
Abstract
Lung cancer and colorectal cancer account for over one-third of all cancer deaths in the United States. MicroRNA-301a (miR-301a) is an activator of both nuclear factor-κB (NF-κB) and Stat3, and is overexpressed in both deadly malignancies. In this work, we show that genetic ablation of miR-301a reduces Kras-driven lung tumorigenesis in mice. And miR-301a deficiency protects animals from dextran sodium sulfate-induced colon inflammation and colitis-associated colon carcinogenesis. We also demonstrate that miR-301a deletion in bone marrow-derived cells attenuates tumor growth in the colon carcinogenesis model. Our findings ascertain that one microRNA—miR-301a—activates two major inflammatory pathways (NF-κB and Stat3) in vivo, generating a pro-inflammatory microenvironment that facilitates tumorigenesis.
Similar content being viewed by others


Introduction
Tumor-promoting inflammation is a recognized hallmark of cancer, as neoplastic epithelial cells recruit ostensibly normal immune inflammatory cells and other stromal cells to create a tumor-promoting microenvironment. Both nuclear factor-κB (NF-κB) and Stat3 activation are important links between inflammation and cancer by regulating distinct functions in cancer cells and surrounding nontumorigenic cells. Within malignant and pre-malignant cells, NF-κB and Stat3 exert important oncogenic functions by promoting tumor cell survival and proliferation and producing cytokines to create a tumor-promoting microenvironment. At the same time, tumor-infiltrating immune cells with activated NF-κB and Stat3 produce cytokines that foster tumor cell growth and attract additional immune cells that sustain tumor-associated inflammation [1].
The study of inflammation-associated lung and colon carcinogenesis has revealed the role of NF-κB activation in both tumor cells and tumor-associated myeloid cells. For example, loss of the NF-κB subunit p65/RelA in mice reduces Kras-driven cellular transformation and lung tumor development [2, 3]. It is demonstrated that NF-κB is activated in both tumor cells and myeloid cells within lung tumors in KrasLA2 mice, but its activation in myeloid cells has a more critical role in tumor progression than in epithelial-derived tumor cells [4]. In colon tumorigenesis induced by azoxymethane and dextran sodium sulfate (AOM/DSS), deletion of IKKβ—a key component in NF-κB activation—in intestinal epithelial cells does not decrease inflammation or affect tumor size, yet it leads to a marked decrease in tumor incidence. However, deleting IKKβ in myeloid cells results in a significant decrease in tumor size [5]. Similar to NF-κB, persistent activation of Stat3 is also critical to inflammation-associated tumorigenesis in the lung [6] and colon [7]. Overexpression of Stat3 in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung [6], whereas loss of Stat3 in pancreatic or intestinal epithelial cells inhibits tumorigenesis [7, 8]. Induced Stat3 ablation in hematopoietic cells in adult mice modulates cytokine production to inhibit tumor-specific immune responses and inflammation [9, 10]. These cytokines, including interleukin (IL)-6, tumor necrosis factor-α (TNF-α), and IL-17, are transactivated by NF-κB and/or Stat3 and are direct mediators in the tumor microenvironment, stimulating tumor and immune cell proliferation, and fostering tumor growth and progression. High expression levels of these cytokines are found in different types of epithelial-derived tumors and is associated with poor prognosis [11].
Over 1000 miRNA genes have been identified in the human genome, rendering them one of the most abundant classes of regulatory molecules. Despite that a substantial number of miRNAs modulate NF-κB or Stat3 activation [12, 13], there have been few investigations of miRNAs using animal models to appraise their roles in the pathobiology of inflammatory-associated cancer. MicroRNA-301a (miR-301a) is overexpressed in lung cancer [14–16] and colon cancer [17], and previous studies indicated that miR-301a is a potential oncogenic miRNA and contributes to tumor formation (18–21). Inhibition of miR-301a reduces anchorage-independent colony formation of lung cancer cells [18] and attenuates xenograft pancreatic tumor growth [19]. In gastric cancer, overexpression of miR-301a promotes cell proliferation [20]. Of strongest relevance to inflammation at the molecular level, miR-301a is an activator of both NF-κB and Stat3 in tumor cells [19], and in inflammatory T cells [21]. Furthermore, NF-κB transactivates the miR-301a gene [19]. Collectively, these in vitro data implicate that miR-301a promotes inflammation during tumorigenesis. In the present study, we report that miR-301a deficiency inhibits the activation of both NF-κB and Stat3, and protects mice from tumorigenesis at the two major cancer sites. Through these genetic and chemical-induced carcinogenesis models, our findings suggest that miR-301a is a key positive regulator of inflammation, and that it can be attenuated to reduce the burden of cancer mortality.
Results
miR-301a deficiency suppresses Kras-driven lung tumorigenesis
To explore the role of miR-301a in inflammation and tumorigenesis, we performed genetic targeting of the miR-301a gene, which is located between exons 1 and 2 of the Ska2 gene in the mouse genome (Supplementary Figure S1A). To delete miR-301a in mouse embryonic stem (ES) cells, we generated a vector that replaces the precursor to miR-301 with a neomycin (NEO)-resistance expression cassette flanked by FLP recombinase target sequences. Targeted ES colonies were selected for the presence of NEO (Supplementary Figure S1B). The ES cells and all mice used were on a pure C57bl/6J (B6) genetic background. To reduce the risk of altering the expression of the host gene Ska2, the NEO resistance cassette was removed by crossing miR-301+/NEO mice with mice carrying the Flpe recombinase gene (Supplementary Figure S1C). Heterozygous (miR-301a+/−) mice were intercrossed to obtain homozygous (miR-301a−/−) mice, which were born at the predicted Mendelian frequency and were viable, with no obvious developmental defects or physiological abnormalities.
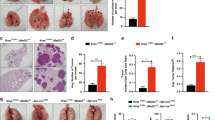
We measured the expression levels of miR-301a in an experimental mouse model of non-small cell lung cancer using KrasLA2 mice with a targeted and latent mutant KrasG12D allele. These animals develop multifocal lung tumors with 100% penetrance, and, less frequently, develop thymic lymphomas and skin papillomas [22]. miR-301a was highly expressed both in lung tumors and in spleens in KrasLA2 mice compared with wild-type (WT) mice as determined by quantitative real-time PCR (qPCR; Figure 1a). Of note, the expression level of miR-301a in spleens represented a 9.4-fold upregulation, whereas that in lung tumors represented a 2.6-fold upregulation. To investigate the role of miR-301a in lung tumor development, we crossed the miR-301a−/− mice with KrasLA2 mice. Compared with KrasLA2 mutant mice, miR-301a−/−;KrasLA2 mice showed significantly fewer lung tumors at 18 weeks, either by visual inspection (Figure 1b) or by histological analysis with hematoxylin and eosin staining (Figures 1c–e). In addition, smaller tumors developed in miR-301a−/−;KrasLA2 mice (Figure 1f). Next, we analyzed whether deletion of miR-301a alters the tumor spectrum. By the age of 18 weeks, all miR-301a−/−;KrasLA2 mice developed lung tumors but showed a decreased incidence of thymic lymphomas (21.4% compared with 32.3% in KrasLA2 mice) and skin papillomas (7.1% compared with 17.6%; Figure 1g). In agreement with a reduced tumor burden, miR-301a−/−;KrasLA2 mice showed significantly increased survival rates compared with their KrasLA2 littermates (Figure 1h; 261±98 days versus 165±64 days; P<0.0001).

