
Abstract
The deregulation of lineage control programs is often associated with the progression of haematological malignancies. The molecular regulators of lineage choices in the context of tyrosine kinase inhibitor (TKI) resistance remain poorly understood in chronic myeloid leukemia (CML). To find a potential molecular regulator contributing to lineage distribution and TKI resistance, we undertook an RNA-sequencing approach for identifying microRNAs (miRNAs). Following an unbiased screen, elevated miRNA182-5p levels were detected in Bcr-Abl-inhibited K562 cells (CML blast crisis cell line) and in a panel of CML patients. Earlier, miRNA182-5p upregulation was reported in several solid tumours and haematological malignancies. We undertook a strategy involving transient modulation and CRISPR/Cas9 (clustered regularly interspersed short palindromic repeats)-mediated knockout of the MIR182 locus in CML cells. The lineage contribution was assessed by methylcellulose colony formation assay. The transient modulation of miRNA182-5p revealed a biased phenotype. Strikingly, Δ182 cells (homozygous deletion of MIR182 locus) produced a marked shift in lineage distribution. The phenotype was rescued by ectopic expression of miRNA182-5p in Δ182 cells. A bioinformatic analysis and Hes1 modulation data suggested that Hes1 could be a putative target of miRNA182-5p. A reciprocal relationship between miRNA182-5p and Hes1 was seen in the context of TK inhibition. In conclusion, we reveal a key role for miRNA182-5p in restricting the myeloid development of leukemic cells. We propose that the Δ182 cell line will be valuable in designing experiments for next-generation pharmacological interventions.
Similar content being viewed by others


Main
The pathways that regulate haematopoietic differentiation are well understood and have served as paradigms in developmental biology.1 With the discovery of microRNAs (miRNAs), there has been an interest in analysing the role of these molecules in haematopoiesis and related disease states.1, 2, 3, 4 Examples of such miRNAs are miRNA223, miRNA486, miRNA144 and miRNA451.6, 7 Specifically, in the context of hematopoietic development and malignancies, a miRNA of particular interest is miRNA182-5p.5, 6, 7, 8, 9 The locus that encodes miRNA182 is located on chromosome 7q32.2 of human genome in a cluster of three miRNAs—MIR182, MIR183 and MIR96.13 The miRNA182 locus has been reported to be abundantly expressed in the retina, central nervous system and normal human embryonic stem cells.14 In hematopoietic development, miRNA182-5p has been shown to regulate clonal expansion of T cells.15 Although miRNA182-5p is clearly of importance in cancer progression and therapeutic resistance,9, 13, 16 its role in lineage distribution remains uncharacterized.
Chronic myeloid leukemia (CML), with its well-defined genetic characterization, has lent itself to a molecular understanding of cancer progression.17, 18 The disease progression is invariably associated with a Philadelphia translocation or 9–22 chromosomal rearrangement, the product of which is an activated tyrosine kinase, Bcr-Abl.19 It has been reported that Bcr-Abl promotes proliferation of multipotent progenitors and myeloid progenitor cells while suppressing erythroid progenitors.20 Over the past decade or so, a considerable body of literature has documented the striking clinical remissions that Gleevec and next-generation tyrosine kinase inhibitor (TKI) have achieved.21, 22, 23 However, there is a small but significant set of CML patients who develop resistance to TKIs in the absence of specific identifiable tyrosine kinase mutations.24, 25 The mechanisms associated with this class of relapse are currently poorly understood.
