: 183–194.
Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Chow WS et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 1998; 4: 1025–1031.
Luo Y, Li S-J, Yang J, Qiu Y-Z, Chen F-P . HMGB1 induces an inflammatory response in endothelial cells via the RAGE-dependent endoplasmic reticulum stress pathway. Biochem Biophys Res Commun 2013; 438: 732–738.
Deegan S, Saveljeva S, Gorman AM, Samali A . Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 2013; 70: 2425–2441.
Ahrens I, Chen Y-C, Topcic D, Bode M, Haenel D, Hagemeyer CE et al. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thromb Haemost 2015; 114: 994–1003.
Rider P, Carmi Y, Voronov E, Apte RN . Interleukin-1α. Semin Immunol 2013; 25: 430–438.
Dinarello CA . Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 2009; 27: 519–550.
Dinarello CA . Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011; 117: 3720–3732.
Aden N, Nuttall A, Shiwen X, de Winter P, Leask A, Black CM et al. Epithelial cells promote fibroblast activation via IL-1alpha in systemic sclerosis. J Invest Dermatol 2010; 130: 2191–2200.
Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S et al. Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut 2014; 63: 598–609.
Kawaguchi Y, Hara M, Wright TM . Endogenous IL-1alpha from systemic sclerosis fibroblasts induces IL-6 and PDGF-A. J Clin Invest 1999; 103: 1253–1260.
Buryskova M, Pospisek M, Grothey A, Simmet T, Burysek L . Intracellular interleukin-1alpha functionally interacts with histone acetyltransferase complexes. J Biol Chem 2004; 279: 4017–4026.
Maier JA, Statuto M, Ragnotti G . Endogenous interleukin 1 alpha must be transported to the nucleus to exert its activity in human endothelial cells. Mol Cell Biol 1994; 14: 1845–1851.
Werman A, Werman-Venkert R, White R, Lee J-K, Werman B, Krelin Y et al. The precursor form of IL-1alpha is an intracrine proinflammatory activator of transcription. Proc Natl Acad Sci USA 2004; 101: 2434–2439.
Pollock AS, Turck J, Lovett DH . The prodomain of interleukin 1alpha interacts with elements of the RNA processing apparatus and induces apoptosis in malignant cells. FASEB J 2003; 17: 203–213.
Zamostna B, Novak J, Vopalensky V, Masek T, Burysek L, Pospisek M . N-terminal domain of nuclear IL-1α shows structural similarity to the C-terminal domain of Snf1 and binds to the HAT/core module of the SAGA complex. PLoS One 2012; 7: e41801.
Lonnemann G, Shapiro L, Engler-Blum G, Müller GA, Koch KM, Dinarello CA . Cytokines in human renal interstitial fibrosis. I. Interleukin-1 is a paracrine growth factor for cultured fibrosis-derived kidney fibroblasts. Kidney Int 1995; 47: 837–844.
Kawaguchi Y, Nishimagi E, Tochimoto A, Kawamoto M, Katsumata Y, Soejima M et al. Intracellular IL-1alpha-binding proteins contribute to biological functions of endogenous IL-1alpha in systemic sclerosis fibroblasts. Proc Natl Acad Sci USA 2006; 103: 14501–14506.
Yin H, Morioka H, Towle CA, Vidal M, Watanabe T, Weissbach L . Evidence that HAX-1 is an interleukin-1 alpha N-terminal binding protein. Cytokine 2001; 15: 122–137.
Kobayashi Y, Yamamoto K, Saido T, Kawasaki H, Oppenheim JJ, Matsushima K . Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc Natl Acad Sci USA 1990; 87: 5548–5552.
Kavita U, Mizel SB . Differential sensitivity of interleukin-1 alpha and -beta precursor proteins to cleavage by calpain, a calcium-dependent protease. J Biol Chem 1995; 270: 27758–27765.
Carruth LM, Demczuk S, Mizel SB . Involvement of a calpain-like protease in the processing of the murine interleukin 1 alpha precursor. J Biol Chem 1991; 266: 12162–12167.
Zheng Y, Humphry M, Maguire JJ, Bennett, MR, Clarke MCH . Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1α, controlling necrosis-induced sterile inflammation. Immunity 2013; 38: 285–295.
Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 1995; 267: 2000–2003.
Keller M, Rüegg A, Werner S, Beer H-D . Active caspase-1 is a regulator of unconventional protein secretion. Cell 2008; 132: 818–831.
Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 2012; 36: 388–400.
Brough D, Le Feuvre RA, Wheeler RD, Solovyova N, Hilfiker S, Rothwell NJ et al. Ca2+ Stores and Ca2+ entry differentially contribute to the release of IL-1 and IL-1 from murine macrophages. J Immunol 2003; 170: 3029–3036.
Mohan SK, Yu C . The IL1alpha-S100A13 heterotetrameric complex structure: a component in the non-classical pathway for interleukin 1alpha secretion. J Biol Chem 2011; 286: 14608–14617.
Mandinova A, Soldi R, Graziani I, Bagala C, Bellum S, Landriscina M et al. S100A13 mediates the copper-dependent stress-induced release of IL-1alpha from both human U937 and murine NIH 3T3 cells. J Cell Sci 2003; 116: 2687–2696.
Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA 2012; 109: E3168–E3176.
Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USA 2010; 107: 2574–2579.
Berda-Haddad Y, Robert S, Salers P, Zekraoui L, Farnarier C, Dinarello CA et al. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1α. Proc Natl Acad Sci USA 2011; 108: 20684–20689.
Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Lüthi AU et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol Cell 2011; 44: 265–278.
Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER . Identification of a membrane-associated interleukin 1 in macrophages. Proc Natl Acad Sci USA 1985; 82: 1204–1208.
Conlon PJ, Grabstein KH, Alpert A, Prickett KS, Hopp TP, Gillis S . Localization of human mononuclear cell interleukin 1. J Immunol 1987; 139: 98–102.
Brody DT, Durum SK . Membrane IL-1: IL-1 alpha precursor binds to the plasma membrane via a lectin-like interaction. J Immunol 1989; 143: 1183–1187.
Kaplanski G, Farnarier C, Kaplanski S, Porat R, Shapiro L, Bongrand P et al. Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood 1994; 84: 4242–4248.
Dower SK, Kronheim SR, Hopp TP, Cantrell M, Deeley M, Gillis S et al. The cell surface receptors for interleukin-1 alpha and interleukin-1 beta are identical. Nature 1986; 324: 266–268.
Weber A, Wasiliew P, Kracht M . Interleukin-1 (IL-1) pathway. Sci Signal 2010; 3: cm1.
Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E et al. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol 2011; 187: 4835–4843.
Eigenbrod T, Park J-H, Harder J, Iwakura Y, Nuñez G . Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol 2008; 181: 8194–8198.
Chen C-J, Kono H, Golenbock D, Reed G, Akira S, Rock KL . Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13: 851–856.
Rider P, Kaplanov I, Romzova M, Bernardis L, Braiman A, Voronov E et al. The transcription of the alarmin cytokine interleukin-1 alpha is controlled by hypoxia inducible factors 1 and 2 alpha in hypoxic cells. Front Immunol 2012; 3: 290.
Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG et al. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol 1987; 139: 1902–1910.
Dinarello CA, Simon A, van der Meer JWM . Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012; 11: 633–652.
Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ . Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood 2010; 115: 3632–3639.
Marquardt L, Ruf A, Mansmann U, Winter R, Schuler M, Buggle F et al. Course of platelet activation markers after ischemic stroke. Stroke 2002; 33: 2570–2574.
Sheremata WA, Jy W, Horstman LL, Ahn YS, Alexander JS, Minagar A . Evidence of platelet activation in multiple sclerosis. J Neuroinflammation 2008; 5: 27.
Karakantza M, Maniatis A, Metallinos CI, Papapetropoulos T, Paschalis C . In vivo platelet activation in ischemic stroke patients. Stroke 2003; 34: e174–e175.
Turner NA, Das A, Warburton P, O'Regan DJ, Ball SG, Porter KE . Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am J Physiol Heart Circ Physiol 2009; 297: H1117–H1127.
Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360: 2426–2437.
Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360: 2438–2444.
