
Abstract
Inflammasomes play essential roles in immune protection against microbial infections. However, excessive inflammation is implicated in various human diseases, including autoinflammatory syndromes, diabetes, multiple sclerosis, cardiovascular disorders and neurodegenerative diseases. Therefore, precise regulation of inflammasome activities is critical for adequate immune protection while limiting collateral tissue damage. In this review, we focus on the emerging roles of post-translational modifications (PTMs) that regulate activation of the NLRP3, NLRP1, NLRC4, AIM2 and IFI16 inflammasomes. We anticipate that these types of PTMs will be identified in other types of and less well-characterized inflammasomes. Because these highly diverse and versatile PTMs shape distinct inflammatory responses in response to infections and tissue damage, targeting the enzymes involved in these PTMs will undoubtedly offer opportunities for precise modulation of inflammasome activities under various pathophysiological conditions.
Similar content being viewed by others
INTRODUCTION
The innate immune system relies on pattern recognition receptors (PRRs) to sense microbial infections and tissue damage and to initiate inflammatory responses.1, 2, 3 Inflammasomes constitute a group of PRRs that have essential roles in immune protection against bacterial, viral, fungal and parasitic infections.4, 5, 6, 7, 8, 9, 10, 11 Dysregulation of inflammasomes is implicated in various human diseases, including autoinflammatory disorders,12 metabolic disorders13, 14 and cancer.15, 16 Each type of inflammasome is characterized by a particular sensor or receptor molecule from the nucleotide-binding domain (NOD), leucine-rich repeat (LRR)-containing protein (NLR) family, or pyrin and HIN domain-containing protein (PYHIN) family. These include the NLRP1,4 NLRP2,17 NLRP3,18 NLRC4,19, 20, 21, 22 NLRP6,23, 24 NLRP7,25 NLRP12,26 AIM227, 28, 29, 30 and IFI1631, 32 inflammasomes. NLRP proteins contain an pyrin domain (PYD) at their N-termini, whereas NLRC4 contains a caspase activation and recruitment domain (CARD).33 The AIM2 and IFI16 proteins are DNA sensors that contain N-terminal PYD domains and C-terminal DNA-binding HIN domains. The adapter molecule apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) consists of N-terminal PYD and a C-terminal CARD domains that facilitate its interaction with both upstream sensor molecules through its PYD domain and downstream caspase-1 through its CARD domain. Assembly of the canonical inflammasome complexes containing caspase-1, as well as the noncanonical inflammasomes containing caspases-4, -5 and -11 that are bound to intracellular LPS,34, 35, 36 leads to activation of these caspases, which promotes maturation and secretion of proinflammatory cytokines, such as IL-1β, IL-18 and IL-1α.9, 11 Activated caspase-1 and caspase-11 also cleave gasdermin D (GSDMD) and other substrates that induce pyroptotic cell death through as-yet unknown mechanisms.37, 38, 39 Not all cell types undergo pyroptosis upon inflammasome activation. For example, activation of the NLRC4 inflammasome in neutrophils infected by Salmonella leads to the production of IL-1β without apparent pyroptosis, thus facilitating clearance of invading pathogens by the neutrophils.40, 41, 42 Furthermore, the AIM2 and NLRP3 inflammasomes are recruited to autophagosomes for degradation, which serves as a mechanism to eliminate activated inflammasomes and terminate inflammatory signaling.43 It is unclear whether other inflammasomes can also be degraded through similar mechanisms.
Because of the highly proinflammatory consequences of inflammasome activation, this process is tightly regulated to limit collateral damage. Prior to the activation of inflammasomes by diverse stimuli or ligands, a priming step, which can be provided by ligands for the Toll-like receptors (TLRs), is necessary. Priming leads to the activation of NF-κB and expression of inflammasome components, such as sensor molecules and the pro-forms of inflammatory cytokines, such as pro-IL1β and pro-IL18.44 Priming also modulates the post-translational modifications (PTMs) of NLRP3 and ASC to facilitate assembly of inflammasome complexes.45, 46, 47 In fact, the PTMs of inflammasome components have emerged as a major regulatory mechanism for inflammatory signaling. Virtually all cellular processes are regulated by a combinatorial post-translational modification ‘code’ that changes the surface features of proteins to endow them with diverse functionalities. Of these modifications, phosphorylation and ubiquitination are among the best characterized.48, 49, 50, 51 PTMs can covalently modify 15 of the 20 amino acids or cause proteolytical cleavage at specific residues to regulate the size, conformation, location, turnover and interaction of target proteins. These PTMs are catalyzed by hundreds of enzymes that are themselves modulated by binding partners or small molecule inhibitors. As a result, targeting these enzymes may offer opportunities for fine-tuning inflammatory signaling processes for therapeutic benefits in infectious diseases and metabolic, autoimmune and autoinflammatory disorders.52, 53
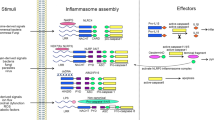
In this review, we focus on recent developments in post-translational modification of inflammasome components, such as sensor/receptor molecules and the adapter ASC, through phosphorylation, ubiquitination, proteolytic processing and other mechanisms (Table 1 and Figure 1). Convergence of these post-translational modifications contributes to comprehensive and balanced immune surveillance mechanisms. We anticipate that knowledge regarding PTMs will expand from these examples to other PTM types and less well-characterized inflammasomes. Because highly diverse and versatile PTMs shape distinct inflammatory responses toward infections and resulting from tissue damage, the role of post-translational modifications as a regulatory mechanism for inflammasome activation will remain an exciting area of research for the foreseeable future. Other mechanisms of inflammasome regulation relevant to NLRP3 were recently reviewed by Jo et al.54

Combinatorial post-translational modifications regulate inflammasomes. Phosphorylation, ubiquitination, de-ubiquitination, proteolytic processing, S-nitrosylation and ADP-ribosylation are indicated according to the keys in the lower left corner. The PTMs that promote activation of inflammasomes are shown as black arrows, whereas those that suppress inflammasome activation are shown as red blocks.
PHOSPHORYLATION
Phosphorylation is the most common post-translational modification and involves hundreds of kinases and phosphatases that dynamically modulate the proteome. It is well established that several inflammasome sensor molecules and the adapter ASC are regulated by distinct phosphorylation events, some of which have been mapped to specific residues of relevant inflammasome components. In most cases, the exact mechanisms by which phosphorylation of these proteins affects their structure and function remain to be clarified.
Phosphorylation of ASC modulates the activation of multiple inflammasomes
ASC is an essential adapter molecule for most inflammasome sensors.55 Phosphorylation of ASC was reported soon after identification of the inflammasome,56 but the role of its phosphorylation in inflammasome activation remained unclear. More than a decade later, it was discovered that in macrophages, ASC phosphorylation was required for activation of the NLRP3 and AIM2 inflammasomes, but not for that of NLRC4.57, 58 Specifically, Syk and Jnk kinase phosphorylate Tyr144 within the CARD domain of murine ASC57 or Tyr146 and Tyr187 in human ASC.58 This phosphorylation event facilitates formation of the ASC speck in the perinuclear area. Interestingly, inhibition of Syk and Jnk kinase activities does not disrupt the interaction between ASC and NLRP3, which suggests that phosphorylation of ASC may impact downstream events such as its ability to migrate to the perinuclear region, self-associate or recruit procaspases-1 through CARD–CARD interactions. This may also explain the fact that the NLRC4 inflammasome is not affected by the ASC-CARD phosphorylation because NLRC4 contains a CARD that can directly engage procaspase-1 in the absence of ASC.20, 21, 59 In contrast to the above studies in macrophages, phosphorylation of ASC does not appear to be important for inflammasome activation in bone marrow-derived dendritic cells.57 The mechanisms for such cell-type specificity and inflammasome selectivity remain to be determined. Furthermore, another study reported that Syk deficiency in macrophages did not reduce activation of the NLRP3 inflammasome.60 The reason for this discrepancy is not clear, but may be related to the different mouse strains used in the studies.
Bruton’s tyrosine kinase (BTK) was shown to interact with both ASC and NLRP3 through its kinase domain and, possibly, to phosphorylate ASC, which promotes activation of the NLRP3 inflammasome, but not the AIM2 inflammasome.61 Importantly, treatment with an FDA-approved BTK inhibitor has a neuroprotective effect in a brain ischemia model in mice, which suggests potential roles for BTK and inflammasomes in brain ischemia.