The reciprocal relationship between proliferation and differentiation determines the steady-state distribution of subsets of hematopoietic cells.26, 27 The two major classes of these subsets are erythroid and myeloid-derived cells. The erythroid class encompasses colony-forming unit (CFU)—megakaryocyte cells (Meg), an intermediate erythroid progenitor—burst-forming unit erythroid (BFU-E) and CFU-E.28 The myeloid compartment includes CFU-Granulocyte, CFU-Macrophage and CFU-GM (granulocyte–macrophage).29 In the absence of a disease, measurements of steady states typically reveal a low percentage (<5%) of myeloid blast cells in the bone marrow.21, 30 In contrast, myeloid leukemias are characterized by a high percentage of myeloid blast cells (upto 50%).31 Similarly, erythroid leukemic conditions display a higher percentage of erythroid blasts.32 A paradoxical observation is the detection of an erythroid burden in the late-stage CML patients.21
The mechanisms of altered expression of miRNA182 in myeloid leukemia remain poorly characterized. With an interest in delineating the role of miRNAs that regulate TKI resistance, we used K562 cells; a multipotent Bcr-Abl-positive CML cell line.33 We undertook a small RNA-sequencing analysis to identify the miRNAs that were potentially upregulated in the context of TKI resistance. We found a striking association between elevated miRNA182-5p level and TKI resistance in CML lines and primary cells. The colony formation assay determined that ectopic miRNA182-5p expression is positively correlated with erythroid differentiation. We embarked on a precise gene-editing approach using CRISPR (clustered regularly interspaced short palindromic repeats) and generated Δ182 cells. The loss of miRNA182 expression by both locked nucleic acid (LNA) anti-miRNA and CRISPR knockout revealed an increase in myeloid differentiation. Further, we examined a role for Hes1, a putative target of miRNA182-5p in regulating percentage of myeloid and erythroid cells (ME%). Collectively, elevated miRNA182-5p expression blocked the myeloid differentiation of K562 cells. This study deciphers the role of miRNA182-5p in a conserved lineage program of leukemic cells and holds promise to the use of miRNA182-5p for therapeutic improvements in parallel to TKI therapy.
Results
High miRNA182-5p expression is associated with TKI resistance in CML cells
To assess the miRNAs that were modulated in the context of resistance to imatinib, Illumina sequencing was performed on RNA extracted from imatinib-treated K562 cells. The K562 cell line retains a rearranged Bcr-Abl gene, with no detectable mutations. Further, this cell line can be induced to differentiate and thus serves as a model for analysing the contribution of distinct lineages to late-stage CML progression.33, 34 In Figure 1a, we showed the expression profile of all the miRNAs from imatinib-treated K562 cells compared with an online available data set from untreated K562 cells (courtesy Professor Alok Bhattacharya, JNU).35 The heatmap revealed that the expression of 83 miRNAs was altered (Supplementary Table 2). Of particular interest was the detection of a twofold increase in miRNA182-5p expression (Figure 1a). Quantitative PCR analysis of miRNA182-5p revealed a twofold increase in both K562 cells (Figure 1b) and KCL22 cells (Supplementary Figure S2A). There was a 160-fold increase of miRNA182-5p expression in K562 cells resistant to imatinib (Figure 1c).

High expression of MiRNA182-5p is associated with TK inhibitor resistance in CML cells. (a) Heatmap of differentially expressed miRNAs between control and imatinib-treated K562 cells. Column labels represent the type of sample: control and imatinib. The red arrow shows miRNA182-5p expression in the heatmap. Range of expression measured was −3- to +3-fold change. (b and c) Expression of miRNA182-5p in K562 cells after imatinib treatment (b) and imatinib-resistant K562 cells (c). Data are shown as mean of three independent experiments. Error bars show s.e. of three independent experiments with P-values of the mean. **P<0.01 and ***P<0.001. (d) Percentage cell proliferation of LNA anti-miRNA182-5p-transfected K562 cells with TK inhibitor. The data are compared with scramble control. Data are shown as mean of three independent experiments. Error bars show s.e. of three independent experiments with P-values of the mean. *P<0.05, ***P<0.001. (e) Expression of miRNA182-5p in clinical samples of CML. The Y-axis shows the fold change in miRNA182-5p expression (log2 scale). Data are presented as mean of three biological triplicates. The normalisation is undertaken to the expression of miRNA182-5p from K562 cell line. CC, CML chronic phase; CN, CML chronic nonresponder; CB, CML blast; NR, nonresponder
To directly explore the role of miRNA182-5p expression in TKI resistance, we used LNA anti-miRNAs in cell proliferation assay. The anti-miRNA182-5p and scramble transfection revealed 37% and 75% rate of proliferation, respectively, after imatinib treatment (Figure 1d). A marked 10-fold increase in the expression of miRNA182 was noted in six CML samples of early nonresponders and late progressors. Out of the 19 samples, three nonresponders revealed an increase in the expression of miRNA182. These data were generated in comparison with miRNA182 expression in K562 cells (Figure 1e).