miR-301a deletion reduces lung tumor development and increases survival in KrasLA2 mice. (a) Expression of miR-301a in lung tumors and spleens from KrasLA2 mice. Total RNA of lung and spleen was extracted from WT and KrasLA2 mutant littermates (n=5 per group) at 9, 13, and 18 weeks of age. (b–d) Representative gross (b) and hematoxylin and eosin (H&E) histological sections (c, d) of lungs isolated from KrasLA2 (n=5) and miR-301a−/−;KrasLA2 (n=6) mice at 18 weeks of age. Images in d show the enlargement of boxed areas from c. Scale bars, 5 mm (b, c) or 500 μm (d). (e) Total number of tumor nodules on the lung surface of KrasLA2 (n=5) and miR-301a−/−;KrasLA2 (n=6) littermates at 18 weeks of age. (f) Distribution of various-sized lung tumors from KrasLA2 (n=5) and miR-301a−/−;KrasLA2 (n=6) mice at 18 weeks of age. (g) Tumor spectrum in KrasLA2 (n=34) and miR-301a−/−;KrasLA2 (n=28) mice. (h) Survival curves of KrasLA2 (n=34) and miR-301a−/−;KrasLA2 (n=28) mice (P<0.0001). Values are means±s.d. **P⩽0.01 indicates the significance of the differences between experimental groups.
miR-301a regulates NF-κB and Stat3 activation in lung tumorigenesis
miR-301a overexpression was associated with inhibition of apoptosis and augmentation of cell proliferation in breast cancer cells and xenografts [23]. We evaluated cell proliferation and apoptosis in lung tumors. There were significantly fewer Ki67+ tumor cells (Figures 2a and b) in miR-301a−/−;KrasLA2 mice compared with KrasLA2 mice. However, there was no significant difference in cell apoptosis between miR-301a−/−;KrasLA2 tumors, and KrasLA2 control tumors, as judged by terminal deoxynucleotidyl transferase dUTP nick end labeling terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL); Figures 2c and d). We examined NF-κB and Stat3 activation in lung tumors. NF-κB and Stat3 activation were evaluated using immunostaining with antibodies against phospho-p65 or phospho-Stat3. Consistent with previous reports, both NF-κB and Stat3 were constitutively activated in KrasLA2 lung tumors [2, 4], as judged by increased phosphorylation of RelA and Stat3. However, significantly less NF-κB and Stat3 activation was found in miR-301a−/−;KrasLA2 lung tumors compared with KrasLA2 lung tumors (Figures 2e and f).

Inhibition of lung tumorigenesis in miR-301a−/−;KrasLA2 mice correlates with reduced NF-κB and Stat3 activation. (a) Cell proliferation in lung adenomas from KrasLA2 (n=5) and miR-301a−/−;KrasLA2 (n=6) at 18 weeks of age as determined by Ki67 staining. The scale bars represent 50 μm. (b) Ki67-positive cell counts in tumor tissue sections. (c) Quantification of apoptotic cells in the lung adenomas from KrasLA2 (n=5) and miR-301a−/−;KrasLA2 (n=6) at 18 weeks of age as determined by TUNEL assay. Scale bars, 200 μm. (d) TUNEL-positive cell counts in tumor tissue sections. (e) Detecting activation of NF-κB and Stat3 in lung adenoma sections from KrasLA2 (n=5) and miR-301a−/−;KrasLA2 (n=6) at 18 weeks of age using antibodies against phospho-p65 and phospho-Stat3. Scale bars, 50 μm. (f) Western blot analysis of p65 and Stat3 to detect NF-κB and Stat3 activation. (g) Relative expression levels of the indicated mRNAs isolated from tumor tissues of WT (n=3), miR-301a−/− (n=3), KrasLA2 (n=5), and miR-301a−/−;KrasLA2 mice (n=5) at 18 weeks of age. (h) IL-6 and TNFα expression in lung adenoma sections from KrasLA2 (n=5) and miR-301a−/−;KrasLA2 (n=6) at 18 weeks of age was determined by immunohistochemistry. Scale bars, 50 μm. Values are means±s.d. **P⩽0.01; *P⩽0.05. NS, not a significant difference between experimental groups.
To better understand how inhibition of NF-κB and Stat3 activation by miR-301a deletion leads to attenuated tumor growth, we first measured the expression of cytokine genes that are transactivated by NF-κB and/or Stat3 in lung tumors from miR-301a−/−;KrasLA2, and KrasLA2 mice at 18 weeks. IL-6 and TNF-α showed significantly lower expression in the lung tumors from miR-301a−/−;KrasLA2 mice compared with those from KrasLA2 mice (Figure 2g). The expression levels of IL-17, IL-1β, Interferon gamma (IFNG), IL-22, and IL-10 (Supplementary Figures S2A–E) showed no significant differences. Next, we measured IL-6 and TNFα expression by using immunohistochemistry staining. As shown in Figure 2h, Both IL-6 and TNFα were constitutively activated in peritumoral lung tissue in KrasLA2 mice, yet such activation was markedly inhibited in miR-301a−/−;KrasLA2 peritumoral lung tissue, mainly in infiltrating inflammatory cells.
To further determine whether miR-301a deletion reduces Kras-driven cell transformation, we used tumor cells transformed from WT and miR-301a−/− mouse embryonic fibroblasts cells (MEFs) using retroviruses expressing SV40 large T antigen and H-Ras. Transformed miR-301a−/− MEFs proliferated at a slower rate than WT counterparts (Supplementary Figure S3A). However, there was no significant difference in anchorage-independent colony formation between transformed WT and miR-301a−/− cells (Supplementary Figures S3B and C). We then examined the tumorigenicity of transformed WT and miR-301a−/− MEFs by subcutaneously injecting them into athymic-immunodeficient mice. As shown in Supplementary Figure S3D, tumors in nude mice inoculated with WT or miR-301a−/− MEFs showed no significant difference in growth.
