
Abstract
MicroRNA miR-34a is recognized as a master regulator of tumor suppression. The strategy of miR-34a replacement has been investigated in clinical trials as the first attempt of miRNA application in cancer treatment. However, emerging outcomes promote the re-evaluation of existing knowledge and urge the need for better understanding the complex biological role of miR-34a. The targets of miR-34a encompass numerous regulators of cancer cell proliferation, survival and resistance to therapy. MiR-34a expression is transcriptionally controlled by p53, a crucial tumor suppressor pathway, often disrupted in cancer. Moreover, miR-34a abundance is fine-tuned by context-dependent feedback loops. The function and effects of exogenously delivered or re-expressed miR-34a on the background of defective p53 therefore remain prominent issues in miR-34a based therapy. In this work, we review p53-independent mechanisms regulating the expression of miR-34a. Aside from molecules directly interacting with MIR34A promoter, processes affecting epigenetic regulation and miRNA maturation are discussed. Multiple mechanisms operate in the context of cancer-associated phenomena, such as aberrant oncogene signaling, EMT or inflammation. Since p53-dependent tumor-suppressive mechanisms are disturbed in a substantial proportion of malignancies, we summarize the effects of miR-34a modulation in cell and animal models in the clinically relevant context of disrupted or insufficient p53 function.
Similar content being viewed by others
Facts
-
MiR-34a expression is lost or decreased in many cancers
-
Re-expression of miR-34a has been investigated in clinical trials as potential treatment of advanced cancers
-
MiR-34a contributes to tumor suppression by repressing over 700 transcripts implicated in cellular proliferation, survival and plasticity
-
MiR-34a expression is governed by p53, but can be regulated by multiple p53-independent mechanisms
Open Questions
-
Can miR-34a exert any pro-tumorigenic effects in a specific context?
-
How can miR-34a modulation affect human immune response and the condition of vital organs? What outcomes of systemic miR-34a delivery can be expected in p53-defective cancer cells and in the microenvironment of p53 wild-type normal cells?
-
Is optimization of the delivery method sufficient to avoid potential adverse effects of miR-34a application?
MicroRNAs (miRNA) are evolutionary highly conserved non-coding RNA molecules, exerting essential functions in a wide range of physiological processes. In cancer, miRNAs exert both pro- and anti-tumorigenic effects by virtue of miRNA-specific and context-dependent mechanisms.1 Consequently, deregulation of miRNA expression was reported in most cancer types and at multiple levels or miRNA expression control.2, 3 The strategy of replacing downregulated miR-34a by intravenous liposome-based delivery has been investigated in phase I clinical trials for advanced stages of multiple solid and hematological malignancies.4, 5 The study was recently terminated and reported immune-related adverse effects in several individuals, implicating an urgent need to improve the tolerability of miR-34a-based therapy. Better understanding of the complex biological function of miR-34a in both normal and cancer cells is indispensable for this achievement.
miR-34a was selected as a candidate tumor suppressor miRNA based on its frequent deregulation in cancer tissues6, 7, 8 and its ability to regulate the expression of multiple targets implicated in tumorigenesis and cancer progression, such as MYC, MET, CDK4/6, NOTCH1, BCL2, CD44 and many other molecules.9, 10 Broad target specificity of miRNAs, resulting from their short binding motifs in target gene sequences, can be advantageous for simultaneous targeting of multiple tumor-promoting transcripts, but at the same time poses a risk of potential adverse effects. Thorough understanding of associated signaling pathways both upstream and downstream of miR-34a is therefore a prerequisite for successful therapeutic application.
Expression of miRNA transcripts is driven from promoter regions that accommodate binding sites of canonical transcription factors. Another level of regulation of miRNA expression by feedback loops was described for multiple miRNA-families including miR-34a.11, 12, 13 Transcription of miR-34a is regulated dominantly by the crucial tumor suppressor p53, by means of binding to multiple canonical p53 binding sites in regions proximal to the MIR34A promoter.14, 15, 16 Importantly, miR-34a is detected likewise in tissues and cells with p53-mutation or deletion, implicating the existence of p53-independent mechanisms of miR-34a expression.17, 18
Mutation or inactivation of the tumor suppressor p53 occurs in a high proportion of tumors,19 affecting cell proliferation, survival and sensitivity to chemotherapy. This is often associated with downregulation of miR-34a expression in both hematologic and solid malignancies.18, 20, 21, 22, 23, 24 Due to the implication of miR-34a in multiple feedback loops, which can be strongly affected by the therapeutic dose of miR-34a mimic, it is important to consider potential effects of miRNA-based anti-cancer therapy in the context of disrupted or insufficient p53 function. Important questions arising from this presumption are: (1) Which p53-independent mechanisms can affect miR-34a expression? (2) What may be the consequences of miR-34a modulation in cancers harboring defects in p53 function?
miR-34 family, biogenesis, targets and expression
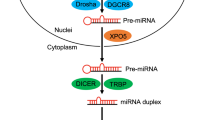
Of the 3 members of miR-34 family, miR-34a is ubiquitously expressed in normal human tissues, while expression of miR-34b/c is characterized by tissue specificity to the testicles, fallopian tubes, lungs or brain.7 In human genome, miR-34a is encoded on chromosome 1p36, while miR-34b and miR-34c are expressed from one common transcript of chromosome 11q23.10, 25 Similarly to the biogenesis of all miRNAs, miR-34a is transcribed as a long hairpin molecule (pri-miRNA), which is subsequently cleaved by an RNase III Drosha to an ~70-nucleotide long stem-loop precursor (pre-miRNA). Following nuclear export, the pre-miRNA is further cleaved by an RNase III Dicer into 22-nucleotide long mature strands, which are incorporated into RNA-induced silencing complex (RISC). This RNA/protein complex mediates downregulation of target transcripts by mRNA degradation or inhibition of translation.9, 10 In case of miR-34a, experiments with synthetic pre-miRNAs revealed that incorporation of both 5p and 3p mature strands into RISC enables specific regulation of different targets.26
MiR-34a is considered to act as a tumor suppressor miRNA, since of the 700 to date experimentally validated miR-34a targets,27 many genes are implicated in the control of cellular proliferation (that is, cyclins, cyclin-dependent kinases, MYCN, NOTCH1, MDMX), apoptosis (BCL2, SIRT1 and BIRC5), senescence (E2F3), cancer stem-like cell phenotype (CD44, NANOG and SOX2), motility (SNAI1, MET and AXIN2) or immune evasion (PD-L1, DGKζ). MiR-34a therefore exerts wide-range effects on cancer progression and metastasis (for overview see references 9, 10, 28, 29 and an updated database of validated miRNA targets27).