The results in Figure 1 suggest an association of miRNA182-5p expression with TKI resistance in CML, particularly in either the later stages of the disease or early nonresponders. Given the contribution of miRNAs in differentiation and development,36, 37 their causality with chemoresistance by modulating the differentiation pathways remain unexplored. To assess the role of miRNA182-5p in lineage distribution, we undertook experiments to overexpress or downregulate miRNA182-5p expression in K562 cells.
MiRNA182-5p modulation and TKI resistance are associated with shift in ME%
To determine whether K562 cells with transient miRNA182-5p perturbation retained their potential to differentiate into erythroid and myeloid cells, we performed methylcellulose colony-forming assay. The percentage of erythroid and myeloid colonies was measured. We assessed the lineage distribution of K562 cells in the context of TKI resistance by treating K562 cells with 0.75 μM concentration of imatinib. The mean number of colonies of BFU-E were 61, 53 and 29, CFU-G were 10, 8 and 1, CFU-E were 16, 11 and 1, and total colony counts were 93, 72, and 33 in RPMI-, DMSO- and imatinib-treated K562 cells, respectively (Supplementary Figure 9e). The graphical representation of this data shown in Supplementary Figure 9f revealed a marginal increase of erythroid cells % (88% versus 93%) in imatinib-treated K562 cells.
Next, to determine the lineage distribution of K562 cells after miRNA182 modulation, we used anti-miRNA182 and mimics-miRNA182 on K562 cells. The mean number of colonies of BFU-E were 39, 36 and 54, CFU-G were 27, 48 and 21, CFU-M were 10, 15 and 7 in Scramble, LNA anti-miRNA182-5p- and mimics-miRNA182-transfected K562 cells, respectively (Figure 2d). The graphical representation of this data shown in Supplementary Figure 2e revealed an increase and decrease in ME% (62% and 33% versus 44%) in LNA anti-miRNA182-5p- and mimics-miRNA182-transfected K562 cells, respectively. The quantitative data for the all the colony types in each condition were provided Figure 2d.

Modulation in the expression of MiRNA182-5p results in a shift of ME% in K562 cells. (a–c) Representative images of colonies formed in methylcellulose CFU assay with scramble- (a), anti-miRNA182-5p- (b) and miRNA182-5p mimics-transfected K562 cells (c). Images are taken at × 4 magnification. Scale bar represents 100 μm. BFU-E, CFU-M, CFU-G and CFU-GM are shown in a2, b1, b2 and a1, respectively. (d) Quantitation of the colony counts and types on day 21 with each condition. Data are shown as number of colonies per five fields. Error bars show s.e. of three independent experiments with P-values of the mean. *P<0.05, **P<0.01 and ***P<0.001. (e) ME% shows the lineage distribution in colony formation assay
The data from Figure 2 and Supplementary Figure 9 suggested that miRNA182-5p expression might augment TKI resistance by altering the lineage distribution. The results in Figure 2 were found analogous to that seen in Supplementary Figure 7. To generate direct evidence that miRNA182-5p regulates ME%, MIR182 genomic locus was deleted using the CRISPR system.
Generation of CRISPR-based knockout system for MIR182 locus
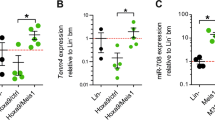
CRISPR/Cas9 system has been used to modify genomic loci of clinical importance.38 A stepwise process to delete the MIR182 locus was undertaken starting with HEK293 to standardise knockout method (Supplementary Figure 5a). To reduce the incidence of off-target effects, three CRISPR plasmids (targeting 129409678, 129410425 and 129410482 according to GRCh37.p13 of human genome assembly) were nucleofected in K562 cell line (Figure 3c). Multiple clones with precise deletion (819 bp, 756 bp and 1497 bp) were detected by PCR amplification of MIR182 locus followed by agarose gel electrophoresis (Figure 3d). To further confirm whether the amplified products corresponded to the MIR182 locus, Sanger sequencing was performed on the purified PCR products. The chromatogram confirmed that MIR182 locus was deleted (Supplementary Figure 6b). MIR182 deleted clones were used to make heterozygous (wt/182) and homozygous lines (Δ182) for further study (Figure 3e). We quantified miRNA182 expression levels by qPCR in K562 cells after MIR182 deletion. There was a significant reduction in miRNA182-5p levels of 66% and 62% in wt/182 and Δ182 cells, respectively, compared with wild-type K562 cells (Supplementary Figure 10g). To assess the proliferation potential of K562 cells after deletion of MIR182 locus, we used WST1 cell proliferation assay. The Δ182 cells and K562 cells displayed 23% and 100% proliferation rate, respectively (Supplementary Figure 10h). The cell proliferation data were analogous to the imaging data seen in Figure 3e. The cumulative results in Figures 3 and 4 and Supplementary Figure 10 revealed that the CRISPR system efficiently deleted the MIR182 locus in K562 cells and a decrease in the rate of proliferation was detected in these cells.

