
Abstract
Due to limited treatment options, pre-eclampsia (PE) is associated with fetal perinatal and maternal morbidity and mortality. During the causes of PE, failure of uterine spiral artery remodeling which might be related to functioning abnormally of trophoblast cells, result in the occurrence and progression of PE. Recently, abnormal expression of long non-coding RNAs (lncRNAs), as imperative regulators involved in human diseases progression (included PE), which has been indicated by increasing evidence. In this research, we found that TUG1, a lncRNA, was markedly reduced in placental samples from patients with PE. Loss-function assays indicated that knockdown TUG1 significantly affected cell proliferation, apoptosis, migration and network formation in vitro. RNA-seq revealed that TUG1 could affect abundant genes, and then explore the function and regulatory mechanism of TUG1 in trophoblast cells. Furthermore, RNA immunoprecipitation and chromatin immunoprecipitation assays validated that TUG1 can epigenetically inhibit the level of RND3 through binding to EZH2, thus promoting PE development. Therefore, via illuminating the TUG1 mechanisms underlying PE development and progression, our findings might furnish a prospective therapeutic strategy for PE intervention.
Similar content being viewed by others

Main
Pre-eclampsia (PE), which is characterized by new-onset hypertension and proteinuria, is a pregnancy-specific syndrome.1 It is the major trigger of fetal morbidity and pregnancy-induced mortality, and it affects 3–5% of pregnancies worldwide, including in developing countries such as China.2, 3
In spite of the recent improvements made in treating PE, its underlying mechanism remains poorly understood. Previous studies have reported that many factors are involved in the pathogenesis of PE, such as oxygen dysregulation, impaired spiral artery remodeling and inappropriate maternal vascular destruction.4, 5 In PE, poor spiral artery remodeling is associated with dysfunctions of extravillous trophoblasts. For example, reduced proliferation,6 induced apoptosis7 and distorted migration and invasion abilities in extravillous trophoblasts8 may prevent them from successfully invading the myometrial spiral arteries.
Over the past decades, despite a focus on the function of protein-coding genes that participate in the pathogenesis of various diseases, long non-coding RNAs (lncRNAs), have received an increasing amount of attention with the development of whole-genome sequencing technologies.9 LncRNAs are RNA molecules longer than 200 nucleotides that do not encode proteins. Among the thousands of lncRNAs that have been revealed by the ENCODE project, only a few have been shown to be endowed with biological functions.10, 11 These lncRNAs are involved in a series of cellular progressions, such as parental imprinting, cell proliferation, apoptosis and metastasis via epigenetic modification, chromatin remodeling and sponging miRNAs.12, 13, 14 Recently, various researches have established that abnormal lncRNA levels might be associated with diverse human diseases, including PE. Previous studies have confirmed that abnormal lncRNAs may affect the proliferation, apoptosis and metastasis of trophoblast cells and stimulate the pathological placental development of PE.15, 16 These resulting data suggest that lncRNAs might have vital roles in the occurrence and progression of PE. Thus, to clarify the connection between PE-associated lncRNAs and their biological functions, a deeper understanding of the molecular mechanism of PE is essential.
Given the importance of lncRNAs in PE, in the present study, we focused on TUG1, a 7598-bp lncRNA gene located on chromosome 22q12.2. We found that the expression of TUG1 is reduced in PE placental tissues compared with levels in controls. Moreover, loss-function assays were conducted to explore the influences of TUG1 in the occurrence and development of PE. We found that TUG1 might result in the impairment of spiral artery remodeling in PE. An experiment was conducted to establish the molecular mechanism by which TUG1 modulates its targets in trophoblast cells. Our results provide a novel understanding of the biological functions of TUG1 and the molecular regulatory mechanisms of its targets in trophoblasts.
Results
Deregulated expression of the TUG1 in PE
We first detected TUG1 expression levels in the placental tissues of a cohort of 52 pairs of PE patients and controls using qRT-PCR. There were no significant differences between the PE cases and controls in terms of gestation weeks or maternal age (P>0.05). Table 1 details the patient's clinical features. In particular, the levels of TUG1 were significantly lower in PE placental samples compared with levels that in control tissues (Figures 1a and b).