In addition to these examples of ASC phosphorylation that enhances inflammasome activation, there are also phosphorylation events that suppress inflammasome activation. Loss of IκB kinase α (IKKα) kinase activity is associated with spontaneous inflammation and lung cancer.62, 63 This prompted an investigation of the role of IKKα in inflammasome activation.64 It turned out that IKKα functions as a negative regulator of the NLRP3, AIM2 and NLRC4 inflammasomes through its interaction with the adapter ASC, by which it reduces the translocation of ASC from the nucleus to the cytoplasm.64 The residues Ser193 in the CARD and Ser16 in the PYD domain of ASC are phosphorylated by IKKα, which is important for the ASC–IKKα association in the nucleus that prevents its translocation to the cytoplasm for inflammasome activation. Mutations of either residue result in enhanced activation of both the NLRP3 and AIM2 inflammasomes. In agreement with the role of IKKα as a negative regulator of inflammasomes, stimulation of the NLRP3 inflammasome with ATP or nigericin suppresses the kinase activity of IKKα and its association with ASC, perhaps through dephosphorylation of IKKα by the serine/threonine phosphatase PP2A. This dephosphorylation event appears to be unique to NLRP3 because activation of the AIM2 inflammasome is not affected by the PP2A knockdown.64 Therefore, activation of the AIM2 inflammasome may be preceded by other mechanisms that inactivate IKKα. In strong contrast to IKKα, the IKK-related kinase (IKKi) phosphorylates ASC at Ser58, which facilitates its nucleus-to-cytoplasm translocation and promotes inflammasome activation.64 Deficiency of IKKi results in reduced activation of both the NLRP3 and AIM2 inflammasomes. How these two related kinases modulate inflammasome activation in completely different manners remains to be determined but may be related to their participation in distinct signaling pathways: a main difference between the functions of IKKα and IKKi is that the former is primarily involved in activation of the NF-κB pathway, whereas the latter is implicated in activation of the interferon response factor (IRF) 3 and 7.65
Multiple kinases regulate the NLRP3 inflammasome
The NLRP3 inflammasome responds to structurally and chemically diverse stimuli from infections and tissue damage.6, 55 Similar to ASC, NLRP3 has been reported to be phosphorylated by Syk, which is essential for the activation of the NLRP3 inflammasome upon infection by fungal, malarial and mycobacterial pathogens. For example, activation of the NLRP3 inflammasome by the fungal pathogens Candida albicans or Aspergillus fumigatus, but not by Salmonella infection or nigericin treatment, was shown to be dependent on Syk kinase, which induces ROS production and potassium efflux.66, 67 Similarly, Syk was phosphorylated and activated by Src kinases upon exposure to hemozoin during Plasmodium infection, perhaps with the effect of initiating immune responses through the NLRP3 inflammasome.68 In addition, activation of the NLRP3 inflammasome by the ESX-1 secretion system and its substrate ESAT-6 from Mycobacterium tuberculosis was reported to be dependent on Syk kinase activity.69 It is not clear from these studies whether Syk directly phosphorylates NLRP3, although conceivably the phosphorylation of ASC by Syk could be one of the mechanisms promoting inflammasome activation, as discussed above.57, 58
Similar to Syk, DAPK kinase was shown to associate with NLRP3 and contribute to NLRP3 inflammasome assembly.70 However, macrophages deficient in DAPK still retained residual caspase-1 activation and IL-1β maturation in response to stimulation by monosodium urate (MSU) and ATP. Because the kinase-inactive DAPK mutant was also capable of enhancing IL-1β secretion, it appears that the role of DAPK in the NLRP3 inflammasome activation is independent of its kinase activity. In agreement with this concept, the NLRP3 LRR was shown to co-IP with the ankyrin repeats, death domain or cytoskeleton-binding region of DAPK rather than with its kinase domain.70 Another kinase implicated in NLRP3 regulation is TAK1. Through screening of natural products and testing of kinase inhibitors, Gong et al.71 identified TAK1 as a kinase that promotes NLRP3 inflammasome activation in a manner independent of its role in the activation of gene expression. It was suggested that TAK1 may regulate the NLRP3 inflammasome through phosphorylation of NLRP3 or other components of the inflammasome complex. Similarly, the ERK1 kinase-mediated phosphorylation events were reported to be essential for priming of the NLRP3 inflammasome.72 However, whether NLRP3 is phosphorylated by these kinases was not addressed in the studies described above.
PKR is a dsRNA-dependent protein kinase. It undergoes autophosphorylation upon inflammasome activation and is broadly implicated in promoting activation of the NLRP3, NLRP1, NLRC4 and AIM2 inflammasomes, but not the NOD1, NLRP12 or NLRX1 inflammasomes.73, 74, 75 PKR is physically associated with NLRP3 in human THP-1 cells, and co-immunoprecipitation experiments in which PKR and NLRP3 were overexpressed in HEK293 cells showed that the PYD, NOD and LRR domains of NLRP3 were associated with PKR.73 By comparing the wild type with the mutant PKR, Lu et al. showed that the kinase activity of PKR is required for the activation of the NLRP3 inflammasome. In contrast, Hett et al.75 demonstrated that known PKR inhibitors did not reduce the activation of NLRP3 or NLRP1 inflammasomes. In agreement with these results, He et al.76 demonstrated that although PKR enhanced the generation of iNOS and bacteria-killing by macrophages, it was not required for activation of the NLRP3, NLRC4 and AIM2 inflammasomes in terms of caspase-1 activation and IL-1β/IL-18 processing. Furthermore, Yim et al.77 showed that the kinase activity of PKR suppressed NLRP3 inflammasome activation by inhibiting the expression of NLRP3 and pro-IL-1β. It was proposed that these apparent discrepancies may be due to the use of different macrophage cells or cell lines, as well as different mouse strains, and to the multiple effects of PKR on protein expression and signaling.77 Clearly, the mechanisms of PKR regulation of inflammasomes remain to be further clarified.
The IL-1R-associated kinases IRAK1 and IRAK4 function downstream of the TLRs in response to priming signals that enhance the transcription of inflammasome components.44 Fernandes-Alnemri et al.78 showed that these two kinases were also implicated in the rapid activation of NLRP3 by Listeria monocytogenes within 45 min of infection. They further demonstrated that this rapid NLRP3 activation was independent of transcription but required generation of reactive oxygen species. Although co-immunoprecipitation experiments have shown that IRAK1 associates with NLRP3, it is not clear whether its kinase activity is required for this rapid NLRP3 activation or whether any of the NLRP3 inflammasome components are phosphorylated by IRAK1.78
Recently, the serine–threonine kinase NEK7 was identified by three independent groups as being essential for activation of the NLRP3 inflammasome, but not the AIM2 or NLRC4 inflammasomes.79, 80, 81 Diverse screening approaches were used by these groups, including forward genetic screening for N-ethyl-N-nitrosourea-induced mutations,79 genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screening in macrophages80 and proteomic approaches using a tagged NLRP3 protein.81 The interaction of NEK7 with NLRP3 is dependent on the LRR and perhaps the NOD domain of NLRP3 and the kinase domain of NEK7. However, mutation of the NEK7 catalytic residues K63, K64 or G43 did not impair IL-1β secretion or the NEK7–NLRP3 interaction.79, 81 The NEK7–NLRP3 association is necessary for the recruitment of ASC by NLRP3 and the formation of ASC oligomers. In addition, activation of the NLRP3 inflammasome is blocked during mitosis, which suggests that NEK7 acts as a switch between inflammasome activation and cell division. Its kinase activity is dispensable for the former but required for the latter.79, 82, 83, 84
The role of phosphorylation in NLRC4 inflammasome activation
NLRC4 is one of the key inflammasome sensors that is responsible for the immune response to bacterial infections.20, 21, 22, 85 Gain-of-function mutations in NLRC4 also lead to autoinflammatory disorders in patients.86, 87, 88 Members of the family of NLR apoptosis-inhibitory proteins (NAIPs) are direct receptors for bacterial flagellin and type III secretion systems.89, 90, 91, 92, 93, 94, 95 Ligand-bound NAIPs induce a wheel-like assembly of the NAIP-NLRC4 inflammasomes that may catalyze the activation of oligomerized caspase-1.96, 97, 98, 99 An important regulatory mechanism for NLRC4 was revealed through the discovery that mouse NLRC4 in macrophages is phosphorylated following infection with Salmonella typhimurium.100 The phosphorylation site Ser533 is conserved among the NLRC4 proteins from different species, which suggests that it is potentially important for the structure or function of NLRC4. Biochemical characterization of kinase activities, screening of kinase inhibitors and in vitro phosphorylation assays identified PKCδ as the kinase that phosphorylates the Ser533 residue in mouse NLRC4.100 Importantly, the S533A mutant was not able to activate the NLRC4 inflammasome, whereas the phosphomimetic NLRC4 S533D mutant induced rapid pyroptosis in macrophages in the absence of infection. However, PKCδ-deficient macrophages retained residual inflammasome activation upon S. typhimurium infection, which suggests that other kinases provided compensatory mechanisms. Matusiak et al.101 further demonstrated that phosphorylation of NLRC4 primed the inflammasome for activation. However, the detection of bacteria by NAIP5 was independent of NLRC4 phosphorylation, although both required the D0 domain of the bacterial flagellin. In contrast to the studies described above, reconstitution of the NAIP–NLRC4 complexes in the presence of bacterial flagellin or needle proteins demonstrated that the phosphorylation of NLRC4 is not required for assembly of the inflammasome signaling complex.93 In addition, crystallization of an apparently inactive form of NLRC4 in which Ser533 was already phosphorylated has been achieved.98 In a third study, PKCδ did not appear to play any role in NLRC4 activation upon Shigella or Salmonella infection.94 The reasons for these apparent contradictory results are not clear, and further studies are required to clarify the mechanisms of NLRC4 phosphorylation and the role of PKCδ in inflammasome activation.