Generation of a CRISPR-based knockout system to delete MIR182 locus. (a) A Cartoon diagram shows the surveyor nuclease assay. (b) Surveyor nuclease assay of each guide RNAs targeting MIR182 locus in HEK293T cells. Percentage indels are shown under each guideRNA. (c) Schematic of three guideRNA approach for targeting MIR182 locus in K562 cells. CAS9+9678, CAS9+0482 and CAS9+0425 are used to delete MIR182 genomic locus from K562 cells. (d) Agarose gel electrophoresis shows PCR amplification of MIR182 locus in CRISPR guide RNAs nucleofected K562 cells. wtMIR182 (1560 bp), Δ182-A (756 bp), Δ182-B (819 bp), Δ182-C (1497 bp). (e) Colonies from Δ182 and wt/182 cells grown in a 96-well plate as single cells. Images are taken at 10 days. Scale bar represents 100 μm. (f) Agarose gel analysis shows off-target sites of CRISPR guide RNAs targeting MIR182 locus by surveyor nuclease assay. The 0425-E site shows additional bands

MIR182 deletion markedly shifts ME% in K562 cells. (a–c) Representative images of colonies formed in methylcellulose CFU assay with wt (a), wt/182 (b) and Δ182 cells (c), respectively. Images are taken at × 4 magnification. Scale bar represents 100 μm. CFU-Gs are shown in a2 and b2. BFU-E and CFU-M are shown in c1 and c3, respectively. (d) Quantitation of the colony counts on day 21 with each condition. Data are shown as number of colonies per five fields. Error bars show s.e. of three independent experiments with P-values of the mean. *P<0.05, **P<0.01 and ***P<0.001. (e) ME% shows the lineage distribution in colony formation assay
MIR182 locus contributes to the maintenance of ME% in K562 cells
As an inverse relationship between proliferation and differentiation had been predicted earlier,39 we assessed whether the loss of MIR182 locus resulted in any specific lineage commitment. The mean number of colonies of BFU-E were 73, 8 and 4, CFU-G were 13, 52 and 29, CFU-M were 1.5, 11 and 10, and total colonies were 88, 73 and 44 in K562, wt/182 and Δ182 cells, respectively (Figure 4d). The graphical representation of this data shown in supplementary Figure 4e revealed an increase of ME% (88% and 92% versus 16%) in wt/182 and Δ182 cells, respectively.
To exclude the possibility of off-targets contributing to lineage shift, miRNA182-5p mimics were transfected in Δ182 cells. The mean number of colonies of BFU-E were 15 and 43, CFU-G were 34 and 7, CFU-M were 10 and 2, and total colonies were 57 and 64 in Δ182 cells and miRNA182-5p mimics-transfected Δ182 cells, respectively (Figure 5e). The graphical representation of this data shown in supplementary Figure 5f revealed an increase of ME% (67% versus 14%) in miRNA182-5p mimics-transfected Δ182 cells, respectively (Supplementary Figure 10I and Figure 5f).

Ectopic expression of miRNA182-5p increases erythroid phenotype in Δ182 cells. (a and d) An image of a Petri plate showing the colonies of Δ182 cells (a) and mimics-miRNA182-5p-transfected Δ182 cells (b). A magnified region of the same is shown in c and d. (e) Quantitation of the colony counts on day 21 with each condition. Data are shown as number of colonies per five fields. Error bars show s.e. of three independent experiments with P-values of the mean. *P<0.05, **P<0.01 and ***P<0.001. ME% shows the lineage distribution in colony formation assay
Figures 4 and 5 showed that Δ182 cells became poised for myeloid differentiation and the phenotype was rescued by ectopic expression of miRNA182-5p. The data from MIR182 knockout were found analogous to that seen in Figure 2. Collectively, Figures 1,2,3,4 and 5 revealed that lineage distribution of K562 cells was associated with TKI resistance. In addition, miRNA182-5p played a central role in determining the lineage distribution. To strengthen the role of miRNA182-5p in above context, we queried for intersecting pathways of miRNA182-5p signalling.
