
Abstract
Whether physiologically induced pluripotent stem cell (iPSC)-derived organs are immunogenic and can be used for transplantation is unclear. Here, we generated iPSC-derived skin, islet, and heart representing three germ layers of the body through 4n complementation and evaluated their immunogenicity and therapeutic efficacy. Upon transplantation into recipient mice, iPSC-derived skin successfully survived and repaired local tissue wounds. In diabetic mouse models, explanted iPSC-derived islets effectively produced insulin and lowered blood glucose to basal levels. iPSC-derived heart grafts maintained normal beating for more than 3 months in syngeneic recipients. Importantly, no obvious immune rejection responses against iPSC-derived organs were detected long after transplantation. Our study not only demonstrates the fundamental immunogenicity and function of iPSC derivatives, but also provides preclinical evidence to support the feasibility of using iPSC-derived skin, islet, and heart for therapeutic use.
Similar content being viewed by others


Introduction
Induced pluripotent stem cells (iPSCs) are derived by ectopic expression of four transcription factors in somatic cells [1]. iPSCs exhibit transcriptional and epigenetic characteristics that are highly similar to those of embryonic stem cells (ESCs). They can undergo self-renewal and maintain pluripotency to differentiate into any cell type [2–6]. Thus, iPSCs hold great promise by providing unlimited autologous cells for regenerative medicine. The therapeutic potential of iPSCs has been extensively evaluated by transplantation of cells differentiated from iPSCs in vitro into different animal models of disease. For example, transplanted iPSC-differentiated cells can effectively treat sickle cell anemia, diabetes, Parkinson's disease, spinal cord injury, myocardial infarction, and so on [7–14]. However, a few studies have reported tumors were formed in iPSC-differentiated cells due to the low purity of differentiated cells and/or unstable in vitro differentiation systems [15–19]. Furthermore, some cells derived from mouse iPSC have been shown to elicit antigen-specific immune rejection responses in a teratoma formation model [20]. By transplantation of differentiated iPSC derivatives, recent studies demonstrated that myocardial and endothelial cells, but not hepatocytes, skin, bone marrow, or neuronal cells, induced immune rejection responses [21–23]. Yet, whether organs derived from iPSCs in vivo are immunogenic and can be safely used for transplantation is still unknown.
Here we established different integration-free iPSC lines and generated iPSC mice by 4n complementation. By transplanting organs isolated from 4n complementation mice into syngeneic recipient mice, we evaluated the survival, 'ground' immunogenicity, and potential therapeutic effects of these physiological iPSC derivatives. iPSC-derived skin, heart, and islet were used to represent the three germ layers of the body and transplanted into different mouse models. The ability of iPSC-derived skin explants to heal skin lesions was examined using a wound healing mouse model, while the effect of iPSC-derived islet on high blood glucose concentrations was assessed in diabetic mice. Vascularized heterotropic transplantation of iPSC-derived heart was also performed. Tumor formation, necrosis, and T-cell infiltration of each graft type was assessed at different time points after transplantation to specifically evaluate survival and immunogenicity of grafts.
Results
Generation of integration-free iPSC lines and tetraploid (4n) complementation mice
Reactivation of integrated genomic reprogramming factors has been previously demonstrated and shown to contribute to tumorigenicity and immunogenicity of iPSC derivatives upon differentiation, which cannot represent the 'ground' characters of iPSC derivatives [2, 20, 24]. To rule out the effects of exogenous gene integration on intrinsic characteristics of iPSCs, we generated non-integrating iPSCs by delivering reprogramming factors into C57BL/6 mouse embryonic fibroblasts (MEFs) with an episomal vector (Figure 1a) [25]. The established iPSCs were alkaline phosphatase-positive and exhibited a cellular morphology similar to that of mouse ESCs (Figure 1b). PCR and southern blotting demonstrated that the iPSCs were free of plasmid integration (Figure 1c and Supplementary Figure S1A), had normal karotypes (Figure 1d), and expressed pluripotent genes (Figure 1e and Supplementary Figure S1B). Gene expression profiling indicated that global gene expression of iPSC lines (iPS-1, iPS-2, and iPS-3) was not obviously different from ESCs (Supplementary Figure S1D). Bisulfite sequencing showed that Oct4 and Nanog promoters of iPSCs were largely demethylated compared with MEFs (Supplementary Figure S1C). The iPSCs could differentiate into embryoid bodies and teratomas in vitro, validating their pluripotency (Figure 1f and g). Furthermore, using 4n complementation, we obtained viable and fertile mice from integration-free iPSC lines with high efficiency, which was comparable to that of normal ESCs (Figure 1h and i, and Supplementary Table 1), indicating the totipotency of these iPSCs.