UBIQUITINATION
Protein ubiquitination and deubiquitination are versatile and dynamic processes that regulate the degradation, trafficking and signaling properties of the proteome in numerous aspects of the biological processes.102, 103, 104, 105 Conjugation of the ubiquitin chains is mediated by an E1–E2–E3 cascade involving ~40 E2 and 600 E3 enzymes, whereas ~100 deubiquitinating enzymes (DUBs) catalyze the removal or editing of ubiquitin chains. The distinct surfaces of the various ubiquitin chain types act as three-dimensional ‘codes’ that are ‘read’ by more than 20 ubiquitin-binding domains to facilitate the assembly of diverse protein interaction networks.106, 107 Thus, the ubiquitination, deubiquitination and ubiquitin recognition events involving inflammasome components constitute highly diverse processes that significantly expand the complexity of inflammatory signaling.
ASC ubiquitination modulates inflammasome activation
In addition to being phosphorylated at multiple sites, ASC is also ubiquitinated. Shi et al.43 reported that activation of the AIM2 inflammasome with poly(dA:dT) resulted in K63-linked polyubiquitination of ASC in macrophages, which led to its recruitment to the autophagy pathway that degrades the inflammasome. This was proposed to be a mechanism to prevent excessive inflammation by eliminating activated inflammasomes. In contrast, Rodgers et al.47 showed that the linear ubiquitin assembly complex (LUBAC) promotes ASC-dependent inflammasome activation through linear ubiquitination of ASC. LUBAC is an ubiquitin ligase complex composed of HOIL-1 l, HOIP and SHARPIN. HOIL-1 l-deficient bone marrow-derived macrophages demonstrated reduced production of mature IL-1β upon stimulation of NLRP3 and AIM2, but NF-κB activation was not affected. Multiple domains of HOIL-1 l, including the HOIP-binding, ubiquitin-binding and ubiquitination ligase domains, are required for robust ubiquitination of ASC, formation of ASC speck, and promotion of NLRP3 inflammasome activation. Of note, the suppressive effects on NLRP3 were more severe than those on AIM2, and the IL-1β secretion resulting from NLRC4 stimulation was not affected by the HOIL-1 l deficiency. These results suggest a specific role for HOIL-1 l in NLRP3 inflammasome regulation. However, in vitro ubiquitination experiments demonstrated that ASC is directly linear ubiquitinated by LUBAC, whereas NLRP3 was not. It is likely that LUBAC also regulates other factors specific for the NLRP3 pathway to modulate its activation.
The apparent contradictions between the studies by Shi and Rodgers regarding the roles (suppressive versus stimulatory) of ASC ubiquitination in inflammasome regulation have not been resolved, although it was suggested that the apparent differences in the ubiquitination of ASC (linear vs K63-linked) between these studies may be the result of cross-detection of linear ubiquitination by K63-ubiquitin antibodies.108 It remains to be determined how NLRP3-specific stimuli trigger linear ubiquitination of ASC to promote inflammasome activation. Importantly, the in vivo role of LUBAC in inflammasome activation was demonstrated through the observation that HOIL-1 l was essential for IL-1β secretion and neutrophil recruitment in an MDP-induced peritonitis model, as well as in an LPS-induced lethal inflammation model that is dependent on the NLRP3 inflammasome.47
Another E3 ubiquitin ligase, TNFR-associated factor 3 (TRAF3), was suggested to target ASC for K63-linked ubiquitination upon infection with RNA viruses, such as vesicular stomatitis virus (VSV) or influenza virus.109 The mitochondrial antiviral signaling protein (MAVS) serves as a platform that recruits both TRAF3 and ASC to promote TRAF3-mediated K63-linked ubiquitination of ASC at residue Lys174. In fact, MAVS also colocalizes with NLRP3 within 2 h of VSV infection, although the MAVS–NLRP3 interaction appears to be independent of the MAVS–TRAF3 association. MAVS-mediated ubiquitination of ASC enhances activation of the NLRP3 inflammasome in response to VSV infection, but not following exposure to crystalline materials, such as MSU crystals. Furthermore, MAVS does not enhance activation of the AIM2 or NLRC4 inflammasomes. Clearly, infections by RNA viruses that activate the RIG-I pathway may stimulate MAVS-mediated ubiquitination of ASC and activation of the NLRP3 inflammasome. Whether RIG-I has a role in this ubiquitination event or whether production of interferon by the RIG-I pathway promotes NLRP3 activation remains to be determined.
Ubiquitination of NLRP3 negatively regulates inflammasome activation
The potential role of ubiquitination in inflammasome regulation was inferred from the observation that the ubiquitin ligase-associated protein SGT1 is associated with a number of NLR proteins, including NLRP2, NLRP3, NLRP4, NLRP12, NLRC4, NOD1 and NOD2.110 In particular, binding of SGT1 to the LRR domain of NLRP3 was essential for maintaining its inactive but signaling-competent state.110 This is analogous to the essential role of plant SGT1 in activation of the nucleotide-binding site/leucine-rich repeat (NBS–LRR) proteins for host protection.111 However, the specific roles of ubiquitination in the functions of any of the NLR/NBS–LRR proteins were not demonstrated in the above study.
Recently, one of the E3 ubiquitin ligases for NLRP3 was identified as the Skp-Cullin-F box (SCF) family member, F-box L2 (FBXL2), which mediates the ubiquitination and proteasomal degradation of NLRP3.112 FBXL2 is in turn regulated by another F-box protein, F-box O3 (FBXO3), through its own ubiquitination and degradation. A small molecule FBXO3 inhibitor, BC-1215, was shown to release the inhibition of FBXL2 and reduce NLRP3-mediated inflammation. FBXL2 binds Trp73 in the PYD domain and Lys689 in the LRR domain of NLRP3, the latter of which is a ubiquitination site. The authors argue that rather than enhancing the expression of NLRP3, the role of LPS priming is to reduce the ubiquitin-mediated proteasomal degradation of NLRP3 via the elevated expression of FBXO3.
Another ubiquitination-mediated process in the regulation of the NLRP3 inflammasome was identified through the dopamine D1 receptor DRD1 pathway.113 The neurotransmitter dopamine activates the DRD1 signaling pathway to produce the second messenger cyclic adenosine monophosphate (cAMP). Binding of cAMP to NLRP3, perhaps in conjunction with activation of other pathways by cAMP, promotes Lys48-linked polyubiquitination of NLRP3 by the E3 ubiquitin ligase MARCH7. The ubiquitinated NLRP3 is subsequently degraded through the autophagy pathway. This ubiquitination-mediated regulation by dopamine and its receptor is specific for the NLRP3 inflammasome and does not affect the AIM2 or NLRC4 inflammasomes. Furthermore, in vivo experiments demonstrated that the dopamine signaling pathway suppressed neurotoxin-induced inflammation (a model of Parkinson’s disease), lipopolysaccharide-induced systemic inflammation and MSU-induced peritonitis.113 This suggests that DRD1 agonists may have therapeutic value in NLRP3-driven inflammatory diseases. The role of ubiquitination in regulating the NLRP3 degradation was further demonstrated through the study of the autophagy chaperone p62. P62 was shown to colocalize with a detergent-insoluble polyubiquitinated protein aggregates containing NLRP3 and to promote the degradation of NLRP3.114 P62 also promotes the degradation of damaged mitochondria through mitophagy, thus reducing the release of NLRP3-activating stimuli from mitochondria.115 In agreement with these results, p62 deficiency in mice results in an increased atherosclerotic plaque burden114 and elevated inflammation in alum-induced peritonitis and fuminant hepatitis.115
In addition to FBXL2 and MARCH7, another E3 ubiquitin ligase, X-linked inhibitor of apoptosis protein (XIAP), has been implicated in regulation of the NLRP3 inflammasome. XIAP is a member of the IAP family that inhibits caspases and suppress apoptotic cell death.116 The C-terminal RING domain of XIAP harbors an E3 ubiquitin ligase activity that is required for NOD2 signaling.117, 118 It was reported that the loss of XIAP or its RING domain resulted in excessive cell death or elevated secretion of IL-1β, although the cell death appeared to be independent of caspase-1, -11 or -8.119 Currently, it is not clear whether XIAP directly ubiquitinates NLRP3 or whether its RING domain mediates ubiquitination of any other inflammasome components.