Hes1 serves as a putative target of miRNA182-5p in regulating ME%
The miRNA-Org, DIANA-microT and target-scan tools predicted a 7-mer conserved binding site for miRNA182-5p on 3′-UTR of Hes1 mRNA (Figure 6a). To directly explore this interaction, anti-miRNA182-5p were transfected in K562 cells and Hes1 expression was quantified by flow cytometry. The histogram showed an increase in Hes1 expression in the anti-miRNA182-5p-transfected cells compared with scramble-transfected K562 cells (Figure 6b). To address the role of Hes1 in lineage distribution, we undertook a loss and gain of function approach. The mean number of colonies of BFU-E were 42 and 67, CFU-G were 21 and 16, CFU-M were 10 and 4, CFU-E were 9 and 10 in control shRNA- and shHes1.1-nucleofected K562 cells, respectively (Figure 6g). The mean number of colonies of BFU-E were 55 and 28, CFU-G were 13 and 40, and CFU-M were 3 and 10 in miGR1-eGFP- and miGR1-Hes1-nucleofected K562 cells, respectively (Figure 6h). The graphical representation of this data shown in Figures 6i and j revealed a decrease and increase of ME% (20% and 60% versus 14%) in shHes1.1- and miGR1-Hes1-nucleofected K562 cells, respectively.

Hes1 serves as a putative target of miRNA182-5p in regulating ME%. Human Hes1 has a 7-mer conserved binding site at its 3′-UTR for miRNA182-5p. (a) Flow cytometric analysis of Hes1 expression in anti-miRNA182-5p-transfected K562 cells. The blue and red histograms represent Hes1 expression in scramble control and miRNA182-5p inhibition, respectively. (c–f) Representative images from the colony formation assay in K562 cells (c), miGR1-Hes1- (d), shRNA1-Hes1- (e) and shRNA2-Hes1-nucleofected K562 cells (f), respectively. Images are taken at × 4 magnification. Scale bar represents 100 μm. (g and h) Quantitation of the colony counts from the shRNA- (g) and MiGR1-Hes1-nucleofected K562 cells (h) on day 21. Data are shown as number of colonies per five fields. Error bars show s.e. of three independent experiments with P-values of the mean. *P<0.05, **P<0.01 and ***P<0.001. (i and j) ME% showing percentage of myeloid and erythroid cells in colony formation assay from shRNA- and MiGR1-Hes1-nucleofected K562 cells
In a translational approach, Hes1 mRNA expression levels were assessed in primary CML cells. The qPCR analysis showed an increase in Hes1 expression in three out of seven cases after anti-miRNA182-5p transfection (Supplementary Figure 4). Collectively, Hes1 served as a putative target of miRNA182-5p in CML and affected the lineage distribution of CML cells. These results were analogous to that seen in Figures 2 and 4.
Extended results
A bioinformatic approach was applied on miRNA-sequencing data to examine miRNA182-5p signalling pathway (Supplementary Figure 3a). The sequencing data revealed an upregulation in expression of miRNA182-5p and miRNA19. In addition, 81 other miRNAs were downregulated in imatinib-treated K562 cells (Supplementary Table 2). These miRNAs were examined for their putative pathways by miRNA-path tool from Diana lab. The pathway analysis predicted TGF-β signalling as one of the candidate pathways upstream to miRNA182-5p regulation (Supplementary Figure 3b). Earlier, inhibition of TGF-β signalling has been shown to downregulate the colony formation ability of leukemia initiating cells.40 To find a direct link between TGF-β and miRNA182 expression, K562 cells were treated with recombinant TGF-β ligand. The treatment resulted in 1.3- and 5.8-fold increase in miRNA182-5p expression with 2 ng and 5 ng ligand concentration, respectively (Supplementary Figure 3c). The 10 ng concentration treatment resulted in the decrease of miRNA182-5p levels to 0.45-fold (Supplementary Figure 3c). Next, a link between TK resistance and TGF-β was explored. The TKI-treated K562 cells showed a marginal increase in TGF-β receptor (Supplementary Figure 3d) and p-Smad3 levels (Supplementary Figure 3e) at the protein level.