Generation of 4n complementation mice using integration-free induced pluripotent stem cells (iPSCs). (a) Integration-free iPSC generation method. (b) Morphology and alkaline phosphatase (AP) staining of integration-free iPSCs. Scale bars, 100 μm. (c) PCR analysis using seven primers spanning the whole episomal plasmid did not detect integration of the reprogramming vector in iPSm lines 1–5. The reprogramming plasmid was used as a positive control. (d) G-banding chromosomal analysis showed iPSCs had normal karotypes. (e) Immunofluorescence of pluripotent markers (Nanog, Oct3/4, Sox2, and SSEA-1) for iPSCs. (F) RT-PCR analysis showed iPSCs were well-differentiated into three germ layers. (g) iPSCs generated teratomas containing all three embryonic germ layers in severe-combined-immune-deficiency mice. (h) iPSC mice generated by 4n complementation. (i) SSLP analysis of mice derived from different iPSC lines.
To evaluate the fundamental immunogenicity and function of iPSC derivatives for transplantation, inbred C57BL/6 mice derived from iPSC lines and ESC lines by 4n complementation were used as organ donors. Skin, islets, and heart were chosen as representatives of the three germ layers of the body (Figure 2a and Supplementary Table S1) to evaluate immunogenicity and function.

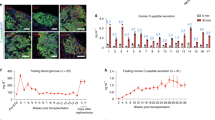
Induced pluripotent stem cell (iPSC)-derived skins were tolerated by hosts and successfully repaired skin wounds. (a) Schematic diagram of iPSm-derived organ transplantation. Skin, islets, and hearts dissociated from iPSC mice were transplanted onto the backs (top), under kidney capsules (middle), and on neck vessels (bottom), respectively. Primed T cells were detected by T-cell proliferation or interferon (INF)-γ release assays. (b) Wound repair by transplantation of iPSm-derived skin. Like ESm and syngeneic skin, iPSm skin effectively survived on recipient mice for more than 100 days after transplantation. Allogeneic skin transplants were rejected within 3 weeks and served as negative controls. Representative images show grafts 20 weeks after transplantation. (c) Survival rate summary of explanted iPSm skin 20 weeks after transplantation. ESm and syngeneic skin transplants served as positive controls; allogeneic skin transplants served as negative controls. (d) H&E staining of iPSm skin isolated from recipient mice 8 weeks after transplantation. Allografts were stained 1 week after transplantation and served as a negative control. As with ESm and syngeneic mice, iPSm skin explants showed normal structures, while extensive tissue necrosis was detected in allografts. (e) T-cell infiltration was hardly detected in iPSm skin explants 8 weeks after transplantation. T cells were identified by immunostaining with anti-CD3, anti-CD4, and anti-CD8 antibodies. Sections from the spleen and allogeneic skin grafts (1 week after transplantation) were used as positive controls. (f) The percentage of proliferating cells (low carboxyfluorescein succinimidyl ester) was quantified and shown as mean±s.e.m. of triplicates (syngeneic: n=3; ESm: n=6; iPSm: n=6; allogeneic: n=3). (g) IFN-γ release assay to detect primed T cells in iPSm skin recipients. The IFN-γ release was quantified and shown as mean±s.e.m. of triplicates (syngenic: n=3; ESm: n=6; iPSm: n=6; allogenic: n=3).
Successful wound repair by iPSC-derived skin transplantation
To test the 'ground' function and immunogenicity of iPSC-derived skin, we performed a skin transplantation assay using a traditional 'tail-to-back' model (Figure 2a and Supplementary Figure S2A), which enables grafts to be distinguished from recipient tissue. Skin samples from the tails of iPSC-derived, ESC-derived, C57BL/6, and allogeneic mice were transplanted onto the backs of wounded C57BL/6 mice. Transplanted skin from iPSm (mice derived from iPSC lines by 4n complementation), ESm (mice derived from ESC lines by 4n complementation), and syngeneic mice successfully survived over 4 months and grew well and even regenerated hairs (Figure 2b and c). In contrast, all transplanted allogeneic skin samples elicited intensive rejection by recipient mice immediately after transplantation and were completely rejected within 2 weeks (Figure 2b–d, Supplementary Figure S2B and S2C).