Similar to the Guan study of the MAVS–ASC interaction, MAVS was reported to promote NLRP3 inflammasome activation through the association of MAVS with NLRP3.120 The residues 2-7 (KMASTR) within the PYD domain of NLRP3 are essential for this interaction. The recruitment of NLRP3 to the mitochondria by MAVS may promote formation of the ASC speck,120 oligomerization of NLRP3 and/or stimulation of the mitochondrial formation of reactive oxygen species.121 The role of ubiquitination was not explored in this study, although, presumably, TRAF3 may be recruited by MAVS, as shown in the Guan study,109 in which TRAF3 was found to be located in close proximity to NLRP3, which harbors multiple potential ubiquitination sites. Similar to the Guan study,109 MAVS is required for optimal stimulation of the NLRP3 inflammasome by poly (I:C)120 or Sendai virus infection,121 but not by crystalline substances, such as alum or MSU.120 These similarities suggest potential cross-regulation of the RIG-I and inflammasome pathways. In agreement with these results, association of MAVS with NLRP3 suppressed the MAVS-induced type I interferon response to Sendai virus infection.121
A20 is an ubiquitin-editing enzyme that is involved in negative feedback regulation of NF-κB signaling.122, 123, 124, 125 Independent of its role in NF-κB regulation, A20 was recently reported to be an inhibitor of the NLRP3, but not NLRC4 or AIM2, inflammasome.126, 127 The significance of this inflammasome regulatory function is borne out by the observations that A20 deficiency in mice leads to pathological inflammation that resembles rheumatoid arthritis126 and that heterozygous germline mutations of A20 in humans lead to early onset systemic inflammation.128 Mechanistically, the suppression of NLRP3 by A20 was dependent on TRIF and RIPK3. Furthermore, A20 may reduce the ubiquitination of pro-IL-1β at its conserved residue K133, thus impacting its maturation by caspases or secretion through the autophagy pathway.127
Ubiquitination of NLRC4 activates the inflammasome
Sug1 is a regulatory protein with ubiquitin ligase activity that is associated with the 26S proteasome and has been identified as a binding partner for NLRC4 through yeast two-hybrid assays.129 Sug1 binds residues 91-253 of NLRC4, which include its CARD-NOD linker region and NOD domain. The Sug1–NLRC4 interaction releases its autoinhibited conformation, followed by ubiquitination and aggregation of NLRC4 that in turn leads to recruitment and activation of caspase-8. However, the site and type of ubiquitination of NLRC4 have not yet been characterized. In addition, it is not clear how the ubiquitinated NLRC4 recruits caspase-8, although a potential adapter molecule that bridges the ubiquitinated NLRC4 and caspase-8 has been proposed.129
Ubiquitination and degradation of IFI16 mediated by viral E3-ubiquitin ligase
IFI16 belongs to the p200 family of interferon-inducible proteins and the PYHIN family.130, 131 As a PRR, it functions as a DNA sensor in the cytosol and nucleus that induces interferon production. It also induces the assembly of inflammasome complexes in response to DNA viruses, such as herpes simplex virus 1 (HSV-1),31 Kaposi’s sarcoma-associated herpesvirus (KSHV)32 and Epstein–Barr virus,132 as well as retroviruses such as HIV-1.133, 134 Interferon production downstream of IFI16 is dependent on the endoplasmic reticulum adapter molecule stimulator of interferon genes (STRING), which is also known as mediator of IRF3 activation (MITA), endoplasmic reticulum IFN stimulator (ERIS) or met-pro-tyr-ser (MPYS),135, 136, 137, 138 and an essential transcription factor IRF-3.139 IFI16 is known to form an inflammasome complex with the adapter ASC in the nucleus and translocate to the cytoplasm to activate caspase-1.32 In addition to directly binding to DNA, IFI16 and its murine ortholog p204 were recently shown to cooperate with another DNA sensor, cyclic GMP–AMP synthase,140 to recognize DNA from HSV-1 and Francisella infection in human foreskin fibroblasts and macrophages, respectively.141, 142 To counter IFI16-mediated immune responses, HSV-1 expresses an immediate-early protein, infected cell protein 0 (ICP0), which contains a RING finger domain with E3-ubiquitin ligase activity. Within a few hours after HSV-1 infection, ICP0 inhibits the nuclear localization of activated IRF-3 and promotes the ubiquitination and proteasome-dependent degradation of IFI16 and p204.143, 144, 145 The specific types of IFI16 ubiquitination have not been characterized.144 ICP0-mediated degradation is specific for IFI16 on the basis that the NLRP3 and AIM2 inflammasomes are not affected.144 In contrast to the studies described above, another report did not observe IFI16 degradation upon expression of ICP0 in the absence of HSV-1 infection,146 which suggests that factors other than ICP0 may be responsible for IFI16 regulation following HSV-1 infection. The reasons for such seemingly contradictory results are not clear but may be attributed to differential degradation of IF16 in various cell types.
Ubiquitination of caspase-1 upon inflammasome activation
While investigating activation of the Nlrp1b inflammasome by the anthrax lethal toxin, Lamkanfi and colleagues noticed the presence of high-molecular-weight species in immunoblots of macrophage cell lysates for caspase-1.147 This was interpreted as indicative of caspase-1 post-translational modifications, which was confirmed by immunobloting for ubiquitin. Importantly, high-molecular-weight caspase-1 species were also present following activation of the NLRP3 inflammasome by ATP or nigericin, the AIM2 inflammasome by Francisella infection and the NLRC4 inflammasome by Salmonella infection. The apparent post-translational modification of caspase-1 required its enzymatic activity on the basis that treatment with the caspase-1 inhibitor YVAD-cmk significantly reduced the formation of these high-molecular-weight species. These results also suggest that caspase-1 ubiquitination occurs downstream of caspase-1 activation. The authors proposed that caspase-1 ubiquitination may represent a negative feedback mechanism that dampens inflammatory signaling by targeting activated caspase-1 for degradation. However, it is currently not clear whether ubiquitinated caspase-1 is destined for degradation because treatment with the proteasome inhibitor MG132 suppressed rather than enhanced caspase-1 activation.147 Future studies are required to clarify the fate of ubiquitinated caspase-1.
Deubiquitination of NLRP3 promotes inflammasome activation
Observation of NLRP3 ubiquitination suggested that deubiquitination was a likely mechanism for inflammasome regulation. Lopez-Castejon et al.148 identified a DUB inhibitor b-AP15 that inhibited activation of the NLRP3 inflammasome and partially inhibited activation of the AIM2 inflammasome. The authors concluded that multiple as-yet uncharacterized DUBs may be required for IL-1β processing and release. Employing an NLRP3 ubiquitination assay, Yuan and colleagues identified a DUB/isopeptidase inhibitor, G5, that suppressed IL-1β secretion downstream of the NLRP3 inflammasome in macrophages, but not downstream of the AIM2 or NLRC4 inflammasomes.46 G5 treatment enhanced polyubiquitination of the NLRP3 NOD and LRR domains, but not the PYD domain, and suppressed the formation of ASC oligomers. Subsequently, the deubiquitination enzyme BRCC3 (mouse)/BRCC36 (human) was identified, which coimmunoprecipitated with the NOD and LRR domains of NLRP3 and deubiquitinated its LRR.46 BRCC3 is a JAMM domain-containing Zn2+ metalloprotease that specifically cleaves Lys63-linked, but not Lys48-linked, polyubiquitin chains.149 Currently, it is unclear how NLRP3 stimuli activate BRCC3. Nonetheless, deubiquitination-dependent activation of NLRP3 may be a potential target for therapeutic modulation of inflammatory pathogeneses that involve NLRP3.
PROTEOLYTIC PROCESSING OF INFLAMMASOME COMPONENTS
In addition to phosphorylation and ubiquitination, cleavage of inflammasome sensor proteins by proteases from bacteria or the host and complex interactions among proteases, their substrates and protease inhibitors have crucial roles in regulating inflammasome activation. In fact, serine protease inhibitors were shown to suppress activation of both NLRC4 and NLRP3 inflammasomes. These results contrast with those for pan-cathepsin inhibitors, which were ineffective, suggesting that proteolytic processing is an integral component of inflammasome regulation.150, 151 In contrast to phosphorylation and ubiquitination, proteolytic processing is an irreversible PTM process that generates new protein isoforms.