Relative TUG1 expression in PE. (a) The relative expression of TUG1 was measured by qRT-PCR. The levels of TUG1 were lower in pre-eclamptic placentas samples (N=52) than in normal placentas (N=52). (b) Results are presented as the fold-change in PE placental samples relative to the control, and TUG1 expression was classified into two groups. (c) TUG1 expression were detected by qRT-PCR in several cell lines and were normalized to that in HTR-8/SVneo. At least three times of biological replicates have been performed and presented (values are mean±S.E.M.; **P<0.01)
Effect of TUG1 on the proliferation, migration and invasion in trophoblast cells
To explore the latent biological function of TUG1 in trophoblast cells, we first assessed the levels of TUG1 in several associated cell lines, such as HTR-8/SVneo, JEG-3, BeWo, WISH and HUVEC-C. We found that the expression of TUG1 in HTR-8/SVneo and JEG-3 cells was lower than in the other cell lines (Figure 1c). Then we knocked down TUG1 expression levels through the transfection of TUG1 siRNAs in HTR-8/SVneo, JEG-3 and HUVEC-C cells. qRT-PCR analysis demonstrated that TUG1 expression was silenced in si-TUG1-1#-treated cells and by si-TUG1-2#-treated cells (Supplementary Figure 1a).
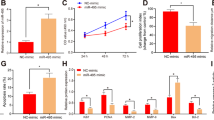
We next conducted an MTT assays that showed that cell proliferation was suppressed when TUG1 levels were silenced in HTR-8/SVneo and JEG-3 cells (Figure 2a). In addition, a colony formation assay was performed, and the resulting data revealed that growth ability was reduced after downregulating TUG1 in HTR-8/SVneo and JEG-3 (Figure 2b). Moreover, EdU staining assay also showed that knockdown of TUG1 reduced the proliferation of these two types of cells (Figure 2c).

Effect of TUG1 on proliferation, migration and invasion in trophoblast cells. (a) MTT assays were used to determine the viability of si-TUG1-transfected trophoblast cells. (b) Colony formation assays were performed to determine the proliferation of si-TUG1-transfected HTR-8/SVneo and JEG-3. Colonies were counted and captured. (c) Proliferating trophoblast cells were labeled with Edu. The Click-it reaction revealed Edu staining (red). Cell nuclei were stained with DAPI (blue). (d) The migration and invasion capacity of the cells transfected with si-TUG1 was significantly lower than that of the negative control and higher in the cells overexpressing TUG1, as determined by transwell assays. All experiments were performed in biological triplicates with three technical replicates (values are mean±S.E.M.; **P<0.01, *P<0.05)
Because cell migration and invasion are essential features of PE progression, we performed transwell assays to examine the roles of TUG1 on the migration or/and invasion abilities of HTR-8/SVneo and JEG-3 cells. Knockdown of TUG1 inhibited the migration and invasion abilities, and the number of cells was significantly reduced compared with that in controls in HTR-8/SVneo and JEG-3 cells (Figure 2d). These results indicate that the knockdown of TUG1 expression inhibits the invasion and migration phenotype in trophoblast cells.
Effect of TUG1 on cell cycle and apoptosis in trophoblasts
To examine whether the effect of TUG1 on cells proliferation reflects changes in the cell cycle, we performed flow cytometry to examine cell cycle progression. The results showed that two effective siRNA transfected cells caused cell accumulation in the G1/G0 phase and/or entered the S phase less often compared with the cells which treated with scramble siRNAs (Figure 3a). Additionally, flow cytometry assays were performed to establish whether the silencing of TUG1 affected the cell apoptosis. The proportion of both early apoptotic and late apoptotic cells were increased following TUG1 knockdown in cells (Figure 3b). These data imply that TUG1 promotes a proliferation phenotype in trophoblasts.

Effect of TUG1 on cycle, apoptosis and network formation in vitro. Trophoblast Cells were treated with specific TUG1 siRNAs. (a) Cell cycle analyses by Flow cytometry in HTR-8/SVneo and JEG-3 cells. (b) Flow cytometry was used to detect the apoptotic rates of cells. LR, early apoptotic cells; UR, terminal apoptotic cells (values are mean±S.E.M.; **P<0.01, *P<0.05). (c) Performing network formation, cells transfected with siRNAs targeting TUG1 showed an increase in node numbers as compared with the negative control. All experiments were performed in biological triplicates with three technical replicates (values are mean±S.E.M.; *P<0.05; **P<0.01)
Effects of TUG1 on network formation ability in vitro
The failure of spiral artery remodeling can result in placental ischemia and hypoxia and further result in the occurrence and development of PE.4, 17 In this study, we explored the effect of TUG1 on the network formation ability of associated cell lines in vitro, and further noticed the personality of TUG1 in process of PE. As shown in Figure 3c, the number of capillary-like networks was significantly reduced after transfection of TUG1 siRNAs and further decreased network formation ability.