In accordance with general downregulation of miRNA expression in malignancies,30 downregulation of miR-34a expression was reported in multiple types of cancer.6, 7, 8, 25, 31 Still, well designed cohort studies are required to establish miR-34a as a prognostic factor.32 Concomitantly, the genomic locus 1p36 encoding miR-34a transcript is lost in certain tumors,33 proposing one of possible mechanisms of miR-34a loss in neuroblastoma.34, 35
p53-dependent regulation of miR-34a expression and function
In physiological conditions, expression of miR-34a is transcriptionally regulated by the key tumor suppressor p53. The function of p53 in the prevention of uncontrolled proliferation of cells with damaged DNA predisposes p53 to be one of the most frequently inactivated proteins in human cancer. Activation of p53 results either in cell cycle arrest enabling repair of minor damage, induction of replicative senescence, or apoptosis. Mechanisms of evasion from p53-mediated tumor suppression encompass selection of cancer cells harboring p53 mutations (nonsense or missense, eventually accompanied by a gain of function) or allelic loss, as well as inactivation by viral or cellular proteins.19, 36
An evolutionary conserved p53-binding site was identified upstream of the miR-34a transcript.14, 15, 37 Additional interaction between p53 and an intronic region of miR-34a was shown to be enhanced by genotoxic stress.37 On the other hand, several feedback loops implicate miR-34a in the regulation of p53 (Figure 1). For example, TP53 mRNA was shown to be targeted by miR-34a through non-canonical response elements in 5′UTR and the coding sequence.38 MDMX, an inhibitor of p53 transactivation, is a direct target repressed by miR-34a.39, 40 From epigenetic mechanisms, miR-34a represses the histone deacetylases SIRT1 and HDAC1, thereby enhancing the ability of p53 to transactivate its target genes.13, 41

Feedback regulation of miR-34a. Upon DNA damage, miR-34a transcription is induced by p53. miR-34a targets p53 directly by interaction with its transcript and indirectly through targeting of p53 inhibitor MDMX (yellow). MiR-34a-mediated inhibition of p53 deacetylase SIRT1 is implicated in apoptosis (green). Feedback inhibition between miR-34a, IL-6R and P-STAT3 is implicated in inflammation, while regulation of PD-L1 – P-Akt axis is important for immune surveillance (red). In EMT, miR-34a is implicated in multiple feedbacks encompassing EMT regulators Snail, ZEB1 or AXL (blue)
Induction of miR-34a expression by genotoxic stress is strongly p53-dependent,6, 15, 37, 42, 43, 44 with only one reported exception in bladder cancer cells.45 However, a correlation between basal level of miR-34a and p53 status was demonstrated only in a proportion of experimental models.13, 14, 17, 46, 47, 48 A lack of significant correlation between p53 mutational status and miR-34a expression was observed in a set of lung cancer patients,42 pancreatic cancer and CSC-like cells49 and colorectal cancer,50 suggesting either that p53 transcriptional activity rather than mutation status is essential for miR-34a expression, or that the basal level of miR-34a expression can be maintained by p53-independent mechanisms as well.
On the other hand, members or the miR-34 family were proposed to be key mediators of p53 tumor suppressor function after DNA damage. In most conditions, miR-34a overexpression inhibited cell proliferation or induced a senescent phenotype,6, 15, 37, 42, 43 while induction of apoptosis after miR-34a overexpression was observed only in certain experimental models.6, 14, 15, 37 The level of miR-34a expression was proposed as a mechanism responsible for cell-fate decision after p53 induction.51 Importantly, miR-34a inhibition consistently desensitized cells to apoptosis induced by genotoxic stress.15, 28, 42, 45, 52
Nevertheless, recent reports show that p53 can exert its function even in the absence of miR-34a. It was demonstrated that the response to genotoxic stress is intact in miR-34 KO cells and animals, but this effect may result from redundancy between miR-34 and miR-449 families, sharing the same seed.17, 38 This hypothesis is supported by facts that miR-34/449 double KO mice exhibited postnatal mortality, infertility and strong respiratory dysfunction caused by defective mucociliary clearance,53 and that deletion of miR-34b/c cluster induces expression of miR-449 family, and vice versa.54 In a Kras-induced mouse lung cancer model, miR-34a deficiency alone does not exhibit a strong oncogenic effect. However, miR-34a deficiency strongly promotes tumorigenesis when p53 is haploinsufficient, suggesting that the defective p53-miR-34 feedback loop can enhance oncogenesis in a specific context.40 Consistently, prostate epithelium-specific inactivation of miR-34 and p53 leads to expansion of the prostate stem cell compartment and development of early invasive adenocarcinomas and high-grade prostatic intraepithelial neoplasia through enhanced MET signaling.55
p53-independent mechanisms of miR-34a regulation
Multiple experimental observations suggest that besides p53-driven miR-34a expression, miR-34a levels can be regulated in a p53-independent manner.42, 49 Mechanisms responsible for p53-independent regulation can either operate simultaneously with p53-dependent control, or establish dominance in case of disrupted p53 function. From the multitude of miR-34a influencing factors, some can be classified as extrinsic (triggered by activation of signaling pathways by external stimuli, such as in the case of factors originating from cell microenvironment, or epithelial–mesenchymal transition (EMT)-associated changes), while factors classified as intrinsic act at the level of intracellular signaling pathways, epigenetic regulation or affect the general process of miRNA biogenesis (Figure 2). While certain miR-34a regulating mechanisms were described in physiologic conditions, other are associated with disease-related processes such as inflammation or oncogene signaling.