Anthrax protease activates the Nlrp1b inflammasome
Both mouse Nlrp1b and rat Nlrp1 were shown to be cleaved by the anthrax lethal factor (LF), a zinc metalloprotease that is part of the anthrax lethal toxin.152, 153, 154, 155 The cleavage site is located in the N-terminal region of these proteins, outside of the predicted NOD domain. This cleavage event was sufficient for Nlrp1b activation, implying that Nlrp1b can serve as a sensor for other proteases from diverse pathogens or even host cells. Mechanistically, protease cleavage may release an autoinhibited conformation of Nlrp1b to facilitate assembly of the inflammasome complex.156 Alternatively, the cleaved Nlrp1b may recruit unknown binding partners that may in turn promote inflammasome activation. Whether such mechanisms apply to human NLRP1 protein remains to be determined. Compared with murine Nlrp1 proteins, human NLRP1 contains an additional PYD domain at its N terminus that may endow it with distinct activation mechanisms. In addition, the LF cleavage sites of murine Nlrp1 proteins have poor sequence conservation: murine Nlrp1b is cleaved at a Lys-Leu sequence, whereas rat Nlrp1 is cleaved at a Pro-Leu sequence. Therefore, it remains to be determined whether the human NLRP1 protein is also cleaved by the anthrax LF protease or other microbial proteases. It is interesting to note that such mammalian immune responses to proteolytic events are analogous to the recognition of modifications of host proteins by plant resistance (R) proteins, the majority of which are also NLR proteins. This suggests that recognition of proteolytic events is an ancient mechanism of immune protection.157, 158, 159
Enterovirus proteases suppress the NLRP3 inflammasome
The enterovirus 71 (EV71) is responsible for many epidemics in the world and is manifested by hand, foot and mouth disease, and neurological complications in children.160 Following-up on the observation that EV71-infected patients, particularly those with neurological complications, showed elevated levels of IL-1β, Wang et al.161 identified NLRP3 as the inflammasome that is activated upon EV71 infection. Conversely, EV71 encodes two proteases, 2A and 3C, that specifically cleave NLRP3 at G493-L494 and Q225-G226, respectively, thus antagonizing the NLRP3 inflammasome, but not other inflammasomes. Both cleavage sites are located within the predicted NOD domain of NLRP3, suggesting that this cleavage may prevent NLRP3 oligomerization and/or assembly of the inflammasome complex. In fact, both of the EV71 proteases have been shown to antagonize innate immune responses through cleavage of antiviral signaling proteins, such as MAVS162 and IRF7.163
Mycobacterial Zmp1 protease reduces inflammasome activation
Infection of macrophages by Mycobacterium tuberculosis (Mtb) leads to formation of granulomas, in which the bacteria become latent, often for the lifetimes of infected individuals.164 Reactivation of latent infections can be triggered under immunosuppressive conditions, and under these conditions, active tuberculosis ensues. IL-1 and the NLRP3165, 166, 167, 168, 169, 170, 171, 172, 173 and AIM2174, 175, 176, 177 inflammasomes have been shown to have important roles in the host response to mycobacterial infection. However, there is controversy regarding the protective role of the NLRP3 inflammasome.69, 167, 170, 178, 179 Mycobacterial species encode a zinc metalloprotease, Zmp1, that was shown to inhibit activation of the NLRP3 and NLRC4 inflammasomes on the basis that compared with the wild type, an M bovis BCG strain deficient in Zmp1 induced significantly higher levels of IL-1β and phagolysosome biogenesis in murine macrophages.180 However, whether Zmp1 cleaves components of the inflammasomes was not clarified. In addition, suppression of the inflammasomes was not reproduced upon infection of human macrophages with Mycobacterium tuberculosis strain H37Rv.69 It is possible that the proteases encoded by different mycobacterial strains may antagonize inflammasomes through distinct mechanisms.
OTHER POST-TRANSLATIONAL MODIFICATIONS OF NLRP3
S-nitrosylation suppresses the NLRP3 inflammasome
Nitric oxide (NO) was shown to inhibit caspases-1, 2, 3, 4, 6, 7 and 8 through S-nitrosylation of the caspase catalytic cysteine residue.181, 182, 183, 184 As a result, NO or its reaction products suppressed caspase-1 activity and inhibited the release of IL-1β and IL-18 from activated macrophages.185 In addition, the NO donor S-nitroso-N-acetylpenicillamine (SNAP) inhibited caspase-1 activity in vitro, and treatment with the inducible NO synthase (iNOS) inhibitor NG-monomethyl-l-arginine significantly enhanced IL-1β secretion from wild type, but not iNOS-deficient, macrophages and mice. Using purified recombinant caspase protein, inhibition of caspase-1 activity was shown to be reversible by treatment with dithiothreitol (DTT), which suggested that the caspase catalytic cysteine residue is S-nitrosylated by NO. However, DTT did not completely reverse the effects of NO treatment when macrophage cell lysates were used. This suggests that modification of the caspase catalytic cysteine residue may not be the sole mechanism underlying the inflammasome regulation mediated by NO.185
In this context, a recent study confirmed that IFN-γ-stimulated NO production mediated the suppression of IL-1β secretion by Mycobacterium tuberculosis-infected macrophages.186 This was proposed as a regulatory mechanism that would reduce the destructive innate inflammatory responses elicited during chronic mycobacterial infection. Mechanistically, NO or SNAP treatment leads to S-nitrosylation of NLRP3, but not AIM2. The resulting S-nitrosylaton results in suppression of ASC oligomerization and IL-1β processing. Related studies reported that secretion of mature IL-1β and IL-18 by ATP or nigericin-stimulated macrophages was significantly reduced upon treatment with SNAP, whereas only partial reduction was observed upon stimulation of the AIM2 or NLRC4 inflammasomes.187, 188, 189 SNAP treatment for 1 h leads to S-nitrosylation of both caspase-1 and NLRP3,187 which implicates direct modification of NLRP3 by S-nitrosylation as the main regulatory mechanism induced by NO. NLRP3 is extremely rich in cysteine residues. Exactly which cysteine residues are the functional targets of S-nitrosylation remains to be determined. Interestingly, a group of naturally occurring compounds, ginsenosides, were shown to reduce S-nitrosylation of NLRP3 by reducing iNOS expression.190 This was proposed as a mechanism by which ginsenosides can protect against LPS-induced endotoxic shock.
Activation of inflammasomes has been shown to promote NO production, perhaps as a negative feedback mechanism. Both activation of the NLRP3 inflammasome upon Leishmania infection191 and stimulation of the NLRC4 inflammasome by intracellular flagellin192 were shown to activate iNOS and the potential S-nitrosylation of inflammasome components. This mitigate the detrimental effects of uncontrolled inflammation.
ADP-ribosylation of NLRP3 triggers robust inflammation and cytopathology
Mycoplasma pneumoniae is a common airway pathogen. Although infection by mycoplasma rarely results in death, the hyperinflammatory conditions that manifest in response to mycoplasma infection have been associated with chronic pulmonary dysfunction, such as bronchial asthma, chronic obstructive pulmonary disease and cystic fibrosis.193 Therefore, immunomodulatory therapeutics in combination with conventional antimicrobial therapeutics are recommended in the management of mycoplasma pneumonia. A mycoplasma toxin observed in community-acquired respiratory distress syndrome (CARDS) is an ADP-ribosyltransferase and was shown to colocalize with NLRP3 and to catalyze its ADP-ribosylation.194 This post-translational modification is essential for NLRP3 activation upon mycoplasma infection of human and mouse macrophages and may account for the hyperinflammatory lung pathologies manifested in the clinic. Importantly, the role of the CARDS toxin as an inflammation-promoting soluble factor is consistent with its ability to trigger allergic reactions in the airway long after the primary infection by mycoplasma subsides.195 As such, the CARDS toxin represents an important therapeutic target for modulating lung inflammation upon mycoplasma infection. The suppressive effect of the CARDS toxin was specific for the NLRP3, but not NLRP1, inflammasome and was dependent on both the N-terminal catalytic domain of the CARDS toxin and its C-terminal domain, which is responsible for its attachment to host cells and subsequent internalization. Although the specific domain of NLRP3 that is modified by the CARDS toxin was not defined, it was suggested that the CARDS toxin may alter the conformation of NLRP3 or its interaction with other partner proteins during assembly of the inflammasome.194 Detailed mechanisms regarding the CARDS toxin–NLRP3 interaction still await future investigations.