Gene expression profiling
To determine TUG1-associated transcriptional changes, we performed RNA transcriptome sequencing of the control and si-TUG1-1#-treated HTR/SVneo cells to reveal latent downstream targets. The cell lines were treated with scramble and si-TUG1-1# for 36 h. Based on the RNA transcriptome sequencing data, the transcript levels of 82 genes exhibited ≥2-fold increases in abundance in HTR/SVneo cells after TUG1 knockdown, while 233 genes exhibited ≤2-fold decreases in abundance (Figures 4a and b; Supplementary tables 2 and 3).

TUG1 knockdown increases the expression of genes involved in cell proliferation and migration. (a) RNA transcriptome sequencing analysis was performed to analyze gene expression profiling in HTR-8/SVneo cells following TUG1 knockdown. The picture showed the all of different expressed gene. (b) GO analysis for all genes with altered expressions between the scrambled siRNA-treated and si-TUG1-treated cells in vitro. Cell growth was among the significant biological processes for genes whose transcripts level were changed in the TUG1-depleted Trophoblast cells. (c,d) qRT-PCR analysis in si-TUG1-treated trophoblast cells reveal altered mRNA level of genes involved in cell proliferation and migration upon TUG1 depletion. (e,f) Western blotting assays of RND3 protein levels after si-TUG1 or si-NC was transfected into HTR-8/SVneo and JEG-3 cells. GAPDH protein was used as an internal control. Values represent the mean±S.E.M. from three independent experiments (values are mean±S.E.M.; **P<0.01, *P<0.05)
The assessment of pathways activated by TUG1 according to the GO and KEGG databases indicated that cell growth, apoptosis and migration were altered in TUG1-depleted cells (Figure 4c). Noticeably, the GO results were fundamentally in accordance with our previous experimental findings. Using qRT-PCR, we confirmed the changes in gene expression that particularly affected the growth of HTR-8/SVneo and JEG-3 cells. Our results showed that RND3 was significantly upregulated after TUG1 knockdown (Figures 4c and d), indicating that the dysregulated genes RND3 may be a crucial downstream mediator of TUG1. Western blotting assay was also performed and further confirmed that the Rnd3 protein levels were significantly increased in TUG1-depleted cells (Figures 4e and f).
TUG1 epigenetically silences RND3 transcription by interacting with EZH2
LncRNAs participate in regulating cell function by inactivating suppressors or activating enhancing factors via interactions with specific RNA-binding proteins (RBPs). To explore the potential biological mechanism of TUG1 activity in trophoblast cells, we performed a molecular/cytoplasm separation, in which GAPDH was used as a cytoplasmic biomarkers and U1 acted as a biomarker of the nucleus. We then detected the subcellular localization of TUG1, which may supply cues for its role in molecular processes in trophoblast cells. The results showed that TUG1 was essentially located in the cell nucleus in HTR-8/SVneo and JEG-3 cells (Figures 5a and b). Moreover, FISH assays were implemented to visualize the expression and subcellular localization of TUG1 in HTR-8/SVneo cells (Supplementary Figure 1b). The data indicated that TUG1 localized in the cytoplasm as well as the nucleus, but that levels of TUG1 were higher than in the cytoplasm.

TUG1 can recruit EZH2 to silence RND3 expression. (a,b) Cell fractionation assays indicated that TUG1 is mostly located in nucleus. GAPDH and U1 acted as the marker of cytoplasm and nucleus, respectively. (c,d) RIP assays established that TUG1 can recruit EZH2. (e,f) The silencing of EZH2 increased RND3 expression at protein levels. (g) The enrichment of EZH2 and H3K27me3 in the promoter regions of RND3 were identified via ChIP assays, and this enrichment was decreased after TUG1 knockdown. Values represent the mean±S.E.M from three independent experiments (values are mean±S.E.M.; **P<0.01, *P<0.05)
Previous studies have reported that lncRNAs can interact with RBPs to exert their regulatory functions. We therefore determined the interaction probabilities of TUG1 with different RBPs through RNA–protein interaction prediction (http://pridb.gdcb.iastate.edu/RPISeq/) and discovered that TUG1 potentially bind with Ezh2 and Lsd1 (RF or SVM score >0.5). Accordingly, we hypothesized that TUG1 may regulate RND3 expression by recruiting particular RBPs in trophoblast cells. Next, we conducted RNA immunoprecipitation (RIP) assay and found that TUG1 can recruit Ezh2 in HTR-8/SVneo and JEG-3 cells (Figures 5c and d). Together, these data confirm the interaction between Ezh2 and TUG1.