Reported mechanisms of p53-independent miR-34a regulation in different biological contexts. Intrinsic/intracellular mechanisms are represented in red, extrinsic mechanisms triggered by intercellular or microenvironmental effects are depicted in blue. Notably, certain molecules are characterized by context-dependent roles in the regulation of miR-34a
Regulation of miR-34a expression in the process of miRNA maturation
miRNA maturation involves two subsequent RNA cleavage steps, mediated by RNase III enzymes Drosha and Dicer, respectively. The assembly, activity and target recognition by the Drosha complex require cooperation with DGCR8 and auxiliary factors, such as the DEAD-box RNA helicases p68 (DDX5) and p72/p82 (DDX17).56 During the second cleavage step, Dicer is associated with auxiliary proteins, such as TAR RNA binding protein and kinase R–activating protein to increase its stability and processing activity.57
The expression and assembly of molecules involved in the miRNA processing machinery was shown to be affected by transcription factors primarily associated with other biological processes, such as components of p53, TGF-β or Hippo signaling pathways.58, 59, 60, 61 The maturation of several miRNAs, but not miR-34a, was enhanced by p53 activation after genotoxic treatment.61 Importantly, transcriptionally inactive p53 mutants interfered with a functional assembly between Drosha complex and RNA helicases DDX5/DDX17, leading to attenuation of miRNA processing activity.61, 62 TAp63, except from directly affecting MIR34A transcription by binding to its promoter, can also enhance miR-34a processing by transcriptional activation of Dicer.60
A specific effect on miR-34a maturation was observed after BRCA1 overexpression, which accelerated the processing of miRNA primary transcripts, thereby increasing the expression of both precursor and mature forms of miR-34a. Despite physical association of BRCA1 with p53, the mechanism was reported in cell lines with disturbed p53 function resulting from immortalization or p53 mutation.63 Modulation of SIRT1 activity influenced miR-34a maturation in keratinocytes with both normal and reduced p53 function.64
Furthermore, miR-34a was identified as one of six miRNAs whose expression was affected by knock-down of RNA helicase DDX3X, exerting key roles in cancer development. DDX3X interacts with Drosha/DGCR8 complex, facilitates pri-miRNA binding and promotes miR-34a maturation.65 In cells with undisturbed RNA processing machinery, cell type-specific differences in miR-34a abundance were attributed to unequal processing of its pri-miRNA, resulting in differences in cell fate after p53 activation.51
Regulation of miR-34a expression by epigenetic mechanisms
The phenomenon of CpG methylation, resulting in inactivation of the surrounding chromatin due to recruitment of histone deacetylases, is important in the maintenance of cell specific expression patterns and is stably inherited to the next cell generation.66, 67 Both miR-34 transcripts contain a CpG island in their promoter region.68 CpG methylation of the MIR34A promoter was observed in almost 80% of primary prostate carcinomas and a variable proportion of other tumors. In cancer cell lines, the frequencies of MIR34A methylation varied from 13% in colon cancer to 43% in melanomas.69 Contrarily, in formalin-fixed, paraffin-embedded tumor samples, MIR34A methylation was the most frequently detected in colorectal cancer (74%). Data from other tumor types also suggest that MIR34A promoter methylation occurs more frequently in vivo than in vitro.70 Importantly, MIR34A promoter methylation correlated with distant metastases in colon cancer patients,71 underscoring the clinical importance of miR-34a implication in cellular plasticity.12
Contrarily, promoter hypomethylation was responsible for induced miR-34a expression in alcoholic liver injury. In normal human hepatocytes and cholangiocytes, induction of miR-34a enhanced cell survival and migration, which in the context of liver cells may be associated with tissue remodeling and regeneration.72 Hypomethylation of an alternative promoter region upstream from MIR34A coding sequence was observed in chronic lymphocytic leukemia compared to healthy B cells, which was accompanied by increased miR-34a expression in malignancy.68
Mechanistically, a lack of miR-34a induction in cells with wild-type (wt) p53 after DNA damage was attributed to chromatin methylation in IGR-39 melanoma cells.69 Restoration of miR-34a expression by chromatin modulators affected expression of miR-34a targets, cell survival and EMT phenotype in pancreatic cancer cell lines.49 Analysis of MIR34A methylation in a wide cohort of primary colorectal cancer samples pointed out a statistically significant correlation of MIR34A methylation and the absence of p53 mutation (evaluated by immunohistochemical score of p53), indicating that the loss of the miR-34a-mediated tumor suppressor function may substitute for loss of the p53 response by p53 mutations in colorectal cancer.70 Similarly, low incidence of p53 mutation together with epigenetic mechanism of miR-34a silencing was reported in diffuse large B-cell lymphoma.73 On the other hand, in the context of disrupted p53, reintroduction of miR-34a induced a senescent phenotype in TP53-null PC3 cells and p53 mutated (R248W) MIA PaCa-2 cells, characterized by a moderate and high level of MIR34A promoter methylation, respectively.69 These findings suggest that the therapeutic strategy of miR-34a reintroduction might be beneficial for patients with epigenetically downregulated miR-34a, regardless of the p53 status in tumor cells.