Discussion
In this study, we examined the patterns and consequences of the expression of miRNA182-5p in the context of CML progression. The expression patterns revealed that the expression of miRNA182-5p was associated with TKI resistance in CML cells. The experiments involving lowering the expression of miRNA182-5p resulted in the change of lineage distribution of K562 cells with an enhanced detection of myeloid cells (Figure 7b). We also noted that the expression of miRNA182-5p correlated with the proliferation potential of CML cells. Collectively, these data suggested that miRNA182-5p upregulation concomitantly contributed to both accumulation of erythroid cells as well as resistance to TKIs (Figure 7a).

MiRNA182-5p-mediated control over differentiation program contributes to TKI resistance of CML cells. (a) A cartoon diagram representing the mechanisms of miRNA182-mediated erythroid differentiation in K562 cells. The TK inhibitor induces the overexpression of miRNA182-5p that results in suppression of Hes1 expression. The cells acquire erythroid phenotype and become TKI resistant. Ectopically overexpressed miRNA182-5p leads to erythroid differentiation of K562 cells. The dotted arrows show correlation based on the expression. The solid arrows show direct interactions. (b) A cartoon diagram representing the myeloid differentiation in K562 cells mediated by the loss of miRNA182-5p expression. First, CRISPR system targeting MIR182 locus abrogates miRNA182-5p expression. This leads to myeloid differentiation of K562 cells. The transfection of anti-miRNA182-5p in K562 cells also induces a myeloid-biased phenotype and TKI sensitivity
Our experimental approach was built on an initial RNA-sequencing data that revealed a twofold increase in miRNA182-5p levels in the context of TKI resistance in a cell line model (Figure 1a). In 6 out of 19 cases, the linkage of TK resistance with miRNA182-5p levels was noted in the clinical context of late progressing CML cases. Thus, miRNA182-5p expression could be a major contributor to late state progression, given the heterogeneity of mechanisms that drive progression at this stage.41 These findings correlated with the patterns of miRNA182-5p expression identified in the context of chemoresistance in other tumors.42, 43, 44 The data from Figure 1 and Supplementary Figure 1 suggested a miRNA182-5p-dependent TKI resistance mechanism independent of Bcr-Abl. Previously, a TGF-β/FOXO signalling was suggested to be contributing to TKI resistance and EMT transition seen in CML stem cells.40, 45, 46 Wnt signalling that has been implicated in CML stem cells also shown to be upstream regulator of expression of miRNA182.47, 48 Given the diversity of regulatory signalling pathways upstream and downstream to miRNA182, different stages of CML might require rewiring of the miRNA182-dependent transcriptional programs. The development of modulators of differentiation might synergise with other therapeutic strategies in the management of late-stage CML.
Two independent studies suggested that the outcomes of TKI therapy might be linked with increased miRNA182-5p levels and a differentiation imbalance.45, 49, 50 The erythroid-biased phenotype in a steady-state condition was increased by ectopic expression of miRNA182-5p in a methylcellulose assay (Figure 2). The ectopic expression might overestimate the contribution of miRNA182-5p expression in lineage decisions. Thus, we used an approach that generated loss of physiological expression of miRNA182-5p by both transient and constitutive means. The data from loss of physiological expression using LNA anti-miRNAs were strengthened by CRISPR-mediated complete loss of miRNA182-5p expression (Figure 5). In the current study, we showed that the differentiation program might contribute to TKI resistance and miRNA182-5p expression was deterministic of the process (Figures 1,2,3,4 and 5). The assay performed on K562 cells that displayed limited differentiation potential could be improved by addressing the question in early progenitor cells such as hematopoietic stem cell and using an animal model to track the cells for a longer time period.
The data from Figure 6 revealed that Hes1 expression was rescued by the loss of miRNA182-5p levels. In addition, perturbation in Hes1 levels resulted in an erythroid–myeloid imbalance in K562 cells. Hematopoietic factors such as GRB2, Rac1 and CEBPA, and apoptotic factors such as FOXO1, PDCD4 and BCL2, respectively, have been shown to be regulated by miRNA182.51, 52, 53 Given the unresolved regulation of Hes1 in myeloid differentiation and leukemias,54, 55 we provided evidence for a post-transcriptional regulatory mechanism in Notch signalling pathway mediated by miRNA182. The data generated in our study would suggest a rethinking of the role of miRNA182 signalling in the context of late-stage CML and particularly TKI resistance.