Based to the RIP results, we also evaluated the relational features of the correlation between TUG1 and Ezh2. Following the knockdown of EZH2 by specific siRNAs, we found significant upregulation of RND3 expression (Figures 5e and f). Many studies have established that Ezh2 acts as a suppressor of RNA transcription through histone modification, namely, H3K27me3. Hence, it is possible that TUG1 represses the expression of RND3 through recruiting Ezh2 protein to the promoter regions of RND3, mediating H3K27me3. Next, we performed ChIP assays and revealed that Ezh2 could directly interact with RND3 promoter regions, leading to the trimethylation of H3K27 at gene promoter regions (Figure 5g). These results indicate that TUG1 can promote trophoblast cells proliferation, apoptosis, and migration partially by repressing RND3 expression.
RND3 expression levels are elevated in PE placental tissues and knockdown RND3 is potentially involved in the biological role of TUG1
According to the above discoveries, we assessed whether TUG1 regulates RND3 expression in patients with PE. For this purpose, we evaluated the expression of RND3 mRNA in 28 patients with PE. qRT-PCR analysis showed that RND3 mRNA expression was elevated in placental samples from PE patients compared with levels in controls (Figure 6a).

Upregulation of RND3 inhibit trophoblast cell proliferation and were involved in the function of TUG1. (a) RND3 expression levels were presented as the fold-change in PE placental tissue samples compare to the control. (b,c) MTT assays were executed used to determine the cell viability for pcDNA3.1-RND3-treated trophoblast cells. Colony formation assays were performed to assess the cell proliferation for pcDNA3.1-RND3-treatted HTR-8/SVneo and JEG-3. (d,e) Cell cycle analyses were utilized to examine after transfecting with siRNAs and/or pcDNA3.1-RND3 in trophoblast cells. (f,g) MTT assays and colony formation assays were implemented to assess the cell viability for si-TUG1 and si-RND3 co-transfected trophoblast cells. (h) Summary diagram describes that RND3 regulates trophoblast cell proliferation. Experiments were performed three times independently (values are mean±S.E.M.; **P<0.01, *P<0.05)
To confirm whether RND3 is involved in the biological function of TUG1, we performed gain- function assays in trophoblast cells. By qRT-PCR, RND3 was significantly upregulated in trophoblast cells treated with GFP-RND3. In contrast, RND3 was meaningfully downregulated in HTR-8/SVneo and JEG-3 cells after effective transfection with RND3 effective siRNAs, respectively. Proliferation assays implied that the overexpression of RND3 inhibited cell growth in HTR-8/SVneo and JEG-3 cells (Figures 6b and c). Next, flow cytometry analysis showed that upregulation of RND3 increased G0/G1 phase accumulation, while the downregulation of RND3 reduced G1/G0 phase arrest (Figures 6d and e). These results showed that RND3 inhibition promotes cell cycle progression and proliferation of HTR-8/SVneo and JEG-3 cells.
To investigate whether RND3 was involved in TUG1-induced HTR-8/SVneo and JEG-3 cell growth, we performed rescue experiments. These two cells were co-transfected with TUG1 and RND3 siRNAs. MTT and colony formation assays were conducted, and indicated that co-transfection moderately rescued si-TUG1-induced proliferation of HTR-8/SVneo and JEG-3 cells (Figures 6f and g). These results revealed that TUG1 can promote trophoblast cell proliferation partially by silencing of RND3 in vitro.
Taken together, our findings confirm that TUG1 binds to Ezh2 and epigenetically inhibit RND3 expression in PE by mediating H3K27 trimethylation in the RND3 promoter regions.
Discussion
Recently, increasing researches has indicated that lncRNAs may have pivotal roles in cellular development and progression of multiple diseases,18, 19 including PE.20, 21, 22 Our previous findings also revealed that the abnormal expression of the lncRNAs SPRY4-IT1 and MEG3 can affect the cellular growth, apoptosis, migration, and invasion of trophoblast cells22, 21 to induce the occurrence and development of PE.
In this study, we focused on another lncRNA, TUG1, which is significantly downregulated in the placental samples of PE patients compared with that levels in controls, indicating that decreased TUG1 levels might be associated with PE development and/or progression. Here, we performed relevant assays and confirmed that the silencing of TUG1 expression exerts a suppressive effect on trophoblasts by inhibiting cell proliferation, invasion, and migration and promoting cell apoptosis. To our knowledge, the endometrial epithelial cells may act upon the trophoblast cells to stimulate the essential cellular steps for the trophoblast transformation of the spiral arteries. A disorder of spiral artery remodeling may lead to the pathogenesis of PE. Then, we executed network formation assays. According to network formation assays (Figure 3c), the tube formation after knockdown of TUG1 were reduced compared with than that in controls. These resulting data imply that the aberrant expression of TUG1 may trigger implications for poor transformation of maternal spiral arteries, contributing to placental abnormalities or PE. Previous studies have reported that TUG1 could affect multiple cell biological functions, such as the normal formation of photoreceptors in the developing retina of rodents23and the growth, proliferation and invasion, and apoptosis of oral squamous cell carcinoma.24, 25, 26, 27 Thus, we hypothesis that TUG1 may have an essential role on the behavior of the trophoblast cells. Because the molecular mechanism remains unclear, we need to further exploration, and screening of valuable biomarkers and therapeutic targets of PE are needed.