To understand the successful transplantation of iPSm skin, skin grafts were isolated and stained with anti-CD3, anti-CD4, and anti-CD8 antibodies 3 months after transplantation. No obvious T-cell infiltration, tissue necrosis, or tumors were detected in iPSm, ESm, or syngeneic mouse skin explants. In contrast, explanted allogeneic skin showed extensive T-cell infiltration accompanied by serious tissue necrosis within 1 week after transplantation (Figure 2d and e). To further investigate whether iPSm skin could elicit antigen-specific immune rejection responses, secondary T-cell proliferation and IFN-γ release assays were performed. Splenic T cells were isolated from mice that received iPSm, ESm, syngeneic, and allogeneic skin, labeled with carboxyfluorescein succinimidyl ester, and re-exposed to cells isolated from explanted skin for 72 h. Compared with the prominent immune rejection response elicited by allogeneic grafts, substantially less T-cell proliferation was detected with iPSm, ESm, or syngeneic skins (Figure 2f, Supplementary Figure S2D and S2E). Consistent with T-cell proliferation, IFN-γ production was markedly stimulated by allogeneic but not syngeneic, iPSm, or ESm skins, indicating limited antigen-primed T cells in iPSm skin recipients (Figure 2g).
Reduction of systematically high glucose by iPSC-derived islet transplantation
iPSm, ESm, C57BL/6, and Balb/c mouse islets were transplanted under the kidney capsule of C57BL/6 recipient mice to investigate their effects on blood glucose and immunogenicity (Figure 2a). Eight weeks after transplantation, all iPSm islet grafts survived in the recipients, whereas none of the allogeneic islets survived over 2 weeks (Figure 3a). Hematoxylin and eosin (H&E) staining of kidney grafts showed that iPSm, ESm, and syngeneic islet structures were well-maintained with no necrosis or tumor formation, while explanted allogeneic islets were completely rejected without any residues within 2 weeks (Figure 3b). Sections from explanted ESm, iPSm, allogeneic, and syngeneic islets were also stained with an anti-CD3 antibody. Compared with allogeneic islets that showed apparent T-cell infiltration, no obvious T-cell infiltration was detected in iPSm, ESm, and syngeneic islets 2 months after transplantation (Figure 3c and Supplementary Figure S3D). To further confirm the immunogenicity of iPSC-derived islets, secondary T-cell response and IFN-γ release assays were performed. Consistent with T-cell infiltration assays, allografts stimulated T-cell proliferation and IFN-γ production markedly, while iPSm, ESm, and syngeneic islets elicited limited responses (Figure 3d and e, and Supplementary Figure S3E).

Induced pluripotent stem cell (iPSC)-derived islets reduced systematically high glucose in diabetic mice with limited immunogenicity. (a) Summary of iPSm islet survival in C57BL/6 hosts 8 weeks after transplantation. (b) Representative images of iPSm islets transplanted under kidney capsules (dot circles indicate grafted islets). (c) Detection of T-cell infiltration in iPSm islets with an anti-CD3 antibody (green). Anti-insulin staining was used to label engrafted islets (red). Scale bars, 50 μm. (d) T-cell proliferation induced by different stimulators was quantified and shown as the mean±s.e.m. (syngeneic: n=3; ESm: n=3; iPSm: n=3; allogeneic: n=3). (e) Interferon (IFN)-γ release was quantified and shown as mean±s.e.m. (syngeneic: n=3; ESm: n=3; iPSm: n=3; allogeneic: n=3). (f) Blood glucose levels were monitored in diabetic mice engrafted with allogeneic, syngeneic, and iPSm islets (iPSm: n=2, yellow; syngeneic: n=3, green; allogeneic: n=3, purple). (g) Glucose tolerance test 8 weeks after islet transplantation. Diabetic mice engrafted with iPSm islets (n=2, green) efficiently responded to high-glucose injection similar to mice transplanted with syngeneic islets (n=3, purple).
To investigate the functional integrity of physiologically derived iPSm islets, we transplanted iPSm islet into diabetic mice. Intriguingly, 2 days after transplantation, the blood glucose of all diabetic recipients transplanted with iPSm and syngeneic islets decreased markedly to basal levels and maintained normal levels for more than 2 months (Figure 3f). Although the blood glucose concentration of diabetic mice treated with allogeneic islets significantly decreased 3–10 days after transplantation, levels increased 15 days after transplantation due to rejection of allogeneic islets (Figure 3c and f). Furthermore, iPSm islet recipients responded to glucose injections normally, and transplanted iPSm islets maintained normal structures 2 months after transplantation, indicating the limited immunogenicity of iPSC-derived islets did not affect grafting or functional integrity (Figure 3f and g; Supplementary Figure S3B and S3C). Taken together, these data support the clinical development and use of iPSC-derived islets for regenerative medicine.