NF-κB is activated upon TNF-α treatment, and Stat3 is activated upon IL-6 treatment in various cell types, including tumor cells and immune cells. Constitutive activity of NF-κB was maintained by activated Stat3 in tumor cells and tumor-associated immune cells [24], and NF-κB activation was also induced by IL-6 [25]. We sought to determine whether genetic deletion of miR-301a inhibits NF-κB or Stat3 activation with IL-6 or TNF-α stimulation in MEFs, tumor cells transformed from MEFs with H-Ras and SV40 large T antigen, CD4+ T cells, splenic CD11b+ cells (including monocytes and macrophages), and CD11c+ cells (including dendritic cells). We found that IL-6 activated both NF-κB and Stat3 in transformed cells, and three types of immune cells, and such activation was markedly inhibited by deletion of miR-301a (Supplementary Figure S4A–E). Furthermore, NF-κB activation by TNF-α was inhibited by miR-301a deficiency in MEFs, CD11b+ cells, and CD11c+ cells but not in transformed cells or in CD4+ T cells (Supplementary Figures S4A–E). These data suggest that miR-301a-mediated NF-κB and Stat3 activation in vitro is dependent on the cellular context.
miR-301a deficiency reduces chemical-induced colon tumorigenesis
Colitis-associated cancer (CAC) is a type of colon cancer that appears in patients suffering from ulcerative colitis, a chronic inflammatory bowel disease. Mouse models of CAC induced by the carcinogen AOM and repeated administration of the irritant DSS were the first in which it was demonstrated that inflammation is essential to carcinogen-induced tumorigenesis. We studied the role of miR-301a in inflammation and colon cancer by treating WT and miR-301a−/− mice with one dose of AOM followed by three cycles of oral DSS administration. All WT mice developed colon tumors, but a quarter of miR-301a−/− mice developed no tumors. All colon tumors developed in the middle and the distal colon (Figure 3a), and loss of miR-301a markedly inhibited tumor growth (Figures 3a and b) and resulted in fewer tumors per animal (Figure 3c). The average tumor size was significantly smaller in miR-301a−/− mice (Figure 3d), and miR-301a−/− mice exhibited a lower frequency of large adenomas than WT mice (Figure 3e). Next, we performed Ki67 staining and found that there was significantly decreased cell proliferation in miR-301a−/− mice compared with WT controls (Figures 3f and g). In agreement with the previous role of miR-301a in lung tumorigenesis, immunohistochemical staining revealed downregulated Stat3 and NF-κB activation in miR-301a−/− mice treated with AOM and DSS (Figures 3h and i). These results demonstrate that miR-301a deficiency inhibits colon carcinogenesis in mice by downregulating Stat3 and NF-κB activation.

Deletion of miR-301a reduces colitis-associated colon cancer development. (a) Study timeline of CAC and representative overview of colon from treated WT (n=10) and miR-301a−/− (n=10) mice. (b) Hematoxylin and eosin (H&E) histological sections of colon tumors from WT (n=10) and miR-301a−/− (n=10) mice. Scale bars, 500 μm (left) and 100 μm (right). (c–e) Tumor number (c), tumor size (d), and tumor size distribution (e) in WT (n=10) and miR-301a−/− (n=10) mice. Values are means±s.d. **P⩽0.01 indicates the significance of the differences between the indicated groups. (f) Cell proliferation in colon tumors from WT (n=10) and miR-301a−/− (n=10) mice as determined by Ki67 staining. Scale bars, 50 μm. (g) Ki67-positive cell counts in tumor sections of WT (n=10) and miR-301a−/− (n=10) mice. Values are means±s.d. **P⩽0.01 indicates the significance of the differences between the indicated groups. (h, i) Stat3 and NF-κB activation in colon tumors from WT (n=10) and miR-301a−/− (n=10) mice was determined by immunohistochemistry by using antibodies against phospho-Stat3 (h) and phospho-p65 (i). Scale bars, 500 μm (left) and 100 μm (right).
miR-301a deficiency decreases the severity of DSS-induced colitis
Inhibition of inflammation leads to reduction of tumorigenesis in the CAC mouse model [5, 26], so we sought to determine whether attenuated tumorigenesis in miR-301a−/− mice is due to suppressed intestinal inflammation. We first measured the expression level of miR-301a in WT mice with drinking water containing 3.5% DSS for 5 days. Compared with controls, the ΔCt values of miR-301a normalized to a control RNA (snoRNA292) were significantly lower in colon from mice with 3.5% DSS (P=0.008; Figure 4a). The difference in ΔCt values represented a 2.8-fold upregulation (using mean values) of miR-301a during colitis. Next, we evaluated the severity of colitis in WT and miR-301−/− mice treated with 3.5% DSS. Compared with WT controls, miR-301a−/− mice showed significantly less body weight loss (Figure 4b) and reduced colon shortening (Figures 4c and d). There were fewer incidences of bloody stool, rectal bleeding, and anal prolapse in miR-301a−/− mice than in WT mice. Furthermore, histopathological assessment of colitis severity scores showed less-damaged colonic mucosa in miR-301a−/− mice than in WT mice when mucin depletion, epithelial degeneration, and epithelial cellular necrosis were evaluated (Figures 4e and f). Intestinal epithelial cell proliferation, as judged by Ki67 staining, was significantly elevated in WT mice upon DSS treatment, while miR-301a deletion resulted in fewer proliferating cells than in WT controls (Figures 4g and h).

miR-301a deficiency inhibits the severity of colitis. (a) Expression of miR-301a during DSS-induced acute colitis. (b) Body weight loss during DSS-induced colitis in WT (n=10) and miR-301a−/− (n=10) mice. (c) Overview of colon shortening in treated mice. (d) Colon length of WT (n=10) and miR-301a−/− (n=10) mice with and without DSS treatment. (e) Colitis severity score 12 days after DSS-treated WT (n=10) and miR-301a−/− (n=10) mice. (f) Representative histological sections of mouse colon stained with hematoxylin and eosin (H&E; n=10 per group). Scale bars, 200 μm. (g) Cell proliferation during DSS-induced colitis in WT (n=10) and miR-301a−/− (n=10) mice as determined by Ki67 staining. Scale bars, 50 μm. (h) Ki67-positive cell counts in colon sections (n=10 per group). Values are means±s.d. **P⩽0.01 indicates the significance of the differences between the indicated groups.