To explore the downstream effects of TUG1 in PE, we performed RNA transcriptome sequencing to reveal latent downstream targets of TUG1. We found a noticeable increase in the expression of the cell suppressor Rnd3. Rnd3, also known as RhoE, is a GTPase that involve in the regulation of various cell biological behaviors including proliferation, migration, invasion, and apoptosis.28, 29, 30, 31, 32 Riento et al.28 has reported that Rnd3 is a suppressor of Rock1, which modulates actin dynamics. Previous studies have demonstrated the function of Rnd3 in cancer occurrence and progression.33, 34 For example, Rnd3 is significantly downregulated in lung cancer cells and can regulate lung cancer cell proliferation through Notch signaling.35 However, the function of Rnd3 in PE has not been studied. In the present study, we also found that the levels of RND3 mRNA and proteins can be repressed by TUG1 and that Rnd3 might act as a suppressor in trophoblasts.
Our research has confirmed EZH2 as an essential regulator in the TUG1-mediated Rnd3 repression network. qRT-PCR and western blot assays established that RND3 were upregulated after the silencing of EZH2 in trophoblast cells. In present research, we revealed high abundance interacting between TUG1 and EZH2 in trophoblast cells, and established that TUG1 could mediate transcriptional regulation of RND3. ChIP-qPCR assays confirmed that TUG1 can recruit Ezh2 protein and downregulated RND3 expression. Our resulting data clarified that RND3 expression is explicitly regulated by PRC2, partly through TUG1. Together, this demonstrates that TUG1 affects the function of trophoblasts partly via the epigenetic regulation of RND3 expression.
In short, our research showed that the lncRNA TUG1 is downregulated in placental samples from PE patients compared with levels in controls. Knockdown of TUG1 resulted in suppressive effects, impairing cellular proliferation, migration, and invasion and inducing apoptosis in trophoblasts, and further affecting the process of spiral artery remodeling. Based on these collective results presented above, we propose that TUG1 bring into play functions, partially by recruiting Ezh2 and inhibiting RND3 expression in PE (Figure 6h).
Materials and methods
Tissue samples and patients
We collected 52 paired placental tissues from PE women and normal pregnancies, who were diagnosed with PE and underwent cesarean deliveries at the Jiangsu Province Hospital in 2015–2016, then all placental tissues were instantly snap frozen with liquid nitrogen and collected at −84 °C before RNA extraction. And all patients’ clinic features were recorded in Table 1. Our research was authorized by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University, China. Informed written consents were gotten from all patients in this study.
Cell culture
We selected three cell lines that were related to pregnancy. HTR-8/SVneo Cells was generous furnished by Prof. Charies Graham, Queen’s University, Canada. And JEG-3, HUVEC-C, WISH and BeWo cells were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). HTR/SVneo, JEG-3, BeWo, WISH and HUVEC-C cell lines were cultured in RPMI 1640, MEM, F12K, RPMI 1640 and ECM (KeyGEN, Nanjing, China), respectively, which added to 10% fetal bovine serum (GIBCO, BRL, Invitrogen, Carisbad, CA, USA),100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen) in humidified air at 37 °C/5% CO2.
Cell transfection
Plasmid vectors (pcDNA-RND3 and empty vector) for transfection were arranged by using DNA Midiprep kits (Qiagen, Hilden, Germany). The specific siRNAs were transfected into cell lines, including si-TUG1, si-RND3 or si-NC. The siRNAs sequences were presented in Supplementary Table 1. These cell lines were cultivated on six-well plates and then treated through using Lipofectamine-2000 (Invitrogen) following the manual. After 48 h transfection, cells were harvested for conducting further experiments.
RNA preparation and qRT-PCR
RNA preparation and qRT-PCR assays were performed as previously reported in Zuo et al.20 1 μg RNA was reverse transcribed in a final volume of 20 μl by using the PrimeScript RT reagent Kit (TaKaRa, Dalian, China). SYBR Premix Ex Taq (TaKaRa, Dalian, China) were used to examine the expression of TUG1, according to the manual. All results were normalized to GAPDH. The relative primers sequences were presented in Supplementary Table 1. The qPCR results were evaluated and afterwards converted to fold changes.