Cavalli G, Dinarello CA . Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology (Oxford, England) 2015; 54: 2134–2144.
Graudal NA, Svenson M, Tarp U, Garred P, Jurik A-G, Bendtzen K . Autoantibodies against interleukin 1alpha in rheumatoid arthritis: association with long term radiographic outcome. Ann Rheum Dis 2002; 61: 598–602.
Coleman KM, Gudjonsson JE, Stecher M . Open-label trial of MABp1, a true human monoclonal antibody targeting interleukin 1α, for the treatment of psoriasis. JAMA Dermatol 2015; 151: 555–556.
Sheedy FJ, Moore KJ . IL-1 signaling in atherosclerosis: sibling rivalry. Nat Immunol 2013; 14: 1030–1032.
Spears LD, Razani B, Semenkovich CF . Interleukins and atherosclerosis: a dysfunctional family grows. Cell Metab 2013; 18: 614–616.
Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003; 23: 656–660.
Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein E-deficient mice. Atherosclerosis 2011; 216: 313–320.
Kamari Y, Werman-Venkert R, Shaish A, Werman A, Harari A, Gonen A et al. Differential role and tissue specificity of interleukin-1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis 2007; 195: 31–38.
Kamari Y, Shaish A, Shemesh S, Vax E, Grosskopf I, Dotan S et al. Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1α. Biochem Biophys Res Commun 2011; 405: 197–203.
Freigang S, Ampenberger F, Weiss A, Kanneganti T-D, Iwakura Y, Hersberger M et al. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol 2013; 14: 1045–1053.
Vakkila J, Lotze MT . Inflammation and necrosis promote tumour growth. Nat Rev Immunol 2004; 4: 641–648.
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA . Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013; 13: 759–771.
DeNardo DG, Johansson M, Coussens LM . Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev 2008; 27: 11–18.
Shalapour S, Karin M . Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest 2015; 125: 3347–3355.
Cataisson C, Salcedo R, Hakim S, Moffitt BA, Wright L, Yi M . IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. J Exp Med 2012; 209: 1689–1702.
Qin Y, Ekmekcioglu S, Liu P, Duncan LM, Lizée G, Poindexter N et al. Constitutive aberrant endogenous interleukin-1 facilitates inflammation and growth in human melanoma. Mol Cancer Res 2011; 9: 1537–1550.
Tjomsland V, Spångeus A, Välilä J, Sandström P, Borch K, Druid H et al. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011; 13: 664–675.
Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M et al. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J Immunol 2009; 183: 4705–4714.
Voronov E, Dotan S, Krelin Y, Song X, Elkabets M, Carmi Y et al. Unique versus redundant functions of IL-1α and IL-1β in the tumor microenvironment. Front Immunol 2013; 4: 177.
Hong DS, Hui D, Bruera E, Janku F, Naing A, Falchook GS et al. MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: an open-label, phase 1 dose-escalation and expansion study. Lancet Oncol 2014; 15: 656–666.
Palmer G, Trolliet S, Talabot-Ayer D, Mézin F, Magne D, Gabay C . Pre-interleukin-1alpha expression reduces cell growth and increases interleukin-6 production in SaOS-2 osteosarcoma cells: Differential inhibitory effect of interleukin-1 receptor antagonist (icIL-1Ra1). Cytokine 2005; 31: 153–160.
Song X, Voronov E, Dvorkin T, Fima E, Cagnano E, Benharroch D et al. Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J Immunol 2003; 171: 6448–6456.
Dvorkin T, Song X, Argov S, White RM, Zoller M, Segal S et al. Immune phenomena involved in the in vivo regression of fibrosarcoma cells expressing cell-associated IL-1alpha. J Leukoc Biol 2006; 80: 96–106.
Voronov E, Weinstein Y, Benharroch D, Cagnano E, Ofir R, Dobkin M et al. Antitumor and immunotherapeutic effects of activated invasive T lymphoma cells that display short-term interleukin 1alpha expression. Cancer Res 1999; 59: 1029–1035.
Carriere V, Roussel L, Ortega N, Lacorre D-A, Americh L, Aguilar L et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA 2007; 104: 282–287.