Regulation of miR-34a expression by members of the p53/p63/p73 family of transcription factors
Homologs of the p53 tumor suppressor, p73 and p63, share a high degree of structural similarity and can bind and activate transcription from the majority of p53-responsive promoters. Transcription from alternative promoters and splicing events give rise to full length, transactivation domain containing (TA) and truncated (ΔN) isoforms of p63 and p73, with distinct functions and physiological roles.74
Direct binding to the p53 consensus DNA-binding sites in MIR34A promoter was demonstrated for both p63 and p73.60, 75, 76, 77 The TAp73-miR-34a axis represents a positive regulation, restricted to miR-34a and implicated in neuronal physiology and pathology.75 Similarly, Trp63 KO MEFs exhibit decreased expression of miR-34a and ectopic expression of the ΔNp63β isoform induces miR-34a.76 On the contrary, ΔNp63 represses miR-34a and miR-34c transcription in murine epidermal cells and enables cell cycle progression in a p53-independent manner,77 suggesting context and isoform-specific effects of p63 on miR-34a expression. Furthermore, TAp63 expression is stimulated by a miR-34a target Oct-4, which contributes to oncogenic transformation in the process of pluripotency induction, indicating the importance of miR-34a regulation by p63 in carcinogenesis.78
Regulation of miR-34a in the context of immune response
Chronic inflammation has long been associated with tumor initiation, progression and invasion.79 Secreted molecules implicated in inflammation such as TNF-α, IL-6 or LPS were described as potent inducers of EMT and key players in cancer progression.80 In accordance with its reported anti-tumorigenic function, miR-34a exerts an anti-inflammatory effect by downregulating TNF-α and IL-6.81 Expression of miR-34a itself can be affected by inflammatory stimuli, as it was found downregulated after LPS stimulation in macrophages81 or upregulated by atherosclerosis-inducing oscillatory shear stress in endothelial cells.82 MiRNA profiling of wt versus TP53 KO mice infected by Corynebacterium parvum demonstrated that inflammation-associated upregulation of miR-34 family members is largely p53-dependent.83 Likewise, dependence on intact p53 function was demonstrated for induction of miR-34a by direct binding of immune response-associated transcription factor NF-кB to MIR34A promoter.84
A p53-independent mechanism of miR-34a regulation in inflammation is exemplified by direct repression of MIR34A gene via a conserved STAT3-binding site in the first intron during IL-6–induced EMT and invasion in colorectal cancer cells harboring p53 mutation11 (Figure 2). Nevertheless, p53-dependent expression of miR-34a was crucial for suppression of tumor progression by inhibiting the IL-6R/STAT3/miR-34a feedback loop (Figure 1).11
As an upstream regulator of PD-L1, CCL22 and DGKζ expression, miR-34a was identified as an important regulator of immune response in cancer.31, 85, 86 MiR-34a overexpression reversed chemotherapeutic agent-induced PD-L1 expression, reduced PD-L1 specific T cell apoptosis and inhibited Treg recruitment in p53-defective models.31, 86
Regulation of miR-34a in the context of EMT
EMT encompasses a series of phenotypic and biochemical changes enabling cell spreading, eventually leading to the formation of metastases or acquisition of chemoresistance.87 Multiple external signals, such as cytokine-triggered signaling pathways or more complex forms of cellular or environmental stress (hypoxia, nutrient deprivation, inflammation), converge on the regulation of several nodal EMT-driving transcription factors (Snail, Slug, Twist, ZEB1, ZEB2), responsible for regulation of intracellular and extracellular molecules characteristic for the mesenchymal phenotype.88 A significant role of miRNAs in the control of EMT is exemplified by a double-negative feedback loop between ZEB1 and ZEB2 transcription factors and miR-200 family.89
A role of miR-34a in the regulation of EMT was described in cancer cell lines, animal models of human cancer as well as in hypoxia-induced EMT in renal tubular epithelial cells, suggesting that miR-34a is a universal regulator of EMT.8, 11, 12, 76, 90, 91, 92 Notably, miR-34a expression was shown to be reciprocally controlled by EMT-regulating molecules. Direct binding of Snail or ZEB1 to E-boxes in the promoter regions of both miR-34 family transcripts proved a direct repression of miR-34 transcription by EMT-inducing transcription factors12 (Figure 2).
Similarly to the previously reported p53/miR-200/ZEB1/2 axis, both Snail and ZEB1-mediated regulation of miR-34a is subject to regulation by double-negative-feedback loops (Figure 1). Mutual negative regulation between miR-34a and Snail was discovered in the context of p53-dependent mesenchymal–epithelial transition. Besides repression of Snail, miR-34a was shown to negatively regulate Slug, ZEB1, ZNF281 as well as several stemness factors, thereby stabilizing the epithelial phenotype.12, 93
In a Kras/Trp53-mutation driven mouse model of human non-small cell lung cancer (NSCLC), ZEB1 drove pro-migratory cytoskeletal processes and metastasis by downregulating the expression of miR-34a. In this case, the repression of miR-34a by ZEB1 was indirect, mediated by repression of ΔNp63 by ZEB1. The finding that ΔNp63 serves as a downstream mediator of ZEB1 completes a feedback circuit initiated by p63, which transcriptionally activates the miR-200b/a/429 cluster94 and, in turn, directly targets ZEB1 (Figure 1).76
Although miR-34a induction was originally observed during p53-dependent restoration of epithelial phenotype, mutual regulation between miR-34a and Snail and prevention of TGF-β-induced EMT by miR-34a overexpression was confirmed in models harboring defects in p53 function.12 Likewise, the discovery of miR-34a regulation by ZEB1 on the background of identical germline Trp53 mutation suggests that this mechanism is independent of intact p53 function.76 In the context of EMT-associated miR-34a regulation in hypoxia or after thyroid hormone treatment, the model cell line derived from adult human kidney established by transduction with human papilloma virus E6/E7 genes suggests independence of p53 function, although this aspect has not been experimentally addressed.91, 92 Nevertheless, direct induction of MIR34A transcription by binding of thyroid hormone receptor to MIR34A promoter region inhibited TGF-β1-induced EMT in renal tubular epithelial cells, pointing to another significant mechanism of miR-34a regulation in the context of EMT.92
Regulation of miR-34a in the context of aberrant cancer-related signaling and cancer therapy
The fact that miR-34a expression is often deregulated in cancer suggests a possibility that the expression of miR-34a itself may be regulated by oncogenes or tumor suppressors. In physiological settings, miR-34a expression was proportional to the expression of tumor suppressor p19Arf in a p53-independent manner, with an implication in mouse development through Pdgfrβ expression.95