Grafted iPSC-derived hearts survived and maintained normal beating patterns in recipients
Vascularized heterotropic heart transplantation was performed to test the viability and immunogenicity of iPSC-derived hearts. Hearts isolated from iPSm, ESm, C57BL/6, and Balb/c mice were transplanted under the neck skin of C57BL/6 recipient mice via anastomosis to the neck vessels using a non-suture cuff technique described previously [26, 27] (Figure 2a). Similar to ESm and syngeneic heart transplants, iPSm hearts maintained normal beating patterns in recipients for over 2 months without any obvious defects, while all transplanted allogeneic hearts terminated beating within 10 days after transplantation (Figure 4a, b and Supplementary Figure S4A). Moreover, extensive necrosis and tissue regression were observed in allogeneic heart transplants. In contrast, transplanted iPSm, ESm, and syngeneic hearts maintained normal structures (Figure 4c), indicating the comparative fundamental function of iPSC-derived heart with that of syngeneic mice.

Induced pluripotent stem cell (iPSC)-derived heart transplantation. (a) Survival curve of iPSm, ESm, syngeneic, and allogeneic mouse hearts engrafted in C57BL/6 recipient mice (n=5). (b) Engrafted iPSm hearts showed similar beating rates as ESm and syngeneic mice. (c) H&E staining of transplanted hearts. (d) T-cell infiltration identified by staining sections with anti-CD3 antibodies (green). No obvious T-cell infiltration was detected in grafted iPSm, ESm, and syngeneic mouse hearts isolated from C57BL/6 recipients. Allografts showed extensive T-cell infiltration and served as positive controls. Scale bars, 50 μm. (e) T-cell proliferation and (f) interferon (INF)-γ release were used to detect the presence of primed T cells in C57BL/6 mice engrafted with iPSm, ESm, syngeneic, and allogeneic hearts (syngeneic: n=3; ESm: n=3; iPSm: n=3; allogeneic: n=3). (g) Expression of the Zg16 and Hormad1 genes in transplanted skin, islets and hearts 8 weeks after transplantation.
To test the intrinsic immunogenicity of iPSC-derived hearts and their potential effects on functional integrity, transplanted iPSm hearts were isolated and stained with an anti-CD3 antibody. Consistent with H&E staining, T cells were hardly detected in iPSm, ESm, and syngeneic heart explants, but had infiltrated allogeneic hearts (Figure 4d). T-cell proliferation and IFN-γ release assays further confirmed that transplanted iPSm hearts did not induce significant immune rejection responses (Figure 4e and f), indicating limited immunogenicity. Taken together, these data indicated that the limited immunogenicity of iPSm hearts did not affect their function, supporting the clinical application of iPSC-derived hearts in the treatment heart disease, failure, and so on.
Discussion
Recent studies have shown certain cells (for example, cardiomyocytes and endothelial cells) differentiated from iPSCs in vitro can elicit immune rejection responses, possibly due to differential presentation of abnormalities induced by reprogramming upon iPSC differentiation [15, 20–23, 28]. However, whether physiologically derived iPSC progeny are immunogenic is relatively unknown. By transplantation of iPSC-derived skin, islet, and heart, we have shown that in vivo iPSC-derived organs can be accepted by the recipient immune system without the need for immunosuppression. In particular, we did not observe extensive infiltration or priming of T cells in mice transplanted with iPSC skin, islet, or heart, indicating limited immunogenicity of these organs upon transplantation. The expression of Hormad1 and Zg16 was not significantly increased in the iPSC skin, islet, or heart (Figure 4g). Previously, the Abe group reported that iPSC-derived skin and bone marrow cells obtained from chimeric mice generated by B6 iPSCs have negligible immunogenicity in the host and are completely immune-tolerated by B6 recipients [21]. However, iPSC-derived tissues from chimeric animals may be able to adapt their immunogenicity to the chimeric host immune system, thereby misrepresenting their intrinsic character. It is possible that abnormal or immunogenic iPSC-derived cells were immune-rejected and/or rendered immune-tolerant by the intrinsic immune system of chimeric B6 mice long before transplantation into other B6 recipients. In contrast to tissues derived from chimeric mice, those isolated from 4n complementation iPSC mice do not have these issues and represent the fundamental nature or 'ground state' of iPSC derivatives.