DISCUSSION
Here we have focused on our evolving understanding of how diverse yet highly specific post-translational modifications regulate inflammasome activation. Because these post-translational modifications are dependent on enzymatic activities, they represent attractive therapeutic targets for modulation of inflammasome activities in a broad range of inflammatory pathologies. It is clear that a repertoire of post-translational modifications is involved in the precise regulation of inflammasomes. These combinatorial modifications regulate inflammasomes by changing the conformations of inflammasome proteins and by controlling their turnover, distribution and location, as well as by regulating the intricate protein–protein interactions essential for inflammasome assembly.
Various post-translational modifications in inflammasome signaling can be cross-regulated by each other, perhaps within the context of different cell types. The most prominent crosstalk is between phosphorylation and ubiquitination, which can positively or negatively regulate each other.51, 196 Phosphorylation and ubiquitination are among the most abundant PTMs involving hundreds of enzymes that catalyze both the attachment and the removal of phosphate/ubiquitin. This highly complex network of inflammasome regulation may contribute to the ability of inflammasomes to respond to various stimuli. How these multiple PTMs spatially and temporarily coordinate during the process of specific inflammasome activation in a particular cell type will be an important topic for further studies. For example, the phosphorylation of ASC by Syk kinase is required for ASC speck formation and caspase-1 activation in macrophages, but not in bone marrow-derived dendritic cells.57, 58 In addition, proteolytic cleavage of GSDMD and the subsequent pyroptotic cell death of macrophages was not observed in neutrophils upon activation of the NLRC4 inflammasome, although mature IL-1β was produced.40, 41, 42 It is clear that to appreciate the full scope of inflammasome regulation, studies of cell-type-specific PTMs and exploration of additional PTMs are important future areas of investigation.
Understanding the mechanisms of PTMs in inflammasome regulation may offer tremendous opportunities for the development of therapeutic agents to target inflammatory conditions, such as autoinflammatory disorders, metabolic disorders, neurodegenerative diseases and cardiovascular disorders. For example, some of the specific kinase inhibitors that are developed for cancer treatment can be repurposed as agents to treat inflammatory disorders. The dopamine signaling pathway, which triggers ubiquitination and inhibition of the NLRP3 inflammasome, ameliorated neuro-inflammation in a mouse model of Parkinson’s disease and systemic inflammation in a mouse model of sepsis.113 This suggests that an agonist of the dopamine D1 receptor or the downstream E3 ubiquitin ligase MARCH7 may provide therapeutic benefit in the above diseases. Similarly, the G5 and b-AP15 inhibitors46, 148 of DUBs that have previously been shown to block caspase-1 activation and IL-1β maturation may be developed as agents to combat diseases that involve excessive activation of the NLRP3 inflammasome, such as cardiovascular disorders.197
References
Janeway CA, Medzhitov R . Innate immune recognition. Annu Rev Immunol 2002; 20: 197–216.
Kumar H, Kawai T, Akira S . Pathogen recognition in the innate immune response. Biochem J 2009; 420: 1–16.
Qian C, Liu J, Cao X . Innate signaling in the inflammatory immune disorders. Cytokine Growth Factor Rev 2014; 25: 731–738.
Martinon F, Burns K, Tschopp J . The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell 2002; 10: 417–426.
Franchi L, Nuñez G . Immunology. Orchestrating inflammasomes. Science 2012; 337: 1299–1300.
Latz E, Xiao TS, Stutz A . Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13: 397–411.
de Zoete MR, Palm NW, Zhu S, Flavell RA . Inflammasomes. Cold Spring Harb Perspect Biol 2014; 6: a016287–a016287.
Lamkanfi M, Dixit VM . Mechanisms and functions of inflammasomes. Cell 2014; 157: 1013–1022.
Guo H, Callaway JB, Ting JPY . Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; 21: 677–687.
Vanaja SK, Rathinam VAK, Fitzgerald KA . Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 2015; 25: 308–315.
Man SM, Kanneganti T-D . Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol 2016; 16: 7–21.
Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E . The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol Mech Dis 2015; 10: 395–424.
Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA . Inflammasomes and metabolic disease. Annu Rev Physiol 2013; 76: 57–78.
Robbins GR, Wen H, Ting JPY . Inflammasomes and metabolic disorders: old genes in modern diseases. Mol Cell 2014; 54: 297–308.
Terlizzi M, Casolaro V, Pinto A, Sorrentino R . Inflammasome: cancer's friend or foe? Pharmacol Ther 2014; 143: 24–33.
Kolb R, Liu G-H, Janowski AM, Sutterwala FS, Zhang W . Inflammasomes in cancer: a double-edged sword. Protein Cell 2014; 5: 12–20.
Minkiewicz J, de Rivero Vaccari JP, Keane RW . Human astrocytes express a novel NLRP2 inflammasome. Glia 2013; 61: 1113–1121.
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J . NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004; 20: 319–325.
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004; 430: 213–218.
Franchi L, Amer A, Body-Malapel M, Kanneganti T-D, Özören N, Jagirdar R et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat Immunol 2006; 7: 576–582.
Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol 2006; 7: 569–575.
Amer A, Franchi L, Kanneganti T-D, Body-Malapel M, Özören N, Brady G et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem 2006; 281: 35217–35223.
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011; 145: 745–757.
Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang J-P, Brown EM et al. NLRP6 Inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014; 156: 1045–1059.
Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012; 36: 464–476.
Vladimer GI, Weng D, Paquette SWM, Vanaja SK, Rathinam VAK, Aune MH et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012; 37: 96–107.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009; 458: 514–518.
Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES . AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458: 509–513.
Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 2009; 10: 266–272.
Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009; 323: 1057–1060.
Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 2010; 11: 997–1004.
Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host and Microbe 2011; 9: 363–375.
Ting JPY, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK et al. The NLR gene family: a standard nomenclature. Immunity 2008; 28: 285–287.
Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014; 514: 187–192.
Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA . Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 2013; 341: 1250–1253.
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013; 341: 1246–1249.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526: 660–665.
Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 2015; 526: 666–671.
He W-T, Wan H, Hu L, Chen P, Wang X, Huang Z et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 2015; 25: 1285–1298.
Chen KW, Groß CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep 2014; 8: 570–582.
Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG et al. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J Immunol 2015; 194: 1763–1775.
Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E . Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat Commun 2016; 7: 10555.
Shi C-S, Shenderov K, Huang N-N, Kabat J, Abu-Asab M, Fitzgerald KA et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13: 255–263.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183: 787–791.
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES . Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 2012; 287: 36617–36622.
Py BF, Kim M-S, Vakifahmetoglu-Norberg H, Yuan J . Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 2013; 49: 331–338.
Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 2014; 211: 1333–1347.
Walsh CT, Garneau-Tsodikova S, Gatto GJ . Posttranslational modification of proteins expanding natures inventory. Angew Chem Int Ed Engl 2005; 44: 7342–7372.
Khoury GA, Baliban RC, Floudas CA . Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep 2011; 1: 90.
Nussinov R, Tsai C-J, Xin F, Radivojac P . Allosteric post-translational modification codes. Trends Biochem Sci 2012; 37: 447–455.
Hunter T . The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell 2007; 28: 730–738.
Chaugule VK, Walden H . Specificity and disease in the ubiquitin system. Biochem Soc Trans 2016; 44: 212–227.
Goru SK, Pandey A, Gaikwad AB . E3 ubiquitin ligases as novel targets for inflammatory diseases. Pharmacol Res 2016; 106: 1–9.
Jo E-K, Kim JK, Shin D-M, Sasakawa C . Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 2015; 13: 148–159.
Schroder K, Tschopp J . The inflammasomes. Cell 2010; 140: 821–832.
Stehlik C, Krajewska M, Welsh K, Krajewski S, Godzik A, Reed JC . The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochem J 2003; 373: 101–113.
Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol 2013; 14: 1247–1255.
Lin YC, Huang DY, Wang JS, Lin YL, Hsieh SL, Huang KC et al. Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J Leukoc Biol 2015; 97: 825–835.
Case CL, Shin S, Roy CR . Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect Immun 2009; 77: 1981–1991.
He Y, Varadarajan S, Muñoz-Planillo R, Burberry A, Nakamura Y, Nuñez G . 3,4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem 2014; 289: 1142–1150.
Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A et al. Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 2015; 6: 7360.
Li N, Wu X, Holzer RG, Lee J-H, Todoric J, Park EJ et al. Loss of acinar cell IKKα triggers spontaneous pancreatitis in mice. J Clin Invest 2013; 123: 2231–2243.
Xiao Z, Jiang Q, Willette-Brown J, Xi S, Zhu F, Burkett S . The pivotal role of IKKα in the development of spontaneous lung squamous cell carcinomas. Cancer Cell 2013; 23: 527–540.
Martin BN, Wang C, Willette-Brown J, Herjan T, Gulen MF, Zhou H et al. IKKα negatively regulates ASC-dependent inflammasome activation. Nat Commun 2014; 5: 4977.