Aberrant oncogene activation can trigger cellular senescence, an anti-tumor mechanism characterized by permanent proliferative arrest. In the context of senescence, miR-34a was identified as a key mediator of c-Myc and E2F repression, mediating indirect downregulation of an entire set of mitotic genes and telomerase activity.6, 96, 97 B-RAF-induced senescence of human fibroblasts was accompanied by p53-independent induction of miR-34a, mediated by an ETS-family transcription factor ELK-1.96 ELK-1 was described as a miR-34a regulator also in a feedback regulation of tyrosine kinase AXL, implicated in cancer invasion, EMT and chemoresistance. This feedback loop is activated by AXL overexpression through JNK-mediated ELK-1 activation, and this subsequently leads to an upregulated expression of miR34a that, in turn, downregulates AXL protein expression (Figure 1).98
In accordance with context- and target cell-dependent pro- and anti-cancer effect of TGF-β signaling, different mechanisms of miR-34a regulation by TGF-β were proposed. While computational analysis of microarray data proposes a positive correlation between miR-34a expression and TGF-β signaling,50 TGF-β inhibited miR-34a expression in hepatocellular carcinoma and through miR-34a-CCL22-Treg axis promoted tumor progression and immune escape in p53-deficient cells.86 Contrarily, miR-34a induction by TGF-β silencing was associated with p53 induction and activation in HeLa cells.99
Retinoid therapy, inducing neuroblastoma cell differentiation and growth inhibition, was shown to induce miR-34a levels.35 Alternatively to TAp73-mediated induction of miR-34a in retinoid-induced neuroblastoma differentiation,75 it is plausible that retinoid-induced downregulation of N-Myc represents another mechanism of miR-34a control.100 Gene expression analysis on a panel of breast tumor samples identified a co-regulation between miR-34a and targets of Myc-associated zinc finger protein MAZ,101 further corroborating the implication of miR-34a in Myc signaling. In experimental models of Myc-driven neuroblastoma (including a p53-null mouse model), miR-34a was found to be repressed by the oncogene Myc. Although it was demonstrated that Myc binds to the MIR34A promoter,102 further studies suggested that epigenetic silencing or chromosomal deletion of the MIR34A genomic locus could be responsible for miR-34a downregulation in neuroblastoma as well.73
Epidermal growth factor receptor (EGFR) and hepatocyte growth factor receptor (MET) are tyrosine kinase receptors that have been implicated in the pathogenesis of NSCLC. Combination of miR-34a and let-7 suppressed p53-deficient tumor growth103 and demonstrated a synergic anti-proliferative effect with EGFR inhibitor erlotinib, but not other commonly used chemotherapeutics.104 MiR-34a also prevented HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells,105 suggesting a benefit of adjuvant miRNA-based therapy in p53-deficient NSCLC.
Effects of miR-34a modulation in the context of deranged p53 function
Modulation of miR-34a levels for therapeutic purpose needs to consider the heterogeneity of target cells and tissues. It was demonstrated that human tumors are complex and non-uniform in terms of expression of many tumor markers106 and individual cancer cells can strongly differ even in p53 expression.107 Studying the effects of miR-34a in models with disrupted p53 function can therefore help to predict possible effects in tumor cells harboring defective p53, especially regarding the feedback regulation between miR-34a and p53 (Figure 1). Tables 1 and 2 summarize experimental outcomes of miR-34a modulation in the context of insufficient p53 response. Altogether, the effects of miR-34a manipulation may be weaker in p53-disrupted cells than in wild-type p53 background,46 but most anti-proliferative and pro-apoptotic effects of miR-34a are maintained regardless of upstream p53 signaling. It is plausible that miR-34a exerts its effects in a complementary and parallel fashion to targets that are directly activated by p53.42
Future outlook
Hundreds of cell- and animal-based studies agree on a tumor-suppressive function of miR-34a and propose restoration of miR-34a expression as a potential therapeutic strategy. The advantage of miR-34a-based therapy is the opportunity to simultaneously repress multiple oncogenic and immune evasion pathways.4, 21, 48 While efficiently inhibiting cancer cell proliferation and survival and potentiating the effect of chemotherapy, miR-34a exhibits low toxicity to normal cells in vitro and in vivo.6, 15, 21, 90 Importantly, miR-34a modulation was shown to affect to some extent miR-34a targets in the context of disrupted p53 function.
Unexpectedly, clinical trials of solid cancer treatment with miR-34a mimic delivered by liposomal nanoparticles noted severe adverse effects of immune character in five patients. These effects may be related to miR-34a-specific modulation of gene expression, but could also originate from a reaction to the liposome-based carrier or delivered double-stranded RNA molecules.108 Different delivery approach or improved dosing schedule, addressing the issues of cellular uptake and in vivo stability, could improve the safety and tolerability of miR-34a application and create an opportunity of combination therapy.
Despite targeting miR-34a containing liposomes to tumor tissues, an effect on tumor-associated immune and stromal cells and their function cannot be avoided.85, 109 Apart from tumor tissue, miR-34a was shown to adversely affect age- and myocardial infarction-associated viability and senescence of cardiomyocytes110 and processes associated with pulmonary fibrosis.111 Investigation of a particular miR-34a ‘targetome’ in different cell types may justify appropriately targeted usage of miR-34a mimics in cancer therapy.
With the perspective of therapeutic miR-34a introduction or re-expression, the existence of multiple feedback regulatory mechanisms of miR-34a urges considering the mechanisms, which may affect both downstream miR-34a targets and upstream miR-34a regulators. On the background of disrupted p53 function encountered in a high proportion of tumors, alternative p53-independent mechanisms of miR-34a regulation merit special attention. Mechanisms inhibiting therapeutic re-expression of miR-34a in the context of EMT, inflammation or oncogene signaling (Figure 2) could be responsible for insufficient therapeutic effect, while aberrant expression of miR-34a in normal cells or massive necrotic cell death observed in miR-34a treated tumors21 may underlie systemic negative effects of therapy. Successful management of side effects on non-tumor cells is indispensable for successful therapeutic application.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Svoronos AA, Engelman DM, Slack FJ . OncomiR or tumor suppressor? The duplicity of MicroRNAs in cancer. Cancer Res 2016; 76: 3666–3670.