Organ transplantation has been widely used in clinics to replace damaged organs like the skin, kidney, heart, pancreas and so on. However, the severe shortage of donor organs and immune rejection are two major obstacles to the expansion of organ transplantation therapies. iPSCs hold great promise in regenerative medicine by potentially providing unlimited autologous resources without concerning on immune rejection. Significant progress has been made in using iPSCs for basic research and disease modeling; however, clinic use of iPSCs is still in its preliminary stage [16, 29–31]. The incomplete differentiation of iPSCs in vitro not only limits the generation of transplantable cells, but also poses a serious teratoma risk due to remaining undifferentiated pluripotent stem cells [16, 30, 31]. In vitro generation of organs from a patient’s own pluripotent stem cells is an ideal strategy proposed to address these issues and an ultimate goal for regenerative medicine. For a long time, regeneration of functional organs is hindered by the complexity of organogenesis. Recent advance in the use of interspecific blastocyst complementation systems for in vivo generation of organs derived from donor pluripotent stem cells is a very promising strategy to address this issue [32, 33]. In the meantime, regeneration of functional organs from adult tissue stem cells or iPSCs has enabled significant advancements in the generation of functional organs for regenerative medicine [34–38]. Given those significant progresses, however, whether iPSC-derived organs can be used for transplantation in theory is still largely unknown. In this context, the current study represents a ‘proof-of-principle’ preclinical experiment evaluating the potential feasibility of using iPSC-derived organs for transplantation.
Another major concern for clinical use of iPSCs is safety. Development of new reprogramming techniques (for example, integration-free and small-molecule induction) significantly reduces the potential cancer risks associated with viral integration [15, 39]. Meanwhile, studies have shown that reprogramming itself induces both genomic and epigenetic instability [2, 20, 24, 40–44]. Although studies using in vitro iPSC derivatives to treat disease models have shown promising clinical results [45], the therapeutic efficacy and safety of physiologically derived iPSC organs have never been explored. Encouragingly, in current study we did not detect tumorigenesis in or around the explanted iPSC-derived organs in our experimental settings. These data support the efficacy and safety of iPSC-derived skin, heart, and islet for therapy.
Materials and Methods
Generation of iPSCs with episomal plasmids
For reprogramming, 6 μg of episomal plasmids containing Oct4, Klf4, Sox2, and Nanog, which were generously provided by Dr. Guoliang Xu [25] were transfected into 1×106 MEFs by a neon (Invitrogen, Carlsbad, CA, USA). Transfected MEFs were plated onto 2 mm×100 mm dishes pre-seeded with a layer of feeder cells. One day after transfection, the MEF medium was replaced with our previously reported ESC medium [6] plus 5 μg ml−1 FGF2 for the first 4 days; the culture medium was changed every other day. Colonies with morphology similar to ESCs appeared approximately 8 days after transfection and were picked ~10 days post transfection.
PCR and southern blot analysis
Primers spanning the whole plasmid were used to detect reprogramming vector integration (Supplementary Table S2). For southern blot, 15 μg of genomic DNA was digested with EcoR V (Takara, Japan), electrophoresed on a 0.7% agarose gel overnight, and then transferred onto a positively-charged nylon membrane (Millipore, Darmstadt, Germany). Hybridization was performed at 67 °C with P32 radioactively labeled cAMP prepared by Prime-a-Gene Labeling System (Promega, Fitchburg, WI, USA). The cDNA of Oct4, Nanog, Sox2, and Klf4 were used as probes (Supplementary Table S2).
Karyotype analysis
Karyotyping was conducted using standard chromosomal analysis protocols. G-binding karyotype analysis was carried out by Peking Union Medical College (Beijing, China).
Immunofluorescence and alkaline-phosphatase staining
Immunostaining was carried out as previously described [6]. In brief, samples were fixed with 4% paraformaldehyde for 30 min at ~25 °C then washed three times with PBS. Non-specific sites were blocked with 500 μl of 2% bovine serum albumin (BSA) plus 0.45% Triton X-100 for 1 h at 20–25 °C. The samples were then incubated with the primary antibody overnight at 4 °C. Primary antibodies included anti-Oct4 (Santa Cruz, Dallas, TX, USA, 1:50), anti-Sox2 (Millipore, 1:200), anti-SSEA1 (Millipore, 1:200), and anti-Nanog (Abcam, Cambridge, UK, 1:100). The next day, samples were washed three times with PBS, incubated with an AlexaFlur 488-conjugated secondary antibody (diluted in 2% BSA plus 4.5% Triton X-100) at 25 °C for 1 h, and then imaged with a LSM780 Meta confocal microscope (Zeiss, Oberkochen, Germany). An alkaline phosphatase staining kit (Beyotime, Shanghai, China) was used to detect iPSC colonies according to the manufacturer’s instructions.