We measured NF-κB and Stat3 activation in WT and miR-301a−/− mice following DSS exposure. Both NF-κB and Stat3 were constitutively activated in treated WT colon, yet such activation was markedly inhibited in miR-301a−/− colon (Figures 5a and b). Careful examination revealed that NF-κB and Stat3 activation were inhibited in both epithelial cells and infiltrating immune cells with miR-301a deletion. The mRNA levels of inflammatory cytokines such as IL-6, TNF-α, and IL-17 were elevated in WT colon treated with DSS, whereas they were downregulated in miR-301a−/− colon compared with WT controls (Figure 5c). As the production of these cytokines secreted from colon mucosa increased progressively during DSS treatment [27], we next prepared serum from mice treated with DSS. A marked increase in IL-6, TNF-α, and IL-17 levels was observed in WT mice, yet their levels decreased significantly in miR-301a−/− mice (Figures 5d–f). Furthermore, we also isolated colon tissues from mice after DSS treatment and examined cytokine secretion ex vivo. Secretion of IL-6, TNF-α, and IL-17 decreased with miR-301a deletion (Figures 5g–i). These data suggest that miR-301a deletion reduces NF-κB and Stat3 activation, downregulating the expression of cytokines during colon inflammation.

Reduced severity of colitis in miR-301a−/− mice correlates with inhibition of NF-κB and Stat3 activation and downregulation of cytokine expression. (a) Colon sections were stained using antibodies against phospho-Stat3 and phospho-p65 to evaluate activated NF-κB and Stat3 (n=10 per group). Scale bars, 50 μm. (b) Western blot analysis of colon tissue lysates for NF-κB and Stat3 activation. (c) Cytokine gene expression in colon tissues of WT and miR-301a−/− mice as determined by qPCR (n=3 for the untreated group and n=5 for the DSS-treated group). (d–f) IL-6 (d), TNF-α (e), and IL-17 (f) levels in the serum of WT (n=5) and miR-301a−/− (n=5) mice as determined by ELISA. (g–i) Levels of IL-6 (g), TNF-α (h), and IL-17 (i) secreted from colon ex vivo as determined by ELISA. Values are means±s.d. *P⩽0.05. **P⩽0.01 indicates the significance of the differences between the indicated groups.
Deletion of miR-301a in bone marrow-derived cells inhibits colon inflammation and tumorigenesis
Bone marrow transplantation allows one to obtain chimeric mice with the same epithelial cells but different immune cells. Owing to the lethal doses of radiation, healthy mice are required as the host. We therefore chose the colon inflammation and tumorigenesis models rather than the lung or pancreas tumorigenesis models in which mice develop neoplasia at earlier ages. We performed bone marrow transplantation using WT mice (with CD45.1) as recipients and bone marrow from either miR-301a−/− or WT mice as donors (both with CD45.2). Four weeks after bone marrow transplantation, mice were analyzed for hematopoietic recovery and the level of chimerism. Flow cytometry analysis of peripheral blood mononuclear cells for the presence of donor-derived leukocytes expressing the CD45.2 isoform showed high levels of chimerism in recipient mice (92.3% in WT mice and 93.3% in miR-301a−/− mice). Both groups of mice were administered DSS, and WT mice transplanted with miR-301a−/− hematopoietic cells showed less-severe colitis, as indicated by less body weight loss, reduced colon shortening, fewer histology alterations, and lower colitis scores (Figures 6a–e). The sustained phosphorylation of NF-κB and Stat3 were reduced when miR-301a−/− bone marrow cells were transplanted into WT mice (miR-301a−/−→WT; Figure 6f). We next treated separate groups of transplanted mice with both AOM and DSS, and found that tumor incidence, total tumor size, and the frequency of larger adenomas were significantly reduced in WT mice with miR-301a−/− bone marrow (Figures 6g–j). In addition, the activation of NF-κB and Stat3 in WT mice transplanted with miR-301a−/− hematopoietic cells was reduced when mice were treated with AOM and DSS (Supplementary Figures S5A and B). Concomitantly, there was less IL-6 and TNFα expression in infiltrating macrophages and less IL-17 expression in T cells in the colonic mucosa of WT mice with miR-301a−/− bone marrow than that of WT mice with WT bone marrow (Supplementary Figures S5C and D). These data support the conclusion that attenuated colon tumor development in miR-301a−/−mice was due to miR-301a function in hematopoietic cells rather than in intestinal epithelial cells, underscoring the importance of miR-301a in immune cells during colon tumorigenesis.

Deletion of miR-301a in hematopoietic cells leads to reduced severity of DSS-induced colitis and colitis-associated colon cancer development. WT→WT, WT mice transplanted with bone marrow from WT mice; miR-301a−/−→WT, WT mice transplanted with bone marrow from miR-301a−/− mice (n=5 each). (a) Body weight during DSS-induced colitis in chimeric mice. (b) Overview of colon shortening in DSS-treated mice. (c) Colon length in mice treated with DSS. (d) Representative histological sections of mouse colon stained with hematoxylin and eosin (H&E). Scale bars, 200 μm. (e) Colitis severity score. (f) Western blot analysis of p65 and Stat3 to determine NF-κB and Stat3 activation. (g) H&E histological sections of colon tumors from chimeric mice treated with AOM/DSS. Scale bars, 500 μm (left) and 200 μm (right). (h–j) Bar graphs showing tumor number (h), tumor size (i), and tumor size distribution (j) in AOM/DSS-treated mice. Values are means±s.d. *P⩽0.05. **P⩽0.01 indicates the significance of the differences between the indicated groups.
miR-301a deficiency in host cells inhibits tumorigenesis in murine syngeneic xenograft models
We assessed the role of miR-301a in tumor stroma using syngeneic xenograft models. Sex- and age-matched WT and miR-301a−/− littermates were challenged with Lewis lung carcinoma (LLC1) cells. Tumors grown in miR-301a−/− mice were significantly smaller than those in WT mice (Figure 7a). Consistent with reduced tumor growth, there was a significant decrease in proliferation at the tumor–stroma border as determined by Ki67 staining in miR-301a−/− mice compared with WT mice (Figure 7b). This observation was not limited to LLC1 cells, as attenuated tumor growth was also found when miR-301a−/− mice were inoculated with B16 melanoma cells (Supplementary Figures S6A and B). Collectively, these data imply that loss of miR-301a in the tumor microenvironment but not in tumor cells (Supplementary Figure S3) impedes tumorigenesis in murine xenografts.

miR-301a deficiency in the tumor microenvironment inhibits tumor growth in mouse xenografts. (a) Tumor growth of LLC1 cells in WT and miR-301a−/− mice (n=10 per group). (b) Cell proliferation in tumors xenografted in WT and miR-301a−/− mice using LLC1 cells as determined by Ki67 staining. Values are means±s.d. Scale bars, 200 μm. (c) Intracellular flow cytometry analysis of phospho-Stat3 (left) and phospho-p65 (right) from single-cell suspensions of spleen in WT and miR-301a−/− mice with (lower) or without (upper) LLC1 tumor implantation. (d) Western blot analysis to determine NF-κB and Stat3 activation in tumors. (e) Expression of cytokine genes in LLC1 tumors grown in WT and miR-301a−/− mice as determined by qPCR. **P<0.01 indicates a significant difference.