Wang L, Yu J, Xu J, Zheng C, Li X, Du J . The analysis of microRNA-34 family expression in human cancer studies comparing cancer tissues with corresponding pericarcinous tissues. Gene 2014; 554: 1–8.
Ohtsuka M, Ling H, Doki Y, Mori M, Calin GA . MicroRNA processing and human cancer. J Clin Med 2015; 4: 1651–1667.
Bader AG . miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 2012; 3: 120.
Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs 2017; 35: 180–188.
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H . Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 2007; 104: 15472–15477.
Liang Y, Ridzon D, Wong L, Chen C . Characterization of microRNA expression profiles in normal human tissues. BMC Genomics 2007; 8: 166.
Qiao P, Li G, Bi W, Yang L, Yao L, Wu D . microRNA-34a inhibits epithelial mesenchymal transition in human cholangiocarcinoma by targeting Smad4 through transforming growth factor-beta/Smad pathway. BMC Cancer 2015; 15: 469.
Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V et al. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids 2014; 3: e194.
Rokavec M, Li H, Jiang L, Hermeking H . The p53/miR-34 axis in development and disease. J Mol Cell Biol 2014; 6: 214–230.
Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest 2014; 124: 1853–1867.
Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011; 10: 4256–4271.
Yamakuchi M, Ferlito M, Lowenstein CJ . miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA 2008; 105: 13421–13426.
Chang T-C, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26: 745–752.
Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007; 26: 731–743.
Wong MY, Yu Y, Walsh WR, Yang JL . microRNA-34 family and treatment of cancers with mutant or wild-type p53 (Review). Int J Oncol 2011; 38: 1189–1195.
Concepcion CP, Han YC, Mu P, Bonetti C, Yao E, D'Andrea A et al. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet 2012; 8: e1002797.
Luan S, Sun L, Huang F . MicroRNA-34a: a novel tumor suppressor in p53-mutant glioma cell line U251. Arch Med Res 2010; 41: 67–74.
Olivier M, Hollstein M, Hainaut P . TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2010; 2: a001008.
Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res 2010; 16: 1119–1128.
Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res 2012; 18: 6260–6270.
Dijkstra MK, van Lom K, Tielemans D, Elstrodt F, Langerak AW, van 't Veer MB et al. 17p13/TP53 deletion in B-CLL patients is associated with microRNA-34a downregulation. Leukemia 2009; 23: 625–627.
Mraz M, Malinova K, Kotaskova J, Pavlova S, Tichy B, Malcikova J et al. miR-34a, miR-29c and miR-17-5p are downregulated in CLL patients with TP53 abnormalities. Leukemia 2009; 23: 1159–1163.
Zenz T, Mohr J, Eldering E, Kater AP, Buhler A, Kienle D et al. miR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009; 113: 3801–3808.
Li XJ, Ren ZJ, Tang JH . MicroRNA-34a: a potential therapeutic target in human cancer. Cell Death Dis 2014; 5: e1327.
Guennewig B, Roos M, Dogar AM, Gebert LF, Zagalak JA, Vongrad V et al. Synthetic pre-microRNAs reveal dual-strand activity of miR-34a on TNF-alpha. RNA 2014; 20: 61–75.
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 2016; 44: D239–D247.
Agostini M, Knight RA . miR-34: from bench to bedside. Oncotarget 2014; 5: 872–881.
Ito Y, Inoue A, Seers T, Hato Y, Igarashi A, Toyama T et al. Identification of targets of tumor suppressor microRNA-34a using a reporter library system. Proc Natl Acad Sci USA 2017; 114: 3927–3932.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D et al. MicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–838.
Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal 2015; 27: 443–452.
Imani S, Zhang X, Hosseinifard H, Fu S, Fu J . The diagnostic role of microRNA-34a in breast cancer: a systematic review and meta-analysis. Oncotarget 2017; 8: 23177–23187.
Bagchi A, Mills AA . The quest for the 1p36 tumor suppressor. Cancer Res 2008; 68: 2551–2556.
Feinberg-Gorenshtein G, Avigad S, Jeison M, Halevy-Berco G, Mardoukh J, Luria D et al. Reduced levels of miR-34a in neuroblastoma are not caused by mutations in the TP53 binding site. Genes Chromosomes Cancer 2009; 48: 539–543.
Welch C, Chen Y, Stallings RL . MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007; 26: 5017–5022.
Junttila MR, Evan GI . p53—a Jack of all trades but master of none. Nat Rev Cancer 2009; 9: 821–829.
Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007; 6: 1586–1593.
Navarro F, Lieberman J . miR-34 and p53: new insights into a complex functional relationship. PLoS ONE 2015; 10: e0132767.
Mandke P, Wyatt N, Fraser J, Bates B, Berberich SJ, Markey MP . MicroRNA-34a modulates MDM4 expression via a target site in the open reading frame. PLoS ONE 2012; 7: e42034.
Okada N, Lin CP, Ribeiro MC, Biton A, Lai G, He X et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev 2014; 28: 438–450.
Zhao J, Lammers P, Torrance CJ, Bader AG . TP53-independent function of miR-34a via HDAC1 and p21(CIP1/WAF1.). Mol Ther 2013; 21: 1678–1686.
Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 2007; 17: 1298–1307.
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130–1134.
Wang X, Dong K, Gao P, Long M, Lin F, Weng Y et al. microRNA-34a sensitizes lung cancer cell lines to DDP treatment independent of p53 status. Cancer Biother Radiopharm 2013; 28: 45–50.
Vinall RL, Ripoll AZ, Wang S, Pan CX, deVere White RW . MiR-34a chemosensitizes bladder cancer cells to cisplatin treatment regardless of p53-Rb pathway status. Int J Cancer 2012; 130: 2526–2538.
He C, Xiong J, Xu X, Lu W, Liu L, Xiao D et al. Functional elucidation of MiR-34 in osteosarcoma cells and primary tumor samples. Biochem Biophys Res Commun 2009; 388: 35–40.
Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 2011; 17: 211–215.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst 2016; 108 https://academic.oup.com/jnci/article-lookup/doi/10.1093/jnci/djv303.