Subcellular fractionation location
Subcellular fractionation location assays were performed as previously reported in Zuo et al.20 We used PARIS Kit (Life Technologies, Carlsbad, CA, USA) to separate of nuclear and cytosolic fractions of trophoblast cells according to the manual. The levels of TUG1, RNA U1and GAPDH in cytoplasm and nuclear fraction was detected by qPCR. The relative rate of TUG1, GAPDH and U1 in cytoplasm or nuclear part was presented as the percentage of the total RNA. U1 was used as nuclear control, and GAPDH as cytoplasm control.
Fluorescence in situ hybridization
Cells were cultivated in 24-well plates, and then fixed by 75% ethanol for 30 min followed by washes with PBS. Next, the fixed cells were further conducted assays, according to the manual. The sequences of probe were listed in Supplementary Table 1.
Cell viability assays
Cell proliferation assays, colony formation assays were performed as previously reported in Zuo et al.20
For EdU assays, cells were cultivated in six-well plates, and 10 μM EdU was added to these wells. And then fixed with 4% formaldehyde for 30–40 min after 2 h. After washing, EdU could be identified with a Click-iTR EdU Kit for 25 min, and the cells were stained with 300 μl DAPI (Invitrogen, Molecular Probes, Eugene, OR, USA) at 25 °C for 15–20 min and imagined by performing a fluorescent microscope (Nikon Corporation, Tokyo, Japan). The relative percentage of Edu-positive cells were examined from three to five subjects in three wells.
Flow-cytometric analysis
Flow cytometry assays were performed as previously reported in Zuo et al.20 After the cells were transfected with siRNAs for 48 h, we harvested the cells and then performed FITC-Annexin V and Propidium iodide (PI) by using the FITC-Annexin V Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manual. Cell cycle analysis were stained with propidium oxide by the Cycle TEST PLUS DNA Reagent Kit (BD Biosciences) following the manual and evaluated by FACScan. The rate of the cells in each phase were assessed.
Cell migration and invasion assays
Cell migration and invasion assays were performed as previously reported in Zuo et al.20 3–5 × 104 cells were cultivated on the top of a membrane precoated with Matrigel (BD Biosciences) for cell invasion assays (without Matrigel for cell migration assays). Cells inside the upper chamber were removed after incubation for 24, 36, 48 and 60 h. While cells on the lower membrane surface were fixed with methanol and then stained with 0.5% Crystal violet solution. Three-five randomly selected fields were counted in per well.
Network formation assay
After 36 h of treated HTR-8/SVneo and HUVEC-C cells with TUG1 siRNAs, then perform Network Formation assays as previously reported in Zou et al.22 The HTR-8/SVneo and HUVEC-C cells were transfected with siRNAs (104 cells/well) and then cultivated in 96-well plates with five duplicate. After 6 h, the images were blinded and the number of cell–cell protracted contacts was counted as representations of the number of capillary-like networks present in each field. Results were presented as the average number of networks at least three separate experiments.
Western blotting assays
HTR-8/SVneo and JEG-3 cells were harvested, and protein extractions was separated from transfected cells by using 10% polyacrylamide gradient SDS gel, and incubated with different antibodies, in which Anti-GAPDH and anti-EZH2 were purchased from Abcam (Hong Kong, China). Anti-RND3 was from Cell Signaling Technology (Boston, MA, USA).
RNA-seq bioinformatic analysis
The mRNA-Seq experiments were executed by Wuhan Genomics Institute (Wuhan, China). mRNA-seq library was settled for sequencing exploiting standard Illumina protocols. Briefly, total RNAs from si-NC, or si-TUG1 1# transfected HTR-8/SVneo, were isolated by using TRIzol reagent (Invitrogen). mRNA extraction was performed by utilizing Dynabeadsoligo (dT) (Invitrogen Dynal). Superscript II reverse transcriptase (Invitrogen) and random hexamer primers were utilized to synthesize double-stranded complementary DNAs. To stablish the mRNA-seq library, the cDNAs were next fragmented via nebulization and the standard Illumina protocol followed.
RNA immunoprecipitation assays
RNA immunoprecipitation assays were performed as previously reported in Zuo et al,20 HTR-8/SVneo and JEG-3 cells were lysed for immunoprecipitation of EZH2 and SUZ12. The supernatants were incubated with protein A/G Sepharose beads coated with antibodies for 8–10 h at 4 °C. After the beads were washed with corresponding wash buffer, the complexes were incubated with 0.1% SDS/0.5 mg/ml Proteinase K (30 min at 55 °C) to remove proteins, respectively. The RNA was subjected by qPCR analysis to demonstrate the presence of TUG1.