Baekkevold ES, Roussigné M, Yamanaka T, Johansen F-E, Jahnsen FL, Amalric F et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol 2003; 163: 69–79.
Moussion C, Ortega N, Girard J-P . The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel 'alarmin'? PLoS One 2008; 3: e3331.
Roussel L, Erard M, Cayrol C, Girard J-P . Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep 2008; 9: 1006–1012.
Ohno T, Oboki K, Kajiwara N, Morii E, Aozasa K, Flavell RA et al. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J Immunol 2009; 183: 7890–7897.
Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G . Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem 2009; 284: 19420–19426.
Cayrol C, Girard J-P . The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA 2009; 106: 9021–9026.
Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C . Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009; 31: 84–98.
Lefrançais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard J-P et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA 2012; 109: 1673–1678.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005; 23: 479–490.
Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA . IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol 2007; 179: 2551–2555.
Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU . IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci USA 2007; 104: 18660–18665.
Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Comms 2015; 6: 8327.
Moulin D, Donzé O, Talabot-Ayer D, Mézin F, Palmer G, Gabay C . Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007; 40: 216–225.
Allakhverdi Z, Smith DE, Comeau MR, Delespesse G . Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol 2007; 179: 2051–2054.
Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol 2008; 181: 4780–4790.
Komai-Koma M, Xu D, Li Y, McKenzie ANJ, McInnes IB, Liew FY . IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol 2007; 37: 2779–2786.
Iwahana H, Yanagisawa K, Ito-Kosaka A, Kuroiwa K, Tago K, Komatsu N et al. Different promoter usage and multiple transcription initiation sites of the interleukin-1 receptor-related human ST2 gene in UT-7 and TM12 cells. Eur J Biochem 1999; 264: 397–406.
Hayakawa H, Hayakawa M, Kume A, Tominaga S-I . Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem 2007; 282: 26369–26380.
Xu D, Chan WL, Leung BP, Huang FP, Wheeler R, Piedrafita D et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med 1998; 187: 787–794.
Walzl G, Matthews S, Kendall S, Gutierrez-Ramos JC, Coyle AJ, Openshaw PJ et al. Inhibition of T1/ST2 during respiratory syncytial virus infection prevents T helper cell type 2 (Th2)- but not Th1-driven immunopathology. J Exp Med 2001; 193: 785–792.
Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK . IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol 2008; 180: 2443–2449.
Alves-Filho JC, Sônego F, Souto FO, Freitas A, Verri WA, Auxiliadora-Martins M et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med 2010; 16: 708–712.
Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol 2009; 183: 5094–5103.
Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 2009; 183: 6469–6477.
Kearley J, Buckland KF, Mathie SA, Lloyd CM . Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am J Respir Crit Care Med 2009; 179: 772–781.
Coyle AJ, Lloyd C, Tian J, Nguyen T, Erikkson C, Wang L et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med 1999; 190: 895–902.
Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T . Anti-IL-33 antibody treatment inhibits airway inflammation in a murine model of allergic asthma. Biochem Biophys Res Commun 2009; 386: 181–185.
Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015; 42: 566–579.
Sethi S, Murphy TF . Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359: 2355–2365.
Mohan A, Chandra S, Agarwal D, Guleria R, Broor S, Gaur B et al. Prevalence of viral infection detected by PCR and RT-PCR in patients with acute exacerbation of COPD: a systematic review. Respirology 2010; 15: 536–542.
Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T et al. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am J Respir Crit Care Med 2011; 183: 734–742.
Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N et al. IL-33 reduces the development of atherosclerosis. J Exp Med 2008; 205: 339–346.
Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res 2010; 107: 650–658.
Xu D, Jiang H-R, Kewin P, Li Y, Mu R, Fraser AR et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci USA 2008; 105: 10913–10918.
Palmer G, Talabot-Ayer D, Lamacchia C, Toy D, Seemayer CA, Viatte S et al. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum 2009; 60: 738–749.
Matsuyama Y, Okazaki H, Tamemoto H, Kimura H, Kamata Y, Nagatani K et al. Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J Rheumatol 2010; 37: 18–25.
Gross SR, Sin CGT, Barraclough R, Rudland PS . Joining S100 proteins and migration: for better or for worse, in sickness and in health. Cell Mol Life Sci 2014; 71: 1551–1579.
Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ et al. Functions of S100 proteins. Curr Mol Med 2013; 13: 24–57.
Leclerc E, Fritz G, Vetter SW, Heizmann CW . Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta 2009; 1793: 993–1007.
Marenholz I, Heizmann CW, Fritz G . S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun 2004; 322: 1111–1122.
Lu J, Esposito G, Scuderi C, Steardo L, Delli-Bovi LC, Hecht JL et al. S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors. PLoS One 2011; 6: e22126.
Esposito G, Imitola J, Lu J, De Filippis D, Scuderi C, Ganesh VS et al. Genomic and functional profiling of human Down syndrome neural progenitors implicates S100B and aquaporin 4 in cell injury. Hum Mol Genet 2008; 17: 440–457.
Gogas H, Eggermont AMM, Hauschild A, Hersey P, Mohr P, Schadendorf D et al. Biomarkers in melanoma. Ann Oncol 2009; 20: vi8–v13.
Díaz-Lagares A, Alegre E, Arroyo A, González-Cao M, Zudaire ME, Viteri S et al. Evaluation of multiple serum markers in advanced melanoma. Tumour Biol 2011; 32: 1155–1161.
Fritz G, Botelho HM, Morozova-Roche LA, Gomes CM . Natural and amyloid self-assembly of S100 proteins: structural basis of functional diversity. FEBS J 2010; 277: 4578–4590.
Leśniak W, Graczyk-Jarzynka A . The S100 proteins in epidermis: topology and function. Biochim Biophys Acta 2015; 1850: 2563–2572.
Duarte-Costa S, Castro-Ferreira R, Neves JS, Leite-Moreira AF . S100A1: a major player in cardiovascular performance. Physiol Res 2014; 63: 669–681.
Miwa N, Uebi T, Kawamura S . S100-annexin complexes—biology of conditional association. FEBS J 2008; 275: 4945–4955.
Mocellin S, Zavagno G, Nitti D . The prognostic value of serum S100B in patients with cutaneous melanoma: a meta-analysis. Int J Cancer 2008; 123: 2370–2376.
Yang Z, Tao T, Raftery MJ, Youssef P, Di Girolamo N, Geczy CL . Proinflammatory properties of the human S100 protein S100A12. J Leukoc Biol 2001; 69: 986–994.
Foell D, Seeliger S, Vogl T, Koch H-G, Maschek H, Harms E et al. Expression of S100A12 (EN-RAGE) in cystic fibrosis. Thorax 2003; 58: 613–617.
Gazzolo D, Michetti F . Perinatal S100B protein assessment in human unconventional biological fluids: a minireview and new perspectives. Cardiovasc Psychiatry Neurol 2010; 2010: 703563.
Dassan P, Keir G, Brown MM . Criteria for a clinically informative serum biomarker in acute ischaemic stroke: a review of S100B. Cerebrovasc Dis 2009; 27: 295–302.
Bloomfield SM, McKinney J, Smith L, Brisman J . Reliability of S100B in predicting severity of central nervous system injury. Neurocrit Care 2007; 6: 121–138.
Foell D, Frosch M, Sorg C, Roth J . Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin Chim Acta 2004; 344: 37–51.
Oesterle A, Hofmann Bowman MA . S100A12 and the S100/calgranulins: emerging biomarkers for atherosclerosis and possibly therapeutic targets. Arterioscler Thromb Vasc Biol 2015; 35: 2496–2507.
Altwegg LA, Neidhart M, Hersberger M, Müller S, Eberli FR, Corti R et al. Myeloid-related protein 8/14 complex is released by monocytes and granulocytes at the site of coronary occlusion: a novel, early, and sensitive marker of acute coronary syndromes. Eur Heart J 2007; 28: 941–948.
Vogl T, Ludwig S, Goebeler M, Strey A, Thorey IS, Reichelt R et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 2004; 104: 4260–4268.
Frosch M, Strey A, Vogl T, Wulffraat NM, Kuis W, Sunderkötter C et al. Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum 2000; 43: 628–637.
Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C . Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem 1997; 272: 9496–9502.