Bisulfite genomic sequencing
Bisulfite treatment of genomic DNA was performed with an EpiTect Bisulfite Kit (Qiagen) according to the manufacturer’s instructions. Oct4 and Nanog promoter regions were amplified with nested primers as previously reported [6]. PCR was performed as follows: 94 °C for 5 min; 35 cycles at 94 °C for 30 s, 59 °C for 45 s, 72 °C for 30 min; and 72 °C for 7 min. PCR products were cloned into pMD18-T vectors (Takara), and 10 randomly selected clones were sequenced.
Reverse transcriptase (RT)-PCR and qualitative (q)-PCR
Total RNA was extracted from tissues or cells using TRIZOL (Invitrogen). cDNA was synthesized with oligo (dT) primers (Promega) and M-MLV reverse transcriptase (Promega) according to the manufacturer’s instructions. RT-PCR was performed with ExTaq (Takara), and qPCR was performed with SYBR Green PCR master mixes (Toyobo, Osaka, Japan). Primers used are listed in Supplementary Table 2.
Simple sequence length polymorphism analysis
The primers used for SSLP were selected from the Mouse Genome Informatics website (http://www.informatics.jax.org/). DNA was extracted from ears of mice. PCR products were separated by 3.5% agarose gels and visualized with ethidium bromide using a gel imager (Bio-Rad, Hercules, CA, USA).
Teratoma formation
Approximately 1×107 iPSCs were injected subcutaneously into the hind limbs of 10-week-old male severe-combined-immune-deficiency beige mice. Four weeks later, teratomas were disassociated, fixed with 4% paraformaldehyde, embedded in paraffin, sectioned, and stained with H&E for histological analysis.
Tetraploid (4n) embryo complementation
Generation of mice by 4n embryo complementation was carried out as previously described [46] and handled according to CMPH and NIH guidelines. In brief, two-cell embryos were collected from oviducts of female CD-1 mice, subjected to electrofusion to produce one-cell 4n embryos, and then cultured in CZB media. Ten to 15 iPSCs were injected into each 4n blastocyst and transferred to CD-1 pseudopregnant recipient females. Embryos derived from 4n blastocyst injection were dissected at birth (embryonic day 19.5).
Skin transplantation
Skin transplantation experiments were performed in accordance with the Chinese Ministry of Public Health Guide and US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male C57BL/6 mice (8 week-old, H2 haplotype: b) were anesthetized by intraperitoneal injection of sodium pentobarbital (40 mg kg−1 body weight). Then skin samples (8 mm×8 mm) from the tails of iPSC-derived (n=33), ESC-derived (n=33), C57BL/6 (n=17), or 129SVJ (H2 haplotype: bc) (n=10) mice were transplanted onto the back of C57BL/6 recipient mice.
Diabetes mouse model and islet transplantation
Diabetes was induced by a single intraperitoneal injection of 180 mg kg−1 streptozocin (Sigma-Aldrich, St Louis, MO, USA) in 0.1 m citrate buffer (pH 4.4). Blood glucose was measured by a One-Touch Glucose Analyzer (FreeStyle, Abbott, Lake Forest, IL, USA). Mice with blood glucose concentrations above 16.7 mm for more than two consecutive days were used for transplantation. Donor islets were isolated from pancreases via bile duct cannulation with 3 ml collagenase P (1 mg ml−1; Roche, Penzberg, Germany) and digested for 20 min at 37 °C. The digested pancreatic tissues were washed and purified using an EZ-Sep lymphocyte separation kit (Dakewe Biotech, Guangzhou, China). Isolated islets were collected manually using a pipette under a microscope and injected under the kidney capsule of recipient mice (iPS group: n=7; ES group: n=5; syngenic group: n=5; allogenic group: n=5). Blood glucose in recipient mice was monitored every other day. Mice were fasted for 16 h before the glucose tolerance test, and blood glucose was measured before and 15, 30, 60, 90, and 120 min after glucose injection (2 g kg−1 weight glucose).
Heart transplantation
Vascularized heterotopic heart transplantation was performed with anastomosis to the vessels of the neck using a non-suture cuff technique as previously described [26, 27] (n=7 each group). Cardiac graft survival was monitored by palpation daily for the first week, and every 3 days after. Recipient death and/or loss of graft function within 48 h after transplantation were considered as technical failures, and these animals were omitted from analysis.