Chromatin immunoprecipitation assays
Trophoblast cells were preserved with formaldehyde and incubated for 10 min to generate DNA–protein cross-links. Then cell lysates were sonicated to generate chromatin fragments of 200–300 bp and immunoprecipitated with EZH2 (Millipore, Billerica, MA, USA), H3K27me3 (Millipore) or IgG as the control. Precipitated chromatin DNA was recovered and analyzed by qRT-PCR.
Statistical analysis
All statistical analyses were executed utilizing SPSS 17.0 software (IBM, Chicago, IL, USA). Furthermore, less than 0.05 considered statistically significant P-values. These resulting data were recounted as the mean±SD. Statistical significance were ascribed at *P<0.05 or **P<0.01.
References
Kurtz WS, Glueck CJ, Hutchins RK, Sisk RA, Wang P . Retinal artery and vein thrombotic occlusion during pregnancy: markers for familial thrombophilia and adverse pregnancy outcomes. Clin Ophthalmol 2016; 10: 935–938.
Powe CE, Levine RJ, Karumanchi SA . Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011; 123: 2856–2869.
Knofler M, Pollheimer J . Human placental trophoblast invasion and differentiation: a particular focus on Wnt signaling. Front Genet 2013; 4: 190.
Zarate A, Saucedo R, Valencia J, Manuel L, Hernandez M . Early disturbed placental ischemia and hypoxia creates immune alteration and vascular disorder causing preeclampsia. Arch Med Res 2014; 45: 519–524.
Pennington KA, Schlitt JM, Jackson DL, Schulz LC, Schust DJ . Preeclampsia: multiple approaches for a multifactorial disease. Dis Model Mech 2012; 5: 9–18.
Redline RW, Patterson P . Pre-eclampsia is associated with an excess of proliferative immature intermediate trophoblast. Hum Pathol 1995; 26: 594–600.
Myatt L . Role of placenta in preeclampsia. Endocrine 2002; 19: 103–111.
de Groot CJ, O'Brien TJ, Taylor RN . Biochemical evidence of impaired trophoblastic invasion of decidual stroma in women destined to have preeclampsia. Am J Obstet Gynecol 1996; 175: 24–29.
Mattick JS . RNA regulation: a new genetics? Nat Rev Genet 2004; 5: 316–323.
Consortium, E. P.. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012; 22: 1760–1774.
Ulitsky I, Bartel DP . lincRNAs: genomics, evolution, and mechanisms. Cell 2013; 154: 26–46.
Rinn JL . lncRNAs: linking RNA to chromatin. Cold Spring Harb Perspect Biol 2014; 6: a018614.
Wilusz JE . Long noncoding RNAs: Re-writing dogmas of RNA processing and stability. Biochim Biophys Acta 2016; 1859: 128–138.
Buckberry S, Bianco-Miotto T, Hiendleder S, Roberts CT . Quantitative allele-specific expression and DNA methylation analysis of H19, IGF2 and IGF2R in the human placenta across gestation reveals H19 imprinting plasticity. PLoS One 2012; 7: e51210.
Chen H, Meng T, Liu X, Sun M, Tong C, Liu J et al. Long non-coding RNA MALAT-1 is downregulated in preeclampsia and regulates proliferation, apoptosis, migration and invasion of JEG-3 trophoblast cells. Int J Clin Exp Pathol 2015; 8: 12718–12727.
Yuan SX, Yang F, Yang Y, Tao QF, Zhang J, Huang G et al. Long noncoding RNA associated with microvascular invasion in hepatocellular carcinoma promotes angiogenesis and serves as a predictor for hepatocellular carcinoma patients' poor recurrence-free survival after hepatectomy. Hepatology 2012; 56: 2231–2241.
Wilusz JE, Sunwoo H, Spector DL . Long noncoding RNAs: functional surprises from the RNA world. Genes Dev 2009; 23: 1494–1504.
Maass PG, Luft FC, Bahring S . Long non-coding RNA in health and disease. J Mol Med 2014; 92: 337–346.
Zuo Q, Huang S, Zou Y, Xu Y, Jiang Z . The Lnc RNA SPRY4-IT1 Modulates Trophoblast Cell Invasion and Migration by Affecting the Epithelial-Mesenchymal Transition. Sci Rep 2016; 6: 37183.
Zhang Y, Zou Y, Wang W, Zuo Q, Jiang Z, Sun M et al. Down-regulated long non-coding RNA MEG3 and its effect on promoting apoptosis and suppressing migration of trophoblast cells. J Cell Biochem 2015; 116: 542–550.
Zou Y, Jiang Z, Yu X, Sun M, Zhang Y, Zuo Q et al. Upregulation of long noncoding RNA SPRY4-IT1 modulates proliferation, migration, apoptosis, and network formation in trophoblast cells HTR-8SV/neo. PLoS One 2013; 8: e79598.