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 2009; 5: e1000639.
Riuzzi F, Sorci G, Donato R . S100B protein regulates myoblast proliferation and differentiation by activating FGFR1 in a bFGF-dependent manner. J Cell Sci 2011; 124: 2389–2400.
Sorci G, Giovannini G, Riuzzi F, Bonifazi P, Zelante T, Zagarella S et al. The danger signal S100B integrates pathogen- and danger-sensing pathways to restrain inflammation. PLoS Pathog 2011; 7: e1001315.
Schiavi P, Laccarino C, Servadei F . The value of the calcium binding protein S100 in the management of patients with traumatic brain injury. Acta Biomed 2012; 83: 5–20.
Savola O, Pyhtinen J, Leino TK, Siitonen S, Niemelä O, Hillbom M . Effects of head and extracranial injuries on serum protein S100B levels in trauma patients. J Trauma 2004; 56: 1229–1234; discussion 1234.
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 1999; 97: 889–901.
Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A . Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J Biol Chem 2007; 282: 4218–4231.
Moroz OV, Burkitt W, Wittkowski H, He W, Ianoul A, Novitskaya V et al. Both Ca2+ and Zn2+ are essential for S100A12 protein oligomerization and function. BMC Biochem. 2009; 10: 11.
Srikrishna G, Nayak J, Weigle B, Temme A, Foell D, Hazelwood L et al. Carboxylated N-glycans on RAGE promote S100A12 binding and signaling. J Cell Biochem 2010; 110: 645–659.
Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 2008; 29: 2035–2043.
Ostendorp T, Leclerc E, Galichet A, Koch M, Demling N, Weigle B et al. Structural and functional insights into RAGE activation by multimeric S100B. EMBO J 2007; 26: 3868–3878.
Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A . S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem 2007; 282: 31317–31331.
Hermani A, De Servi B, Medunjanin S, Tessier PA, Mayer D . S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res 2006; 312: 184–197.
Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G . S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res 2011; 9: 133–148.
Björk P, Björk A, Vogl T, Stenström M, Liberg D, Olsson A et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol 2009; 7: e97.
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MAD et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 2007; 13: 1042–1049.
Fassl SK, Austermann J, Papantonopoulou O, Riemenschneider M, Xue J, Bertheloot D et al. Transcriptome assessment reveals a dominant role for TLR4 in the activation of human monocytes by the alarmin MRP8. J Immunol 2015; 194: 575–583.
Yan WX, Armishaw C, Goyette J, Yang Z, Cai H, Alewood P et al. Mast cell and monocyte recruitment by S100A12 and its hinge domain. J Biol Chem 2008; 283: 13035–13043.
Cornish CJ, Devery JM, Poronnik P, Lackmann M, Cook DI, Geczy CL . S100 protein CP-10 stimulates myeloid cell chemotaxis without activation. J Cell Physiol 1996; 166: 427–437.
Kerkhoff C, Sorg C, Tandon NN, Nacken W . Interaction of S100A8/S100A9-arachidonic acid complexes with the scavenger receptor CD36 may facilitate fatty acid uptake by endothelial cells. Biochemistry 2001; 40: 241–248.
Hoppmann S, Steinbach J, Pietzsch J . Scavenger receptors are associated with cellular interactions of S100A12 in vitro and in vivo. Int J Biochem Cell Biol 2010; 42: 651–661.
Robinson MJ, Tessier P, Poulsom R, Hogg N . The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem 2002; 277: 3658–3665.
Lim SY, Raftery MJ, Goyette J, Hsu K, Geczy CL . Oxidative modifications of S100 proteins: functional regulation by redox. J Leukoc Biol 2009; 86: 577–587.
Lim SY, Raftery MJ, Geczy CL . Oxidative modifications of DAMPs suppress inflammation: the case for S100A8 and S100A9. Antioxid Redox Signal 2011; 15: 2235–2248.
Ma W, Lee SE, Guo J, Qu W, Hudson BI, Schmidt AM et al. RAGE ligand upregulation of VEGF secretion in ARPE-19 cells. Invest Ophthalmol Vis Sci. 2007; 48: 1355–1361.