To validate the function of miR-301a in tumor-infiltrating immune cells, we isolated CD11b+ myeloid and CD4+ T cells from tumors developed in WT mice xenografted with LLC1 cells or from spleens from xenografted or uninoculated mice. We found that miR-301a was highly expressed in tumor-associated CD11b+ myeloid cells (12.8-fold; Supplementary Figure S7A) and CD4+ T cells (2.7-fold; Supplementary Figure S7B) compared with the splenic CD11b+ and CD4+ counterparts from unxenografted mice. We next measured the activation of NF-κB and Stat3 in immune cells from spleens of WT and miR-301a−/− mice, with or without LLC1 injection, or from LCC1-xenografted tumors using flow cytometry. As shown in Figure 7c, splenic cells from WT mice subcutaneously injected with LLC1 tumor cells displayed increased NF-κB and Stat3 activation, and such activation was significantly inhibited in miR-301a−/− mice. Similar patterns were observed in the tumor-infiltrating immune cells isolated from xenografted tumors. When tumor-associated immune cells were separately measured, reduced NF-κB and Stat3 activation were found in macrophages (Gr1–CD11b+), myeloid-derived suppressor cells (Gr1+CD11b+), dendritic cells (CD11c+), and CD4+ T cells (CD4+) isolated from tumors in miR-301a−/− mice compared with WT controls (Supplementary Figure S7C). We also performed western blot analysis with protein extracts from LCC1 tumors, and found higher IκBα protein levels and reduced Stat3 phosphorylation, an indication of reduced activation of both NF-κB and Stat3, in tumors from miR-301a−/− mice than those from WT mice. This is likely due to elevated expression of Nkrf and Pias3, respective inhibitors to NF-κB and Stat3, and validated miR-301a target genes [19, 21] (Figure 7d). We determined the mRNA levels of Nkrf and Pias3 in CD11b+ cells and CD4+ T cells isolated from LLC1 tumors using qPCR, and we found that the both Nkrf and Pias3 were upregulated in CD11b+ cells and CD4+ T cells isolated from tumors grown in miR-301a−/− mice compared with those from WT mice (Supplementary Figure S7D). Finally, we examined whether miR-301a regulates cytokine gene expression in LLC1-xenografted tumors in WT and miR-301a−/− mice. IL-6, TNF-α, and IL-17 (Figure 7e), but not IFNG, IL-10, IL-12A, IL-22, and IL-23 (Supplementary Figure S7E), were downregulated in miR-301a−/− mice challenged with LLC1 compared with WT controls. Taken together, these results support that miR-301a is upregulated in tumor-associated immune cells during tumorigenesis and that miR-301a activates both NF-κB and Stat3 through targeting Nkrf and Pias3 and elevates cytokine production and tumor cell proliferation.
Discussion
Among the estimated 570 000 annual cancer deaths in the United States, cancers at the sites of lung and colon account for 157 000 and 51 000 deaths, respectively; combined, they cause ~30–40% of all cancer-related deaths [28]. The two types of deadliest cancers share certain features. First, Kras is one of the most frequently mutated oncogenes in both lung and colon cancers; it is mutated in ~25–50% of lung cancers and ~50% of colon cancers, and Kras-activation-based mouse models have been developed to recapitulate the pathobiology of lung cancer. In rodent colon carcinogenesis induced by AOM/DSS, Kras mutation is observed in most colon tumors from rats and less frequently in those from mice [29]. Kras activation constitutively induces the activation of NF-κB and Stat3 [2, 30, 31]. Second, inflammation is a significant risk factor for these cancers. Epidemiological studies have consistently linked the presence of chronic obstructive pulmonary disease to the development of lung cancer [32]. Long-standing ulcerative colitis predisposes to the development of CAC [33]. Third, the pro-inflammatory miRNA miR-301a, which activates both NF-κB and Stat3, is overexpressed in these two types of cancer. In the present work, we investigated the role of miR-301a in tumorigenesis at the sites of lung using a genetic model and of colon using a chemical-induced model. Our results show that deletion of miR-301a in KrasLA2 mice reduces multiplicity, incidence, and size of lung tumors, and miR-301a deficiency increases animal survival due to the moderated tumor burden. We also demonstrated that miR-301a-null mice are protected from DSS-induced colitis and AOM/DSS-induced carcinogenesis. These studies imply that miR-301a overexpression contributes to the etiology of the two deadliest solid malignancies.
Both NF-κB and Stat3 control the dialog between the tumor cell and its microenvironment by regulating several cytokine and inflammatory immune mediators. IL-6 and TNF-α are two pro-inflammatory cytokines that contribute to pronounced immune cell infiltration and create a tumor-promoting microenvironment; inactivated NF-κB or Stat3 in myeloid cells reduced tumor growth and decreased the production of IL-6 and TNF-α in the tumor microenvironment [34–36]. IL-17 is a pleiotropic pro-inflammatory cytokine and is generally considered to promote non-small cell lung cancer and colon cancer [37–40], although it was found to inhibit tumorigenesis in a xenograft model using MC38 murine colon cancer cells [41]. Thus, the net effects of IL-17 in tumorigenesis vary and are dependent on the context. Nonetheless, NF-κB, Stat3, and these three cytokines are interconnected: IL6 activates Stat3, TNF-α and IL-17 activate NF-κB, while NF-κB and Stat3 control the production of all three cytokines with the exception of TNF-α, which is not transactivated by Stat3. In our study, we found that there is higher expression of IL-6, TNF-α, and IL-17 during tumorigenesis at the sites of lung and colon, excluding IL-17 in lung tumors, where fewer CD4+ T cells are found. Without exception, upregulation of these cytokines is inhibited when miR-301a is deleted. These results underscore the importance of miR-301a activation of both NF-κB and Stat3 in controlling cytokine expression and promoting tumorigenesis.
Previously, miR-301a was shown to be an activator of NF-κB in epithelial-derived tumor cells [19] and macrophages [42], as well as an activator of Stat3 in T cells [21]. We dissected the role of miR-301a in tumor cells using Ras-driven MEF transformation and in immune cells using bone marrow transplantation. First, miR-301a-null transformed cells grew similar sizes as their WT counterparts when both are inoculated into the same immunodeficient host, yet the same syngeneic tumor cells (either LCC1 or B16) grew faster in WT mice than in miR-301a-null mice. Second, we replaced the bone morrow of WT mice with that from miR-301-null mice or their WT littermates, and found that WT mice possessing miR-301-null bone marrow develop less-severe colon inflammation upon DSS treatment and fewer and smaller colon tumors upon AOM/DSS treatment than WT mice possessing WT bone marrow. Collectively, these data support the conclusion that miR-301a promotes tumorigenesis primarily by its effect on immune cells (and potentially other types of cells in tumor stroma) rather than by its direct effect on Ras-transformed cells (Figure 8).