Nalls D, Tang SN, Rodova M, Srivastava RK, Shankar S . Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011; 6: e24099.
Hiyoshi Y, Schetter AJ, Okayama H, Inamura K, Anami K, Nguyen GH et al. Increased microRNA-34b and -34c predominantly expressed in stromal tissues is associated with poor prognosis in human colon cancer. PLoS ONE 2015; 10: e0124899.
Paris R, Henry RE, Stephens SJ, McBryde M, Espinosa JM . Multiple p53-independent gene silencing mechanisms define the cellular response to p53 activation. Cell Cycle 2008; 7: 2427–2433.
Takeda Y, Venkitaraman AR . Micro(mi) RNA-34a targets protein phosphatase (PP)1gamma to regulate DNA damage tolerance. Cell Cycle 2015; 14: 3830–3841.
Song R, Walentek P, Sponer N, Klimke A, Lee JS, Dixon G et al. miR-34/449 miRNAs are required for motile ciliogenesis by repressing cp110. Nature 2014; 510: 115–120.
Wu J, Bao J, Kim M, Yuan S, Tang C, Zheng H et al. Two miRNA clusters, miR-34b/c and miR-449, are essential for normal brain development, motile ciliogenesis, and spermatogenesis. Proc Natl Acad Sci USA 2014; 111: E2851–E2857.
Cheng CY, Hwang CI, Corney DC, Flesken-Nikitin A, Jiang L, Oner GM et al. miR-34 cooperates with p53 in suppression of prostate cancer by joint regulation of stem cell compartment. Cell Rep 2014; 6: 1000–1007.
Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N et al. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004; 432: 235–240.
Gurtner A, Falcone E, Garibaldi F, Piaggio G . Dysregulation of microRNA biogenesis in cancer: the impact of mutant p53 on Drosha complex activity. J Exp Clin Cancer Res 2016; 35: 45.
Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A . Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell 2010; 39: 373–384.
Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD et al. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014; 156: 893–906.
Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin YL et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010; 467: 986–990.
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K . Modulation of microRNA processing by p53. Nature 2009; 460: 529–533.
Garibaldi F, Falcone E, Trisciuoglio D, Colombo T, Lisek K, Walerych D et al. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex. Oncogene 2016; 35: 3760–3770.
Kawai S, Amano A . BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J Cell Biol 2012; 197: 201–208.
Herbert KJ, Cook AL, Snow ET . SIRT1 modulates miRNA processing defects in p53-mutated human keratinocytes. J Dermatol Sci 2014; 74: 142–149.
Zhao L, Mao Y, Zhao Y, He Y . DDX3X promotes the biogenesis of a subset of miRNAs and the potential roles they played in cancer development. Sci Rep 2016; 6: 32739.
Jones PA . Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 2012; 13: 484–492.
Smith ZD, Meissner A . DNA methylation: roles in mammalian development. Nat Rev Genet 2013; 14: 204–220.
Baer C, Claus R, Frenzel LP, Zucknick M, Park YJ, Gu L et al. Extensive promoter DNA hypermethylation and hypomethylation is associated with aberrant microRNA expression in chronic lymphocytic leukemia. Cancer Res 2012; 72: 3775–3785.
Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Körner H et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle (Georgetown, Tex) 2008; 7: 2591–2600.
Vogt M, Munding J, Gruner M, Liffers ST, Verdoodt B, Hauk J et al. Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch 2011; 458: 313–322.
Siemens H, Neumann J, Jackstadt R, Mansmann U, Horst D, Kirchner T et al. Detection of miR-34a promoter methylation in combination with elevated expression of c-Met and beta-catenin predicts distant metastasis of colon cancer. Clin Cancer Res 2013; 19: 710–720.
Meng F, Glaser SS, Francis H, Yang F, Han Y, Stokes A et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am J Pathol 2012; 181: 804–817.
Craig VJ, Cogliatti SB, Imig J, Renner C, Neuenschwander S, Rehrauer H et al. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 2011; 117: 6227–6236.
Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G . p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2010; 2: a004887.
Agostini M, Tucci P, Killick R, Candi E, Sayan BS, Rivetti di Val Cervo P et al. Neuronal differentiation by TAp73 is mediated by microRNA-34a regulation of synaptic protein targets. Proc Natl Acad Sci USA 2011; 108: 21093–21098.
Ahn Y-H, Gibbons DL, Chakravarti D, Creighton CJ, Rizvi ZH, Adams HP et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest 2012; 122: 3170–3183.
Antonini D, Russo MT, De Rosa L, Gorrese M, Del Vecchio L, Missero C . Transcriptional repression of miR-34 family contributes to p63-mediated cell cycle progression in epidermal cells. J Invest Dermatol 2010; 130: 1249–1257.
Ng WL, Chen G, Wang M, Wang H, Story M, Shay JW et al. OCT4 as a target of miR-34a stimulates p63 but inhibits p53 to promote human cell transformation. Cell Death Dis 2014; 5: e1024.
Grivennikov SI, Greten FR, Karin M . Immunity, inflammation, and cancer. Cell 2011; 140: 883–899.
Gao F, Liang B, Reddy ST, Farias-Eisner R, Su X . Role of inflammation-associated microenvironment in tumorigenesis and metastasis. Curr Cancer Drug Targets 2014; 14: 30–45.
Jiang P, Liu R, Zheng Y, Liu X, Chang L, Xiong S et al. MiR-34a inhibits lipopolysaccharide-induced inflammatory response through targeting Notch1 in murine macrophages. Exp Cell Res 2012; 318: 1175–1184.
Fan W, Fang R, Wu X, Liu J, Feng M, Dai G et al. Shear-sensitive microRNA-34a modulates flow-dependent regulation of endothelial inflammation. J Cell Sci 2015; 128: 70–80.
Mathe E, Nguyen GH, Funamizu N, He P, Moake M, Croce CM et al. Inflammation regulates microRNA expression in cooperation with p53 and nitric oxide. Int J Cancer 2012; 131: 760–765.
Li J, Wang K, Chen X, Meng H, Song M, Wang Y et al. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol Biol 2012; 13: 4.