Rohde D, Schön C, Boerries M, Didrihsone I, Ritterhoff J, Kubatzky KF et al. S100A1 is released from ischemic cardiomyocytes and signals myocardial damage via Toll-like receptor 4. EMBO Mol Med 2014; 6: 778–794.
Bi H, Yang Y, Huang J, Li Y, Ma C, Cong B . Immunohistochemical detection of S100A1 in the postmortem diagnosis of acute myocardial infarction. Diagn Pathol 2013; 8: 84.
Schneider M, Kostin S, Strøm CC, Aplin M, Lyngbaek S, Theilade J et al. S100A4 is upregulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc Res 2007; 75: 40–50.
Wu Y, Li Y, Zhang C, Wang X. A,Y, Cui W, Li H et al. S100a8/a9 released by CD11b+Gr1+ neutrophils activates cardiac fibroblasts to initiate angiotensin II-induced cardiac inflammation and injury. Hypertension 2014; 63: 1241–1250.
Tsoporis JN, Izhar S, Leong-Poi H, Desjardins JF, Huttunen HJ, Parker TG . S100B interaction with the receptor for advanced glycation end products (RAGE): a novel receptor-mediated mechanism for myocyte apoptosis postinfarction. Circ. Res. 2010; 106: 93–101.
Xie J, Méndez JD, Méndez-Valenzuela V, Aguilar-Hernández MM . Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal 2013; 25: 2185–2197.
Tsoporis JN, Izhar S, Proteau G, Slaughter G, Parker TG . S100B-RAGE dependent VEGF secretion by cardiac myocytes induces myofibroblast proliferation. Journal of Molecular and Cellular Cardiology 2012; 52: 464–473.
Goyette J, Yan WX, Yamen E, Chung YM, Lim SY, Hsu K et al. Pleiotropic roles of S100A12 in coronary atherosclerotic plaque formation and rupture. J Immunol 2009; 183: 593–603.
Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, Kim G et al. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler Thromb Vasc. Biol 2011; 31: 337–344.
Gawdzik J, Mathew L, Kim G, Puri TS, Hofmann Bowman MA . Vascular remodeling and arterial calcification are directly mediated by S100A12 (EN-RAGE) in chronic kidney disease. Am J Nephrol 2011; 33: 250–259.
Burton DGA, Matsubara H, Ikeda K . Pathophysiology of vascular calcification: Pivotal role of cellular senescence in vascular smooth muscle cells. Exp Gerontol 2010; 45: 819–824.
McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H et al. S100A8 and S100A9 in human arterial wall. Implications for atherogenesis. J Biol Chem 2005; 280: 41521–41529.
Farris SD, Hu JH, Krishnan R, Emery I, Chu T, Du L et al. Mechanisms of urokinase plasminogen activator (uPA)-mediated atherosclerosis: role of the uPA receptor and S100A8/A9 proteins. J Biol Chem 2011; 286: 22665–22677.
Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 2013; 17: 695–708.
Mortensen OH, Nielsen AR, Erikstrup C, Plomgaard P, Fischer CP, Krogh-Madsen R et al. Calprotectin—a novel marker of obesity. PLoS One 2009; 4: e7419.
Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V et al. Increased levels of calprotectin in obesity are related to macrophage content: impact on inflammation and effect of weight loss. Mol Med 2011; 17: 1157–1167.
Perera C, McNeil HP, Geczy CL . S100 Calgranulins in inflammatory arthritis. Immunol Cell Biol 2010; 88: 41–49.
Odink K, Cerletti N, Brüggen J, Clerc RG, Tarcsay L, Zwadlo G et al. Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature 1987; 330: 80–82.
Baillet A, Trocmé C, Berthier S, Arlotto M, Grange L, Chenau J et al. Synovial fluid proteomic fingerprint: S100A8, S100A9 and S100A12 proteins discriminate rheumatoid arthritis from other inflammatory joint diseases. Rheumatology (Oxford, England) 2010; 49: 671–682.
Wittkowski H, Foell D, af Klint E, De Rycke L, De Keyser F, Frosch M et al. Effects of intra-articular corticosteroids and anti-TNF therapy on neutrophil activation in rheumatoid arthritis. Ann Rheum Dis 2007; 66: 1020–1025.
Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H et al. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther 2006; 8: R69.
van Lent PLEM, Grevers LC, Blom AB, Arntz OJ, van de Loo FAJ, van der Kraan P et al. Stimulation of chondrocyte-mediated cartilage destruction by S100A8 in experimental murine arthritis. Arthritis Rheum 2008; 58: 3776–3787.
Schelbergen RFP, Blom AB, van den Bosch MHJ, Sloetjes A, Abdollahi-Roodsaz S, Schreurs BW et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on toll-like receptor 4. Arthritis Rheum 2011; 64: 1477–1487.
Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y et al. RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun 2002; 3: 123–135.
Kane D, Roth J, Frosch M, Vogl T, Bresnihan B, FitzGerald O . Increased perivascular synovial membrane expression of myeloid-related proteins in psoriatic arthritis. Arthritis Rheum 2003; 48: 1676–1685.
Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im H-J et al. Articular chondrocytes express the receptor for advanced glycation end products: potential role in osteoarthritis. Arthritis Rheum 2005; 52: 2376–2385.
Yammani RR, Carlson CS, Bresnick AR, Loeser RF . Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum 2006; 54: 2901–2911.
Madsen P, Rasmussen HH, Leffers H, Honoré B, Celis JE . Molecular cloning and expression of a novel keratinocyte protein (psoriasis-associated fatty acid-binding protein [PA-FABP]) that is highly up-regulated in psoriatic skin and that shares similarity to fatty acid-binding proteins. J Invest Dermatol 1992; 99: 299–305.
Broome A-M, Ryan D, Eckert RL . S100 protein subcellular localization during epidermal differentiation and psoriasis. J Histochem Cytochem 2003; 51: 675–685.
Nukui T, Ehama R, Sakaguchi M, Sonegawa H, Katagiri C, Hibino T et al. S100A8/A9, a key mediator for positive feedback growth stimulation of normal human keratinocytes. J Cell Biochem 2008; 104: 453–464.
Wolf R, Howard OMZ, Dong H-F, Voscopoulos C, Boeshans K, Winston J et al. Chemotactic activity of S100A7 (Psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol 2008; 181: 1499–1506.
Zheng Y, Niyonsaba F, Ushio H, Ikeda S, Nagaoka I, Okumura K et al. Microbicidal protein psoriasin is a multifunctional modulator of neutrophil activation. Immunology 2008; 124: 357–367.
Jinquan T, Vorum H, Larsen CG, Madsen P, Rasmussen HH, Gesser B et al. Psoriasin: a novel chemotactic protein. J Invest Dermatol 1996; 107: 5–10.
Bresnick AR, Weber DJ, Zimmer DB . S100 proteins in cancer. Nat Rev Cancer 2015; 15: 96–109.
Nasser MW, Elbaz M, Ahirwar DK, Ganju RK . Conditioning solid tumor microenvironment through inflammatory chemokines and S100 family proteins. Cancer Lett 2015; 365: 11–22.
Leclerc E, Vetter SW . The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim Biophys Acta 2015; 1852: 2706–2711.
Acknowledgements
We acknowledge research support provided by the NIH (RO1-HL112661), the Deutsche Forschungsgemeinschaft (SFB670, 645, 704, 1123, TRR57, 83, Exc1023) and the European Research Council (ERC InflammAct).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Bertheloot, D., Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol 14, 43–64 (2017). https://doi.org/10.1038/cmi.2016.34
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2016.34
Keywords
This article is cited by
-
HMGB1 prefers to interact with structural RNAs and regulates rRNA methylation modification and translation in HeLa cells
BMC Genomics (2024)
-
Tissue-resident macrophages exacerbate lung injury after remote sterile damage
Cellular & Molecular Immunology (2024)
-
S100 proteins in cardiovascular diseases
Molecular Medicine (2023)
-
Association of HMGB1 levels in synovial fluid with the severity of temporomandibular joint osteoarthritis
BMC Musculoskeletal Disorders (2023)
-
IL-33/ST2 axis of human amnion fibroblasts participates in inflammatory reactions at parturition
Molecular Medicine (2023)