Schematic of the effects of miR-301a on inflammation and tumor formation.
In summary, we have shown that miR-301a is critical for NF-κB and Stat3 activation in the tumor microenvironment in multiple mouse tumorigenesis models. Our results suggest that specific pharmacological inhibition of miR-301a in tumor-associated immune cells is an effective tool against deadly solid malignancies such as lung cancer and colon cancer. It is notable that many key components of NF-κB and Stat3 signaling are essential to embryonic viability in mice [43, 44], and excessive and prolonged inhibition of NF-κB and Stat3 could be detrimental to the whole organism, owing to their roles in innate and adaptive immunity. miR-301a is expressed at extremely low levels in major tissues of adult mice [45], and except for being overexpressed in cancers and transiently expressed during embryonic development, miR-301a levels remain low in most human tissues under physiological conditions [46]. Moreover, mice without miR-301a appear to have no developmental or physiological abnormalities, which is surprising since miR-301a is such a critical regulator of two major inflammation pathways. This implicates that there is unknown functional redundancy of Nkrf and/or Pias3, as we note that mice with NKRF deletion are viable and are indistinguishable from WT mice under physiological conditions or challenged with various pathogens [47]. Nonetheless, targeting miR-301a may be superior to targeting NF-κB and Stat3 in therapeutic development against inflammation-associated cancers.
Materials and methods
Lung tumorigenesis
KrasLA2 mice maintained in a C57BL/6J (B6) background were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Generation of miR-301a−/− mice in the C57BL/6J background was described in supplemental experimental procedures. We performed qPCR to determine the mRNA levels of SKA2 (the host gene) and miR-301a in lung and colon tissues from WT and miR-301a−/− littermates, and found that there was no difference in SKA mRNA levels, and that miR-301a was undetectable in miR-301a−/− tissues. miR-301a−/− mice were bred with KrasLA2 mice to obtain miR-301a+/−; KrasLA2 mice, which were intercrossed to generate miR-301a−/−; KrasLA2, and KrasLA2 mice. Mice were killed at the indicated experimental time or when significant tumor burden or distress was found. For survival analysis, 34 KrasLA2 mice and 28 miR-301a−/−; KrasLA2 mice were analyzed weekly after 8 weeks of age and were killed when exhibiting shortness of breath or other signs of mortal sickness.
CAC models
CAC was induced with AOM/DSS as described [26]. Briefly, mice were injected intraperitoneally with 10 mg kg−1 AOM (Sigma, St. Louis, MO, USA) on the first day. After 5 days, mice were fed with drinking water containing 2.5% DSS for 5 days. Animals were then given regular drinking water for 14 days, followed by two additional DSS treatment cycles. On day 140, colons were collected, and tumors were counted and measured under a dissecting microscope.
Acute colitis
Acute colitis was induced by following a standard DSS-induced inflammatory bowel disease protocol. Eight-week-old WT and miR-301a−/− male mice (n=10 per group) were fed with drinking water containing 3.5% DSS for 5 days, followed by recovery for 5 days on normal drinking water. Mice were monitored twice weekly based on three parameters: stool consistency, fecal blood, and anal prolapse. Histopathological alterations in the colon were examined on hematoxylin and eosin-stained sections. The evaluation of colitis and severity score was performed as described [48]. Grade 0, normal colonic mucosa; Grade 1, shortening and loss of the basal one-third of the actual crypts with mild inflammation and edema in the mucosa; Grade 2, loss of the basal two-thirds of the crypts with moderate inflammation in the mucosa; Grade 3, loss of all crypts with severe inflammation in the mucosa but with the surface epithelium still remaining; and Grade 4, loss of all crypts and the surface epithelium with severe inflammation in the mucosa, muscularis propria, and submucosa.
Bone marrow transplantation
For bone marrow transplantation, 12-week-old recipient male WT CD45.1 mice (B6.SJL-Ptprca Pep3b/BoyJ; Jackson Laboratory) were conditioned with 950-cGy total body irradiation from a cesium source (Gamma-cell 40; Nordion, Ottawa, ON, Canada). Both the recipient and donor mice were maintained on normal chow throughout the experiment. Twenty-four hours after irradiation, mice were transplanted with bone marrow cells (1×107 cells in 0.1 ml phosphate-buffered saline (PBS) per animal) isolated from the tibias and femurs of 11-week old male WT and knockout (KO) donor mice carrying CD45.2 through the retro-orbital plexus with a 27-gauge needle (n=5 each). After 4 weeks, recipient mice were characterized for hematopoietic recovery and chimerism by determining the relative percentages of CD45.2 cells in peripheral blood. Co-staining with markers of T cells (CD3), B cells (B220), granulocytes (Gr-1), and monocytes (CD11b) was used to determine the levels of chimerism in lineage-specific cells. Peripheral blood was obtained through retro-orbital bleeding for leukocyte counts using a HemaVet 950 hematology system (Drew Scientific Inc., Oxford, CT, USA), and nucleated cells were analyzed by flow cytometry (LSRII, BD Biosciences, Franklin Lakes, NJ, USA) to determine the percentage of CD45.2 cells. Antibodies from eBioscience (Santa Clara, CA, USA) were used for flow cytometry analysis (CD11c PE-Cyanine7, 25-0114; Gr-1 Alexa Fluor700, 56-5931; CD3-APC-eFluor780, 47-0032; CD4-eFluor650, 95-0041; and CD11b-eFluor605, 93-0112). Antibodies from Cell Signaling (Danvers, MA, USA) (p-Stat3, no. 4323s; p-p65, no. 4886s) were used to analyze Stat3 and NF-κB activation using flow cytometry.