The use of both the cell line that we have generated and clinical analysis should offer an opportunity to create novel therapies for the relapsing CML cases in the years ahead. As a wider implication, we also validate that gene editing of specific locus in established cell lines is a useful approach to create reagents for phenotypic analysis in the context of drug resistance.
Materials and methods
Cell culture
K562 and KCL22 cells were obtained from Dr. Soumen Chakraborty lab (ILS, Bhubaneswar, India). The cells were cultured in 10% RPMI media with foetal calf serum (v/v) and 0.1 mg/ml PenStrep at 37 °C and 5% CO2 conditions. Cells were passaged every 2–3 days and used for study till passage 20. The cells were tested for mycoplasma routinely and found negative consistently. Short-term TKI treatment was performed at inhibitory concentration 50 (IC50) −0.75 μM of imatinib Mesylate (purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA) and incubated for 48 h. The dead cells were washed and live cells were scored for Bcr-Abl activity by measuring the expression of p-Crkl, a downstream target molecule of Bcr-Abl tyrosine kinase (Supplementary Figures 1a and b). TKI-resistant K562 cell line was generated by gradual increase in the dose of TKI. The first treatment dose was 0.05 μM TKI, and gradually TKI dose was increased every week. The treatment was continued for 3 months till achievement of 5 μM and the cells were then characterized as TKI-resistant K562 cells. The TKI-resistant cells were then studied for further analysis.
Isolation of mononuclear cells from peripheral blood and bone marrow
Bone marrow from CML patients was isolated and mononuclear cells were separated from the whole blood by Ficoll Histo-paque density gradient method. RBCs were lysed from the mononuclear cells by treatment with RBC lysis buffer containing ammonium chloride. The cells were washed with PBS twice. The cells were counted and their viability was checked by the trypan blue method with every thawing. One million mononuclear cells were cultured overnight in a six-well culture plate at 37 °C and 5% CO2. This initial 24 h culture helped the cells to revive and expand before they were being subjected to any treatment or used in further experiments.
Primary cell culture of peripheral blood cells and bone marrow
Primary cells were cultured in serum free IMDM media supplemented with 1% glutamine (100 mM) and 1% penicillin–streptomycin (100 mM). A five growth factor cocktail comprising 100 ng/ml Flt3-ligand, 100 ng/ml stem cell factor, 20 ng/ml each of interleukin IL-3, IL-6 and granulocyte–macrophage colony-stimulating factor was added to the media. Growth factors were purchased from Peprotech (NJ, USA).
Small RNA-sequencing
K562—the cell line from a late-stage CML patient expressed BCR-ABL fusion gene b3-a2.56 Cells were treated with 0.2 μM concentration of TKI for 72 h. Total RNA was isolated from inhibitor-treated and -untreated cells, and ligated with 3′ and 5′ adapters for small RNAs. The adapter-conjugated RNA was reverse-transcribed with 3′ adapter-specific primers, and amplified by PCR. The PCR products were run on a 8% polyacrylamide gel, 100 bp band was cut and eluted for purification. The purified cDNA from drug-treated cells was given for sequencing on Illumina hiseq platform (San Diego, USA). The miRNA sequence data were mapped on to human genome (version hg18) and mature miRNAs were identified using RNA fold and miRDeep to confirm their stability. Sequences were read as frequency, that is, how many times the miRNA had been read and normalized as TPM (transcripts per million). MiRNAs from drug-treated K562 cells were compared with miRNA data set from untreated K562 cells (Professor Alok Bhattacharya, JNU).
Gene expression analysis
In brief, total RNA was isolated from CML cells using Trizol method following the manufacturer’s instructions. An amount of 20 ng RNA was used to synthesize a specific cDNA of hsa-miRNA182-5p using stem-loop miRNA specific RT primer. qRT-PCR was performed using Sybr green dye (purchased from Kappa, Boston, MA, USA) on Applied Biosystems 7500 fast detection system (Carlsbad, CA, USA). Expression of miRNA was normalized using the expression of the housekeeping genes 18 s r-RNA. Relative quantification of miRNAs was calculated with the delta delta ΔΔCt method. Data are shown as mean of three independent experiments. Error bars represent s.e. of three independent experiments with P-values of the mean. *P<0.05, **P<0.01 and ***P<0.001. The Bcr-Abl/Abl ratio was determined for each patient sample (Supplementary Table 1).