Young TL, Matsuda T, Cepko CL . The noncoding RNA taurine upregulated gene 1 is required for differentiation of the murine retina. Curr Biol 2005; 15: 501–512.
Liu Q, Liu H, Cheng H, Li Y, Li X, Zhu C et al. Downregulation of long noncoding RNA TUG1 inhibits proliferation and induces apoptosis through the TUG1/miR-142/ZEB2 axis in bladder cancer cells. Onco Targets Ther 2017; 10: 2461–2471.
Wang H, Yu Y, Fan S, Luo L . Knockdown of long noncoding RNA TUG1 inhibits the proliferation and cellular invasion of osteosarcoma cells by sponging MiR-153. Oncol Res 2017 doi:10.3727/096504017X14908298412505.
Li G, Song H, Chen L, Yang W, Nan K, Lu P et al. TUG1 promotes lens epithelial cell apoptosis by regulating miR-421/caspase-3 axis in age-related cataract. Exp Cell Res 2017; 356: 20–27.
Yun-Bo F, Xiao-Po L, Xiao-Li L, Guo-Long C, Pei Z, Fa-Ming T et al. LncRNA TUG1 is upregulated and promotes cell proliferation in osteosarcoma. Open Med 2016; 11: 163–167.
Riento K, Guasch RM, Garg R, Jin B, Ridley AJ . RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol 2003; 23: 4219–4229.
Katoh H, Harada A, Mori K, Negishi M . Socius is a novel Rnd GTPase-interacting protein involved in disassembly of actin stress fibers. Mol Cell Biol 2002; 22: 2952–2964.
Lin X, Liu B, Yang X, Yue X, Diao L, Wang J et al. Genetic deletion of Rnd3 results in aqueductal stenosis leading to hydrocephalus through up-regulation of Notch signaling. Proc Natl Acad Sci USA 2013; 110: 8236–8241.
Ongusaha PP, Kim HG, Boswell SA, Ridley AJ, Der CJ, Dotto GP et al. RhoE is a pro-survival p53 target gene that inhibits ROCK I-mediated apoptosis in response to genotoxic stress. Curr Biol 2016; 26: 2221–2222.
Xia H, Li M, Chen L, Leng W, Yuan D, Pang X et al. Suppression of RND3 activity by AES downregulation promotes cancer cell proliferation and invasion. Int J Mol Med 2013; 31: 1081–1086.
Wang H, Wang Y, Liang B, He F, Li Y, Che J et al. The Rho GTPase RhoE exerts tumor-suppressing effects in human esophageal squamous cell carcinoma via negatively regulating epidermal growth factor receptor. J Cancer Res Ther 2016; 12: 60–63.
Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016; 12: 515–528.
Tang Y, Hu C, Yang H, Cao L, Li Y, Deng P et al. Rnd3 regulates lung cancer cell proliferation through notch signaling. PLoS One 2014; 9: e111897.
Acknowledgements
The present work was supported by the National Scientific Foundation of China (No. 81270710, NO. 81470065, NO. 81401229, NO. 81702266 and NO.81771603), the traditional Chinese medicine project of Jiangsu Province (NO. ZX2016D2) and the project of construction capacity for birth defect screening and diagnosis laboratory in Jiangsu (BM2015020). This work was partly supported by China Postdoctoral Science Foundation; Grant number (2017M610339), and the Natural Science Foundation of Jiangsu Province (project number: BK20161061).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xu, Y., Ge, Z., Zhang, E. et al. The lncRNA TUG1 modulates proliferation in trophoblast cells via epigenetic suppression of RND3. Cell Death Dis 8, e3104 (2017). https://doi.org/10.1038/cddis.2017.503
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2017.503
This article is cited by
-
LncRNA LINC01123 promotes malignancy of ovarian cancer by targeting hsa-miR-516b-5p/VEGFA
Genes & Genomics (2024)
-
Propofol Promoted the Cell Growth and Epithelial Mesenchymal Transformation of the HTR-8/SVneo Cells through Targeting the METTL3 Mediated ZEB2
Reproductive Sciences (2024)
-
Downregulated DUXAP8 lncRNA impedes trophoblast cell proliferation and migration by epigenetically upregulating TFPI2 expression
Reproductive Biology and Endocrinology (2023)
-
LncRNA-TUG1 Downregulation is Correlated with the Development of Progressive Chronic Kidney Disease Among Patients with Congestive Heart Failure
Molecular Biotechnology (2022)
-
LncRNA Tug1 maintains blood–testis barrier integrity by modulating Ccl2 expression in high-fat diet mice
Cellular and Molecular Life Sciences (2022)

{kind=link}