Histological analysis
Samples isolated from recipient mice were embedded in either OCT (ZLI-9302) or formalin, and then examined by H&E or immunohistochemical staining using anti-CD3 (1:200), anti-CD4 (1:500), anti-CD8 (1:200), and anti-insulin antibodies (BD Pharmingen, San Jose, CA, USA).
Secondary T-cell response assay
CD4+ and CD8+ T cells were isolated from the spleens of recipient C57BL/6 mice grafted with iPSm skin, islets, and heart for 12 weeks using CD4+ and CD8+ T-Cell Isolation Kits (MACS 130-095-248; MACS 130-095-236). The isolated T cells (responders) were labeled with carboxyfluorescein succinimidyl ester (Sigma) and co-cultured with the cells digested from the transplanted grafts (stimulators) for 72 h. Proliferation of CD4+ and CD8+ T cells was analyzed by flow cytometry (Caliber, BD Pharmingen). carboxyfluorescein succinimidyl ester-stained CD4+ and CD8+ T cells without stimulation were used as a control. CD3-positive T cells were selected to calculate the proliferation of CD4+ and CD8+ T cells (n=3). Culture mediums were used to determine INF-γ release using an INF-γ ELISA kit (EMC101g.96; n=3) (Thermo, Waltham, MA USA).
References
Takahashi K, Yamanaka S . Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126: 663–676.
Okita K, Ichisaka T, Yamanaka S . Generation of germline-competent induced pluripotent stem cells. Nature 2007; 448: 313–317.
Wernig M, Meissner A, Foreman R et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007; 448: 318–324.
Boland MJ, Hazen JL, Nazor KL et al. Adult mice generated from induced pluripotent stem cells. Nature 2009; 461: 91–94.
Kang L, Wang J, Zhang Y, Kou Z, Gao S . iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell 2009; 5: 135–138.
Zhao XY, Li W, Lv Z et al. iPS cells produce viable mice through tetraploid complementation. Nature 2009; 461: 86–90.
Hanna J, Wernig M, Markoulaki S et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 2007; 318: 1920–1923.
Wernig M, Zhao JP, Pruszak J et al. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson's disease. Proc Natl Acad Sci USA 2008; 105: 5856–5861.
Alipio Z, Liao W, Roemer EJ et al. Reversal of hyperglycemia in diabetic mouse models using induced-pluripotent stem (iPS)-derived pancreatic beta-like cells. Proc Natl Acad Sci USA 2010; 107: 13426–13431.
Sharp J, Frame J, Siegenthaler M, Nistor G, Keirstead HS . Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants improve recovery after cervical spinal cord injury. Stem Cells 2010; 28: 152–163.
Tsuji O, Miura K, Okada Y et al. Therapeutic potential of appropriately evaluated safe-induced pluripotent stem cells for spinal cord injury. Proc Natl Acad Sci USA 2010; 107: 12704–12709.
Kriks S, Shim JW, Piao J et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 2011; 480: 547–551.
Lee AS, Xu D, Plews JR et al. Preclinical derivation and imaging of autologously transplanted canine induced pluripotent stem cells. J Biol Chem 2011; 286: 32697–32704.
Rhee YH, Ko JY, Chang MY et al. Protein-based human iPS cells efficiently generate functional dopamine neurons and can treat a rat model of Parkinson disease. J Clin Invest 2011; 121: 2326–2335.
Zhao T, Xu Y . p53 and stem cells: new developments and new concerns. Trends Cell Biol 2010; 20: 170–175.
Ben-David U, Benvenisty N . The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer 2011; 11: 268–277.
Vazquez-Martin A, Cufi S, Lopez-Bonet E et al. Metformin limits the tumourigenicity of iPS cells without affecting their pluripotency. Sci Rep 2012; 2: 964.
Liu Z, Tang Y, Lu S et al. The tumourigenicity of iPS cells and their differentiated derivates. J Cell Mol Med 2013; 17: 782–791.
Ohnishi K, Semi K, Yamamoto T et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014; 156: 663–677.
Zhao T, Zhang ZN, Rong Z, Xu Y . Immunogenicity of induced pluripotent stem cells. Nature 2011; 474: 212–215.
Araki R, Uda M, Hoki Y et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 2013; 494: 100–104.