Histology and immunohistochemistry analyses
To evaluate tumor burden and incidence, tissues and tumors were collected at the indicated dates and perfused with 10% neutral-buffered formalin. After 3 days fixation, tissues were paraffin-embedded, sectioned (5 μm), and stained with hematoxylin or eosin for morphological analyses. For immunohistochemical staining, deparaffinized and rehydrated sections were boiled in Na-citrate buffer (10 mM, pH 6.0) for 10 min for antigen retrieval. Sections were incubated with primary antibodies for 2 h at room temperature. The antibodies used in this study are listed in Supplementary Table S2. Tissue sections were developed using the Ultra Vision Detection System Anti-Rabbit (Thermo Fisher Scientific, Waltham, MA, USA). For cases in which cells per microscopic area were counted, 1500 cells were counted per unit area (n=6) in each group to calculate the percentage of positively stained cells. For immunofluorescence staining to measure NF-κB and Stat3 in myeloid cells, antibodies against phospho-p65, phospho-Stat3, and CD11b/c were used. The CD11b/c antibodies react with macrophages, granulocytes, and dendritic cells. After rinsing with PBS, the sections were incubated with Alexa Fluor 633 goat anti-rabbit antibody (Invitrogen) for 1 h at room temperature. Sections were finally incubated with CD11b/c antibodies for 1 h, followed by 1 μg ml−1 4′, 6-diamidino-2-phenylindole (Sigma) for 5 min. Antibodies from Abcam (Cambridge, MA, USA) (p-p65, ab28856; CD31, ab28364; CD11b/c, ab1211; IL-6, ab6672; IL-17, ab79056; CD3, ab16669; and TNFα, ab6671) and Cell Signaling (p-Stat3, no. 9145s; Ki67, no. 12202S) were used in these immunohistochemistry analyses. Slides were analyzed by confocal microscopy (Olympus, Center Valley, PA, USA). For the TUNEL assay, deparaffinized sections were incubated with 20 μg ml−1 proteinase K for 15 min at room temperature, washed with PBS, and incubated with TUNEL reaction mixture (Roche Applied Science) for 1 h at 37 °C in a humidified atmosphere. After staining with 4′, 6-diamidino-2-phenylindole, images were acquired using an Olympus IX51 microscope and analyzed using cellSens Dimension software (Olympus).
ELISA for cytokine expression
To determine cytokine secretion ex vivo, colon tissues were cut into small pieces, weighed, and incubated with Dulbecco's Modified Eagle's medium (DMEM) medium (~50 mg ml−1) at 37 °C for 48 h. IL-6, TNF-α, and IL-17a concentrations in culture supernatants were measured using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol.
Mouse primary cell preparation
MEFs were prepared from E13.5 embryos according to an established protocol [49, 50]. CD4+ T cells, CD11b+ cells, and CD11c+ cells were isolated from spleens using MicroBeads (Miltenyi Biotec, San Diego, CA, USA). Bone marrow-derived macrophages were prepared using a documented protocol [51]. Briefly, 8-week-old male mice were killed and the hind legs were removed. The femurs were separated and flushed with PBS using a 3-ml syringe and a 23-gauge needle. Bone marrow was passed through a pipette several times to make a single-cell suspension. Cells were washed with PBS twice before plating into a culture dish. Cells were maintained in DMEM supplemented with 10% fetal bovine serum and 10 ng ml−1 murine granulocyte macrophage colony-stimulating factor (R&D Systems) and incubated at 37 °C in 5% CO2.
MEF transformation, soft agar, and xenograft assay
Low-passage MEFs were transformed using retrovirus prepared with pBABE-puro SV40 LT and pWZL Blast H-Ras (Addgene, Cambridge, MA, USA). After antibiotic selection, cells were split into six-well plates at 1×105 cells per plate to evaluate cell proliferation. Cell numbers were determined using an automated cell counter (Countess, Invitrogen, Waltham, MA, USA). For soft agar assays, 5×103 cells were suspended in complete medium containing 0.3% Noble agar and seeded into a six-well plate containing 0.6% Noble agar in the wells. Medium was changed twice per week. After 21 days, colonies were stained with 0.005% crystal violet and counted by using a dissecting microscope. For in vivo tumor formation, 5×105 cells were injected subcutaneously into athymic nude mice (Foxn1nu/Foxn1nu; Jackson Laboratory). For syngeneic xenografts, 1×105 cells were injected subcutaneously into WT or miR-301a−/− mice. Tumor size was measured every 3 days. Tumor volume was calculated by the formula of width2×length/2.
RNA preparation and qPCR
To measure cytokine expression, tissues or tumors were dissected from mice, and total RNA was extracted using Trizol (Invitrogen) and run on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) 2100 to ensure that the A260/A280 ratio was in the range of 1.8–2.2 and the rRNA ratio (28S/18S) was >0.9. RNA was reverse transcribed with the iScript Reverse Transcription Supermix kit (Bio-Rad, Hercules, CA, USA). Real-time PCR was performed using the iQ SYBR Green Supermix (Bio-Rad) and CFX96 Real-Time PCR detection system (Bio-Rad). The primers (Supplementary Table S1) used for real-time PCR were designed based on the Universal ProbeLibrary (Roche, Roche Life Science, Pleasanton, CA, USA) with ACTB as reference.
Western blotting
For western blotting analyses, tissues and cells were lysed with RIPA buffer (Cell Signaling), supplemented with protease inhibitor (Calbiochem, Billerica, MA, USA). Soluble proteins were subjected to SDS-polyacrylamide gel electrophoresis before being transferred to polyvinylidene difluoride (PVDF) membranes for blotting (SuperSignal West Dura Chemiluminescent, Thermo Fisher Scientific, Waltham, MA, USA). Antibodies were purchased from Abcam (p-Stat3, no. 9145s; p-p65, ab28856) and Cell Signaling (Stat3, no. 9139s; p65, no. 3034s; IκBα, no. 4814).
Statistical analysis
Statistical analysis was carried out using SPSS16.0 software (SPSS Inc. Chicago, IL, USA). An unpaired two-tailed Student’s t-test was performed for two group comparisons where data were normally distributed; and a one-way analysis of variance test was performed for multiple group comparisons or nonparametric data. For the survival analysis, Kaplan–Meier curves were analyzed by log rank test. The statistical significance was set at a P-value <0.05. Unless otherwise indicated, values were presented as mean±s.d.
All experimental procedures involving animals in this study were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Louisville.
References
Grivennikov SI, Greten FR, Karin M . Immunity, inflammation, and cancer. Cell 2010; 140: 883–899.
Basseres DS, Ebbs A, Levantini E, Baldwin AS . Requirement of the NF-κB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res 2010; 70: 3537–3546.
Finco TS, Westwick JK, Norris JL, Beg AA, Der CJ, Baldwin AS Jr . Oncogenic Ha-Ras-induced signaling activates NF-κB transcriptional activity, which is required for cellular transformation. J Biol Chem 1997; 272: 24113–24116.
Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M . Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010; 17: 89–97.
Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004; 118: 285–296.
Li Y, Du H, Qin Y, Roberts J, Cummings OW, Yan C . Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res 2007; 67: 8494–8503.
Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009; 15: 103–113.
Fukuda A, Wang SC, Morris JP, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011; 19: 441–455.
Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 2005; 11: 1314–1321.