Shin J, Xie D, Zhong XP . MicroRNA-34a enhances T cell activation by targeting diacylglycerol kinase zeta. PLoS ONE 2013; 8: e77983.
Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012; 22: 291–303.
Hanrahan K, O'Neill A, Prencipe M, Bugler J, Murphy L, Fabre A et al. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol Oncol 2017; 11: 251–265.
Singh A, Settleman J . EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010; 29: 4741–4751.
Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell 2011; 22: 1686–1698.
Dong P, Xiong Y, Watari H, Hanley SJ, Konno Y, Ihira K et al. MiR-137 and miR-34a directly target Snail and inhibit EMT, invasion and sphere-forming ability of ovarian cancer cells. J Exp Clin Cancer Res 2016; 35: 132.
Du R, Sun W, Xia L, Zhao A, Yu Y, Zhao L et al. Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS ONE 2012; 7: e30771.
Lu X, Chen Z, Liang H, Li Z, Zou X, Luo H et al. Thyroid hormone inhibits TGFbeta1 induced renal tubular epithelial to mesenchymal transition by increasing miR34a expression. Cell Signal 2013; 25: 1949–1954.
Hahn S, Jackstadt R, Siemens H, Hunten S, Hermeking H . SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J 2013; 32: 3079–3095.
Knouf EC, Garg K, Arroyo JD, Correa Y, Sarkar D, Parkin RK et al. An integrative genomic approach identifies p73 and p63 as activators of miR-200 microRNA family transcription. Nucleic Acids Res 2012; 40: 499–510.
Iqbal N, Mei J, Liu J, Skapek SX . miR-34a is essential for p19(Arf)-driven cell cycle arrest. Cell Cycle 2014; 13: 792–800.
Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M et al. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ 2009; 17: 236–245.
Xu X, Chen W, Miao R, Zhou Y, Wang Z, Zhang L et al. miR-34a induces cellular senescence via modulation of telomerase activity in human hepatocellular carcinoma by targeting FoxM1/c-Myc pathway. Oncotarget 2015; 6: 3988–4004.
Cho CY, Huang JS, Shiah SG, Chung SY, Lay JD, Yang YY et al. Negative feedback regulation of AXL by miR-34a modulates apoptosis in lung cancer cells. RNA 2016; 22: 303–315.
Dogar AM, Towbin H, Hall J . Suppression of latent transforming growth factor (TGF)-beta1 restores growth inhibitory TGF-beta signaling through microRNAs. J Biol Chem 2011; 286: 16447–16458.
Cetinkaya C, Hultquist A, Su Y, Wu S, Bahram F, Pahlman S et al. Combined IFN-gamma and retinoic acid treatment targets the N-Myc/Max/Mad1 network resulting in repression of N-Myc target genes in MYCN-amplified neuroblastoma cells. Mol Cancer Ther 2007; 6: 2634–2641.
Peurala H, Greco D, Heikkinen T, Kaur S, Bartkova J, Jamshidi M et al. MiR-34a expression has an effect for lower risk of metastasis and associates with expression patterns predicting clinical outcome in breast cancer. PLoS ONE 2011; 6: e26122.
Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 2008; 40: 43–50.
Kasinski AL, Slack FJ . miRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53-induced mouse model of lung adenocarcinoma. Cancer Res 2012; 72: 5576–5587.
Stahlhut C, Slack FJ . Combinatorial action of MicroRNAs let-7 and miR-34 effectively synergizes with erlotinib to suppress non-small cell lung cancer cell proliferation. Cell Cycle 2015; 14: 2171–2180.
Zhou JY, Chen X, Zhao J, Bao Z, Zhang P, Liu ZF . MicroRNA-34a overcomes HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells partly by targeting MET. Cancer Lett 2014; 351: 265–271.
Marusyk A, Polyak K . Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 2010; 1805: 105–117.
Mirchandani D, Zheng J, Miller GJ, Ghosh AK, Shibata DK, Cote RJ et al. Heterogeneity in intratumor distribution of p53 mutations in human prostate cancer. Am J Pathol 1995; 147: 92–101.
Robbins M, Judge A, Ambegia E, Choi C, Yaworski E, Palmer L et al. Misinterpreting the therapeutic effects of small interfering RNA caused by immune stimulation. Hum Gene Ther 2008; 19: 991–999.
Rao DS, O'Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D . MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 2010; 33: 48–59.
Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013; 495: 107–110.
Shetty SK, Tiwari N, Marudamuthu AS, Puthusseri B, Bhandary YP, Fu J et al. p53 and miR-34a feedback promotes lung epithelial injury and pulmonary fibrosis. Am J Pathol 2017; 187: 1016–1034.
Acknowledgements
This work was supported by grant 15-11707S from the Czech Science Foundation; by Ministry of Health of the Czech Republic grants 15-33999A, 17-28518A and 15-28628A, all rights reserved (KS); by project Mobility no. 7AMB16AT022 from the Ministry of Education, Youth and Sports; project HistoPARK (CZ.1.07/2.3.00/20.0185), project LQ1605 from the National Program of Sustainability II (MEYS CR); and by the European Union - project ICRC-ERA-HumanBridge (No. 316345).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Agostini
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Slabáková, E., Culig, Z., Remšík, J. et al. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis 8, e3100 (2017). https://doi.org/10.1038/cddis.2017.495
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2017.495
This article is cited by
-
Acacetin inhibited non-small-cell lung cancer (NSCLC) cell growth via upregulating miR-34a in vitro and in vivo
Scientific Reports (2024)
-
Clinical importance of serum miRNA levels in breast cancer patients
Discover Oncology (2024)
-
Restoring microRNA-34a overcomes acquired drug resistance and disease progression in human breast cancer cell lines via suppressing the ABCC1 gene
Breast Cancer Research and Treatment (2024)
-
Targeted therapy using nanocomposite delivery systems in cancer treatment: highlighting miR34a regulation for clinical applications
Cancer Cell International (2023)
-
MicroRNA regulation of AMPK in nonalcoholic fatty liver disease
Experimental & Molecular Medicine (2023)