Guha P, Morgan JW, Mostoslavsky G, Rodrigues NP, Boyd AS . Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell 2013; 12: 407–412.
de Almeida PE, Meyer EH, Kooreman NG et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat Commun 2014; 5: 3903.
Tong M, Lv Z, Liu L et al. Mice generated from tetraploid complementation competent iPS cells show similar developmental features as those from ES cells but are prone to tumorigenesis. Cell Res 2011; 21: 1634–1637.
Wang Y, Chen J, Hu JL et al. Reprogramming of mouse and human somatic cells by high-performance engineered factors. EMBO Rep 2011; 12: 373–378.
Matsuura A, Abe T, Yasuura K . Simplified mouse cervical heart transplantation using a cuff technique. Transplantation 1991; 51: 896–898.
Qi Z, Riesbeck K, Ostraat O, Tufveson G, Ekberg H . Single dose anti-CD4 monoclonal antibody for induction of tolerance to cardiac allograft in high- and low-responder rat strain combinations. Transpl Immunol 1997; 5: 204–211.
Cao J, Li X, Lu X, Zhang C, Yu H, Zhao T . Cells derived from iPSC can be immunogenic - yes or no? Protein Cell 2014; 5: 1–3.
Wu SM, Hochedlinger K . Harnessing the potential of induced pluripotent stem cells for regenerative medicine. Nat Cell Biol 2011; 13: 497–505.
Knoepfler PS . Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cell 2009; 27: 1050–1056.
Cunningham JJ, Ulbright TM, Pera MF, Looijenga LH . Lessons from human teratomas to guide development of safe stem cell therapies. Nat Biotechnol 2012; 30: 849–857.
Kobayashi T, Yamaguchi T, Hamanaka S et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell 2010; 142: 787–799.
Usui J, Kobayashi T, Yamaguchi T, Knisely AS, Nishinakamura R, Nakauchi H . Generation of kidney from pluripotent stem cells via blastocyst complementation. Am J Pathol 2012; 180: 2417–2426.
Shackleton M, Vaillant F, Simpson KJ et al. Generation of a functional mammary gland from a single stem cell. Nature 2006; 439: 84–88.
Stingl J, Eirew P, Ricketson I et al. Purification and unique properties of mammary epithelial stem cells. Nature 2006; 439: 993–997.
Leong KG, Wang BE, Johnson L, Gao WQ . Generation of a prostate from a single adult stem cell. Nature 2008; 456: 804–808.
Spence JR, Mayhew CN, Rankin SA et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011; 470: 105–109.
Takebe T, Sekine K, Enomura M et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013; 499: 481–484.
Lu X, Zhao T . Clinical therapy using iPSCs: hopes and challenges. Genomics Proteomics Bioinformatics 2013; 11: 294–298.
Kim K, Doi A, Wen B et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010; 467: 285–290.
Liu L, Luo GZ, Yang W et al. Activation of the imprinted Dlk1-Dio3 region correlates with pluripotency levels of mouse stem cells. J Biol Chem. 2010; 285: 19483–19490.
Polo JM, Liu S, Figueroa ME et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol 2010; 28: 848–855.
Stadtfeld M, Apostolou E, Akutsu H et al. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 2010; 465: 175–181.
Lister R, Pelizzola M, Kida YS et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011; 471: 68–73.
Kamao H, Mandai M, Wakamiya S et al. Objective qevaluation of the degree of pigmentation in human induced pluripotent stem cell-derived RPE. Invest Ophthalmol Vis Sci 2014; 55: 8309–8318.
Nagy A . Manipulating the Mouse Embryo: a Laboratory Manual. Cold Spring Harbor Laboratory Press, 2003.
Acknowledgements
This study was supported in part by grants from the China National Basic Research Program 2012CBA01300 (to QZ), 2012CB966900 (to TZ), and 2012CBA01303 (to ZQ); the Strategic Priority Research Program of the Chinese Academy of Sciences XDA01020101 (to QZ) and XDA01040108 (to TZ); and the National Natural Science Foundation of China Program 31271592 (to TZ) and 31371516 (to WL).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, L., Cao, J., Wang, Y. et al. Immunogenicity and functional evaluation of iPSC-derived organs for transplantation. Cell Discov 1, 15015 (2015). https://doi.org/10.1038/celldisc.2015.15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/celldisc.2015.15
This article is cited by
-
Exploring the promising potential of induced pluripotent stem cells in cancer research and therapy
Molecular Cancer (2023)
-
High autophagic flux guards ESC identity through coordinating autophagy machinery gene program by FOXO1
Cell Death & Differentiation (2017)