Hacker H, Karin M . Regulation and function of IKK and IKK-related kinases. Sci STKE 2006; 2006: re13.
Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009; 459: 433–436.
Saïd-Sadier N, Padilla E, Langsley G, Ojcius DM . Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One 2010; 5: e10008.
Shio MT, Tiemi Shio M, Eisenbarth SC, Savaria M, Vinet AF, Bellemare M-J et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog 2009; 5: e1000559.
Wong K-W, Jacobs WR . Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell Microbiol 2011; 13: 1371–1384.
Chuang Y-T, Lin Y-C, Lin K-H, Chou T-F, Kuo W-C, Yang K-T et al. Tumor suppressor death-associated protein kinase is required for full IL-1β production. Blood 2011; 117: 960–970.
Gong Y-N, Wang X, Wang J, Yang Z, Li S, Yang J et al. Chemical probing reveals insights into the signaling mechanism of inflammasome activation. Cell Res 2010; 20: 1289–1305.
Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES et al. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J Immunol 2014; 192: 3881–3888.
Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012; 488: 670–674.
Boriushkin E, Wang JJ, Li J, Bhatta M, Zhang SX . p58IPK suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages. Sci Rep 2016; 6: 25013.
Hett EC, Slater LH, Mark KG, Kawate T, Monks BG, Stutz A et al. Chemical genetics reveals a kinase-independent role for protein kinase R in pyroptosis. Nat Chem Biol 2013; 9: 398–405.
He Y, Franchi L, Nuñez G . The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur J Immunol 2013; 43: 1147–1152.
Yim HC, Wang D, Yu L, White CL, Faber PW, Williams BR et al. The kinase activity of PKR represses inflammasome activity. Cell Res 2016; 26: 367–379.
Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES . Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol 2013; 191: 3995–3999.
Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 2015; 17: 250–258.
Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E et al. A Genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J Biol Chem 2015; 291: 103–109.
He Y, Zeng MY, Yang D, Motro B, Nuñez G . NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016; 530: 354–357.
Yissachar N, Salem H, Tennenbaum T, Motro B . Nek7 kinase is enriched at the centrosome, and is required for proper spindle assembly and mitotic progression. FEBS Lett 2006; 580: 6489–6495.
Kim S, Lee K, Rhee K . NEK7 is a centrosomal kinase critical for microtubule nucleation. Biochem Biophys Res 2007; 360: 56–62.
O'Regan L, Fry AM . The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol Cell Biol 2009; 29: 3975–3990.
Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA . Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. The Journal of Experimental Medicine 2007; 204: 3235–3245.
Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 2014; 46: 1140–1146.
Romberg N, Moussawi Al K, Nelson-Williams C, Stiegler AL, Loring E, Choi M et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 2014; 46: 1135–1139.
Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K . An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med 2014; 211: 2385–2396.
Kofoed EM, Vance RE . Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 2011; 477: 592–595.
Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011; 477: 596–600.
Lightfield KL, Persson J, Brubaker SW, Witte CE, Moltke von J, Dunipace EA et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 2008; 9: 1171–1178.
Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA, Cutting Edge . Mouse NAIP1 detects the type III secretion system needle protein. J Immunol 2013; 191: 3986–3989.
Yang J, Zhao Y, Shi J, Shao F . Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA 2013; 110: 14408–14413.
Suzuki S, Franchi L, He Y, Muñoz-Planillo R, Mimuro H, Suzuki T et al. Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcδ. PLoS Pathog 2014; 10: e1003926.
Fortier A, Doiron K, Saleh M, Grinstein S, Gros P . Restriction of Legionella pneumophila replication in macrophages requires concerted action of the transcriptional regulators Irf1 and Irf8 and nod-like receptors Naip5 and Nlrc4. Infect Immun 2009; 77: 4794–4805.
Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science 2015; 350: 399–404.
Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 2015; 350: 404–409.
Hu Z, Yan C, Liu P, Huang Z, RuiMa, Zhang C et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 2013; 341: 172–175.
Halff EF, Diebolder CA, Versteeg M, Schouten A, Brondijk THC, Huizinga EG . Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J Biol Chem 2012; 287: 38460–38472.
Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012; 490: 539–542.
Matusiak M, Van Opdenbosch N, Walle LV, Sirard J-C, Kanneganti T-D, Lamkanfi M . Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc Natl Acad Sci USA 2015; 112: 1541–1546.
Zinngrebe J, Montinaro A, Peltzer N, Walczak H . Ubiquitin in the immune system. EMBO Rep 2013; 15: 28–45.
Malynn BA, Ma A . Ubiquitin makes its mark on immune regulation. Immunity 2010; 33: 843–852.
Alfano C, Faggiano S, Pastore A . The ball and chain of polyubiquitin structures. Trends Biochem Sci 2016; 41: 371–385.
Clague MJ, Heride C, Urbé S . The demographics of the ubiquitin system. Trends Cell Biol 2015; 25: 417–426.
Husnjak K, Dikic I . Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem 2012; 81: 291–322.
Scott D, Oldham NJ, Strachan J, Searle MS, Layfield R . Ubiquitin-binding domains: mechanisms of ubiquitin recognition and use as tools to investigate ubiquitin-modified proteomes. Proteomics 2015; 15: 844–861.
Matsumoto ML, Dong KC, Yu C, Phu L, Gao X . Engineering and structural characterization of a linear polyubiquitin-specific antibody. J Mol Biol 2012; 418: 134–144.
Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y et al. MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J Immunol 2015; 194: 4880–4890.
Mayor A, Martinon F, De Smedt T, Pétrilli V, Tschopp J . A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol 2007; 8: 497–503.
Peart JR, Lu R, Sadanandom A, Malcuit I, Moffett P, Brice DC et al. Ubiquitin ligase-associated protein SGT1 is required for host and nonhost disease resistance in plants. Proc Natl Acad Sci USA 2002; 99: 10865–10869.
Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL et al. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J Biol Chem 2015; 290: 18124–18133.
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015; 160: 62–73.
Sergin I, Bhattacharya S, Emanuel R, Esen E, Stokes CJ, Evans TD et al. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci Signal 2016; 9: ra2.
Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016; 164: 896–910.
Holcik M, Gibson H, Korneluk RG . XIAP: apoptotic brake and promising therapeutic target. Apoptosis 2001; 6: 253–261.
Damgaard RB, Nachbur U, Yabal M, WW-L Wong, Fiil BK, Kastirr M et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 2012; 46: 746–758.
Vandenabeele P, Bertrand MJM . The role of the IAP E3 ubiquitin ligases in regulating pattern-recognition receptor signalling. Nat Rev Immunol 2012; 12: 833–844.
Yabal M, Müller N, Adler H, Knies N, Groß CJ, Damgaard RB et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep 2014; 7: 1796–1808.
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN . The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013; 153: 348–361.
Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol 2013; 191: 4358–4366.
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP et al. Failure to regulate TNF-induced NF-kappa B and cell death responses in A20-deficient mice. Science 2000; 289: 2350–2354.
Coornaert B, Carpentier I, Beyaert R . A20: central gatekeeper in inflammation and immunity. J Biol Chem 2009; 284: 8217–8221.
Harhaj EW, Dixit VM . Regulation of NF-κB by deubiquitinases. Immunol Rev 2012; 246: 107–124.
Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 2015; 528: 370–375.
Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014; 512: 69–73.
Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA et al. A20 Restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015; 42: 55–67.
Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet 2016; 48: 67–73.
Kumar Y, Radha V, Swarup G . Interaction with Sug1 enables Ipaf ubiquitination leading to caspase 8 activation and cell death. Biochem J 2010; 427: 91–104.
Veeranki S, Choubey D . Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: regulation of subcellular localization. Mol Immunol 2012; 49: 567–571.
Schattgen SA, Fitzgerald KA . The PYHIN protein family as mediators of host defenses. Immunol Rev 2011; 243: 109–118.
Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L et al. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol 2013; 87: 8606–8623.
Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Jin T et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci USA 2013; 110: E4571–E4580.
Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ et al. IFI16 DNA sensor Is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2013; 343: 428–432.
Ishikawa H, Barber GN . STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008; 455: 674–678.
Zhong B, Yang Y, Li S, Wang Y-Y, Li Y, Diao F et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008; 29: 538–550.
Sun W, Li Y, Chen L, Chen H, You F, Zhou X et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 2009; 106: 8653–8658.
Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B et al. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol 2011; 187: 2595–2601.
Au WC, Moore PA, Lowther W, Juang YT, Pitha PM . Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc Natl Acad Sci USA 1995; 92: 11657–11661.
Sun L, Wu J, Du F, Chen X, Chen ZJ . Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339: 786–791.
Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM et al. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci USA s 2015; 112: E1773–E1781.