Yu H, Pardoll D, Jove R . STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9: 798–809.
Lin W-W, Karin M . A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 2007; 117: 1175–1183.
Ma X, Becker Buscaglia LE, Barker JR, Li Y . MicroRNAs in NF-κB signaling. J Mol Cell Biol 2011; 3: 159–166.
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K . STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010; 39: 493–506.
Mascaux C, Laes JF, Anthoine G, et al. Evolution of microRNA expression during human bronchial squamous carcinogenesis. Eur Resp J 2009; 33: 352–359.
Miko E, Czimmerer Z, Csánky E, et al. Differentially expressed micrornas in small cell lung cancer. Exp Lung Res 2009; 35: 646–664.
Du L, Schageman J, Irnov, et al. MicroRNA expression distinguishes SCLC from NSCLC lung tumor cells and suggests a possible pathological relationship between SCLCs and NSCLCs. J Exp Clin Canc Res 2010; 29: 75.
Liu L, Nie J, Chen L, et al. The oncogenic role of microRNA-130a/301a/454 in human colorectal cancer via targeting Smad4 expression. PLoS One 2013; 8: e55532.
Cao G, Huang B, Liu Z, Zhang J, Xu H, Xia W, et al. Intronic miR-301 feedback regulates its host gene, ska2, in A549 cells by targeting MEOX2 to affect ERK/CREB pathways. Biochem Biophys Res Commun 2010; 396: 978–982.
Lu Z, Li Y, Takwi A, et al. miR-301a as an NF-kB activator in pancreatic cancer cells. EMBO J 2011; 30: 57–67.
Wang M, Li C, Yu B, et al. Overexpressed miR-301a promotes cell proliferation and invasion by targeting RUNX3 in gastric cancer. J Gastroenterol 2013; 48: 1023–1033.
Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW . microRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci USA 2012; 109: E1248–E1257.
Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001; 410: 1111–1116.
Shi W, Gerster K, Alajez NM, et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res 2011; 71: 2926–2937.
Lee H, Herrmann A, Deng JH, et al. Persistently activated Stat3 maintains constitutive NF-κB activity in tumors. Cancer Cell 2009; 15: 283–293.
Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV . IL-6 induces NF-κB activation in the intestinal epithelia. J Immunol 2003; 171: 3194–3201.
Liang J, Nagahashi M, Kim EY, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013; 23: 107–120.
Perše M, Cerar A . Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol 2012; 2012: 13.
Jemal A, Siegel R, Xu J, Ward E . Cancer Statistics, 2010. CA Cancer J Clin 2010; 60: 277–300.
Rosenberg DW, Giardina C, Tanaka T . Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009; 30: 183–196.
Norris JL, Baldwin AS Jr . Oncogenic Ras enhances NF-κB transcriptional activity through Raf-dependent and Raf-independent mitogen-activated protein kinase signaling pathways. J Biol Chem 1999; 274: 13841–13846.
Kim BY, Gaynor RB, Song K, Dritschilo A, Jung M . Constitutive activation of NF-κB in Ki-ras-transformed prostate epithelial cells. Oncogene 2002; 21: 4490–4497.
Houghton AM . Mechanistic links between COPD and lung cancer. Nat Rev Cancer 2013; 13: 233–245.
Eaden JA, Abrams KR, Mayberry JF . The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 2001; 48: 526–535.
Yoshimura A . Signal transduction of inflammatory cytokines and tumor development. Cancer Sci 2006; 97: 439–447.
Grivennikov SI, Karin M . Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 2010; 21: 11–19.
Bollrath J, Greten FR . IKK/NF-κB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep 2009; 10: 1314–1319.
Numasaki M, Watanabe M, Suzuki T, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol 2005; 175: 6177–6189.
Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL . Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci USA 2010; 107: 5540–5544.
Hyun YS, Han DS, Lee AR, Eun CS, Youn J, Kim HY . Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012; 33: 931–936.
Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H . IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 2009; 206: 1457–1464.
Kryczek I, Wei S, Szeliga W, Vatan L, Zou W . Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 2009; 114: 357–359.
Huang L, Liu Y, Wang L, et al. Down-regulation of miR-301a suppresses pro-inflammatory cytokines in Toll-like receptor-triggered macrophages. Immunology 2013; 140: 314–322.
Li Q, Antwerp DV, Mercurio F, Lee K-F, Verma IM . Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science 1999; 284: 321–325.
Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA 1997; 94: 3801–3804.
Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007; 129: 1401–1414.
Chiang HR, Schoenfeld LW, Ruby JG, et al. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 2010; 24: 992–1009.
Froese N, Schwarzer M, Niedick I, Frischmann U, Köster M, Kröger A, et al. Innate immune responses in NF-kB-repressing factor-deficient mice. Mol Cell Biol 2006; 26: 293–302.
Suzuki R, Kohno H, Sugie S, Nakagama H, Tanaka T . Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 2006; 27: 162–169.
Ma X, Kumar M, Choudhury SN, et al. Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis. Proc Natl Acad Sci USA 2011; 108: 10144–10149.
Xu C, Inokuma MS, Denham J, et al. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechol 2001; 19: 971–974.
Weischenfeldt J, Porse B . Bone marrow-derived macrophages (BMM): isolation and applications. CSH protocols 2008; 2008: 1–6.
Acknowledgements
We wish to thank Drs Lisa L Sandell and Jin Chen for their assistance in tissue processing and staining. This study is supported by an National Institutes of Health (NIH) R01 grant (CA138688) to YL and a grant (no. 81270547) from National Natural Science Foundation of China to XM. YL, DJC, SS, and AB are supported by the Diabetes and Obesity Center funded by National Institute of General Medical Sciences (NIGMS) (P20 GM103492).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ma, X., Yan, F., Deng, Q. et al. Modulation of tumorigenesis by the pro-inflammatory microRNA miR-301a in mouse models of lung cancer and colorectal cancer. Cell Discov 1, 15005 (2015). https://doi.org/10.1038/celldisc.2015.5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/celldisc.2015.5
This article is cited by
-
Role of the miR-301a/Fra-2/GLIPR1 axis in lung cancer cisplatin resistance
Signal Transduction and Targeted Therapy (2023)
-
miR-301a promotes lung tumorigenesis by suppressing Runx3
Molecular Cancer (2019)
-
Elevated miR-301a expression indicates a poor prognosis for breast cancer patients
Scientific Reports (2018)
-
Malignant Transformation of Human Bronchial Epithelial Cells Induced by Arsenic through STAT3/miR-301a/SMAD4 Loop
Scientific Reports (2018)
-
Neutrophil-to-Lymphocyte Ratio Predicts Outcome in Limited Disease Small-cell Lung Cancer
Lung (2017)