Storek KM, Gertsvolf NA, Ohlson MB, Monack DM . cGAS and Ifi204 cooperate to produce type I IFNs in response to francisella infection. J Immunol 2015; 194: 3236–3245.
Orzalli MH, Deluca NA, Knipe DM . Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci USA 2012; 109: E3008–E3017.
Johnson KE, Chikoti L, Chandran B . Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J Virol 2013; 87: 5005–5018.
Kalamvoki M, Roizman B . HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc Natl Acad Sci USA 2014; 111: E611–E617.
Cuchet-Lourenço D, Anderson G, Sloan E, Orr A, Everett RD . The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol 2013; 87: 13422–13432.
Van Opdenbosch N, Gurung P, Vande Walle L, Fossoul A, Kanneganti T-D, Lamkanfi M . Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat Commun 2014; 5: 3209.
Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch SL et al. Deubiquitinases regulate the activity of caspase-1 and interleukin-1 secretion via assembly of the inflammasome. J Biol Chem 2013; 288: 2721–2733.
Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, Cohen RE . K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J 2009; 28: 621–631.
Franchi L, Chen G, Marina-Garcia N, Abe A, Qu Y, Bao S et al. Calcium-independent phospholipase A2 beta is dispensable in inflammasome activation and its inhibition by bromoenol lactone. J Innate Immun 2009; 1: 607–617.
Hilbi H, Puro RJ, Zychlinsky A . Tripeptidyl peptidase II promotes maturation of caspase-1 in Shigella flexneri-Induced macrophage apoptosis. Infect Immun 2000; 68: 5502–5508.
Chavarría-Smith J, Vance RE . Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 2013; 9: e1003452.
Boyden ED, Dietrich WF . Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 2006; 38: 240–244.
Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 2012; 8: e1002638.
Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I et al. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One 2012; 7: e49741.
Liao KC, Mogridge J . Expression of Nlrp1b inflammasome components in human fibroblasts confers susceptibility to anthrax lethal toxin. Infect Immun 2009; 77: 4455–4462.
Axtell MJ, Staskawicz BJ . Initiation of RPS2-specified disease resistance in Arabidopsis is coupled to the AvrRpt2-directed elimination of RIN4. Cell 2003; 112: 369–377.
Mackey D, Belkhadir Y, Alonso JM, Ecker JR, Dangl JL . Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell 2003; 112: 379–389.
Maekawa T, Kufer TA, Schulze-Lefert P . NLR functions in plant and animal immune systems: so far and yet so close. Nat Immunol 2011; 12: 817–826.
Huang P-N, Shih S-R . Update on enterovirus 71 infection. Curr Opin Virol 2014; 5: 98–104.
Wang H, Lei X, Xiao X, Yang C, Lu W, Huang Z et al. Reciprocal Regulation between Enterovirus 71 and the NLRP3 inflammasome. Cell Rep 2015; 12: 42–48.
Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J et al. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 2013; 9: e1003231.
Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J . Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol 2013; 87: 1690–1698.
Flynn JL, Chan J . Immunology of tuberculosis. Annu Rev Immunol 2001; 19: 93–129.
Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A et al. Cutting edge: caspase-1 independent IL-1 production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 2010; 184: 3326–3330.
Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 2011; 35: 1023–1034.
McElvania-TeKippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS One 2010; 5: e12320.
Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014; 511: 99–103.
Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol 2010; 12: 1046–1063.
Carlsson F, Kim J, Dumitru C, Barck KH, Carano RAD, Sun M et al. Host-detrimental role of Esx-1-mediated inflammasome activation in mycobacterial infection. PLoS Pathog 2010; 6: e1000895.
Chen C-C, Tsai S-H, Lu C-C, Hu S-T, Wu T-S, Huang T-T et al. Activation of an NLRP3 inflammasome restricts Mycobacterium kansasii infection. PLoS One 2012; 7: e36292.
Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ . ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol 2008; 10: 1866–1878.
Kurenuma T, Kawamura I, Hara H, Uchiyama R, Daim S, Dewamitta SR et al. The RD1 locus in the Mycobacterium tuberculosis genome contributes to activation of caspase-1 via induction of potassium ion efflux in infected macrophages. Infect Immun 2009; 77: 3992–4001.
Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M et al. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol 2012; 24: 637–644.
Saiga H, Nieuwenhuizen N, Gengenbacher M, Koehler A-B, Schuerer S, Moura-Alves P et al. The recombinant BCG ΔureC::hly vaccine targets the AIM2 inflammasome to induce autophagy and inflammation. J Infect Dis 2014; 211: 1831–1841.
Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 2015; 17: 799–810.
Shah S, Bohsali A, Ahlbrand SE, Srinivasan L, Rathinam VAK, Vogel SN et al. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN- and AIM2 inflammasome-dependent IL-1 production via its ESX-1 secretion system. J Immunol 2013; 191: 3514–3518.
Dorhoi A, Nouailles G, Jörg S, Hagens K, Heinemann E, Pradl L et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol 2011; 42: 374–384.
Briken V, Ahlbrand SE, Shah S . Mycobacterium tuberculosis and the host cell inflammasome: a complex relationship. Front Cell Infect Microbiol 2013; 3: 62.
Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 2008; 3: 224–232.
Li J, Billiar TR, Talanian RV, Kim YM . Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun 1997; 240: 419–424.
Kim YM, Talanian RV, Billiar TR . Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem 1997; 272: 31138–31148.
Dimmeler S, Haendeler J, Nehls M, Zeiher AM . Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med 1997; 185: 601–607.
Mannick JB, Miao XQ, Stamler JS . Nitric oxide inhibits Fas-induced apoptosis. J Biol Chem 1997; 272: 24125–24128.
Kim YM, Talanian RV, Li J, Billiar TR . Nitric oxide prevents IL-1beta and IFN-gamma-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1beta-converting enzyme). J Immunol 1998; 161: 4122–4128.
Mishra BB, Rathinam VAK, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1β. Nat Immunol 2012; 14: 52–60.
Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I et al. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol 2012; 189: 5113–5117.
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011; 34: 213–223.
Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 2013; 23: 201–212.
Yoon S-J, Park J-Y, Choi S, Lee J-B, Jung H, Kim T-D et al. Ginsenoside Rg3 regulates S-nitrosylation of the NLRP3 inflammasome via suppression of iNOS. Biochem Biophys Res Commun 2015; 463: 1184–1189.
Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva ALN, Mineo TWP et al. Inflammasome-derived IL-1β production induces nitric oxide-mediated resistance to Leishmania. Nat Med 2013; 19: 909–915.
Buzzo CL, Campopiano JC, Massis LM, Lage SL, Cassado AA, Leme-Souza R et al. A novel pathway for inducible nitric-oxide synthase activation through inflammasomes. J Biol Chem 2010; 285: 32087–32095.
Waites KB, Talkington DF . Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev 2004; 17: 697–728.
Bose S, Segovia JA, Somarajan SR, Chang T-H, Kannan TR, Baseman JB . ADP-ribosylation of NLRP3 by Mycoplasma pneumoniae CARDS toxin regulates inflammasome activity. mBio 2014; 5: e02186–14.
Medina JL, Coalson JJ, Brooks EG, Winter VT, Chaparro A, Principe MFR et al. Mycoplasma pneumoniae CARDS toxin induces pulmonary eosinophilic and lymphocytic inflammation. Am J Respir Cell Mol Biol 2012; 46: 815–822.
Nguyen LK, Kolch W, Kholodenko BN . When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun Signal 2013; 11: 52.
Marchetti C, Toldo S, Chojnacki J, Mezzaroma E, Liu K, Salloum FN et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J Cardiovasc Pharmacol 2015; 66: 1–8.
Acknowledgements
The work was supported by the Case Research Institute and University Hospitals Case Medical Center fund and the National Institutes of Health (AR069908 to T.S.X.). We thank Dongni Ma for assistance with preparing Figure 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Yang, J., Liu, Z. & Xiao, T. Post-translational regulation of inflammasomes. Cell Mol Immunol 14, 65–79 (2017). https://doi.org/10.1038/cmi.2016.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2016.29
Keywords
This article is cited by
-
NLRP10 promotes AGEs-induced NLRP1 and NLRP3 inflammasome activation via ROS/MAPK/NF-κB signaling in human periodontal ligament cells
Odontology (2024)
-
MafB regulates NLRP3 inflammasome activation by sustaining p62 expression in macrophages
Communications Biology (2023)
-
cFLIPS regulates alternative NLRP3 inflammasome activation in human monocytes
Cellular & Molecular Immunology (2023)
-
Structural basis of NLR activation and innate immune signalling in plants
Immunogenetics (2022)
-
Migraine and neuroinflammation: the inflammasome perspective
The Journal of Headache and Pain (2021)