Methylcellulose colony formation assay
K562 cells were counted for cell viability and an equal number of live cells were seeded in 15% FCS with IMDM culture media mixed with 1% methylcellulose. Colonies were determined for their lineage based on morphological characteristics.57, 58 Clonal colonies emerged after 3 weeks were counted and scored for contribution to granulocyte, macrophage, erythrocyte and megakaryocyte lineages. The CFU-Es were progenitors with one or two clusters with 10–100 hemoglobinized erythroblasts in each cluster. The BFU-E colonies were characterized as small (3–8 clusters), intermediate (10–16 clusters) or large (>16 clusters) of primitive erythroid progenitors. The CFU-Gs were progenitors of homogeneous population of granulocytes and exhibit the presence of refractive granules in their cytoplasm. The CFU-Ms were clonogenic progenitors of macrophages that display ground glass appearance. The CFU-GEMM displayed heterogeneous population of granulocytes, erythrocyte, macrophage and megakaryocyte. The CFU-GM displayed heterogeneous population of granulocyte and macrophage. Erythroid colonies displayed haemoglobin production by their red brown colour. The granulocytic colonies displayed large refractive granules. Images were taken under fluorescence microscope (Nikon Eclipse TE2000-S; Meville, NY, USA). Colonies of 5–10 different areas were counted per dish. The images were taken at × 4 and × 10. The myeloid cells were calculated by adding CFU-G and CFU-M colony numbers. The erythroid cells were calculated by adding CFU-E and BFU-E colonies. The ME% was used to show a shift in lineage distribution. Data are shown as mean of three independent experiments. Error bars represent as s.e. of three independent experiments with P-values of the mean. *P<0.05, **P<0.01 and ***P<0.001.
% of myeloid–erythroid cells (ME%)=((CFU-G+CFU-M)/(CFU-E+BFU-E)) × 100.
LNA anti-miR
LNA-based oligonucleotides are designed to block miRNA182-5p expression. In LNA oligos, the furanose ring of selected ribose sugars is chemically locked into an RNA-mimicking conformation by the introduction of an O2′, C4′-methylene bridge. The LNA oligos were purchased from GeneX India Biosciences Pvt Ltd (Eurogentec, Belgium).
Scramble Tm-32 °C
5′-CATCGTCGATCGTAGCGCA-3′
Anti-miRNA182-5p Tm-42 °C
5′-AGTGTGAGTTCTACCATTGCCAAA-3′
Anti-hsa-let7c-5p Tm-33 °C
5′-AACCATACAACCTACTACCTCA-3′. Bold letters represent LNA modification.
The LNA anti-miRs bind to their complementary miRNA and prevent them from binding to their mRNA sequences. Hence, translational block of target mRNA is released in anti-miRNA-transfected cells. Transfection of K562 cells with anti-miRNA182-5p was performed using Lipofectamine2000 as per manufacturer’s protocol. Three different concentrations used were unit 1 for 10 nM, 2 for 20 nM and 5 for 50 nM concentration. Scramble oligos were used along with the treatment. After 24 h, cells were cultured with TKI at 0.75 μM for another 48 h. Absorbance was measured after addition of WST1 cell proliferation reagent (Roche, IN, USA) and normalised to 100 after 72 h post transfection of the scramble control. Data are shown as mean of three independent experiments. Error bars represent as s.e. of three independent experiments with P-values of the mean. *P<0.05, **P<0.01 and ***P<0.001.
MiRNA mimics
MiRNA mimics were purchased from Sigma (St. Louis, USA). The transfection of K562 cells was performed according to the manufacturer’s protocol. Three different concentrations were used 1 for 10 nM, 2 for 20 nM and 5 for 50 nM concentration. Scramble oligos were used along with the treatment.
Hes1 knockdown and overexpression
In brief, K562 cells were nucleofected with Hes1 overexpression and knockdown plasmids on Amaxa 4d nucleofector from Lonza and incubated for 3 days. Hes1 was overexpressed by miGR1-Hes1 plasmid. Hes1 knockdown was performed by shRNA against Hes1 open-reading frame. ShRNA-Hes1 plasmids were a kind gift from Dr Adolo Ferrando, Columbia University. MigR1 control and MigR1-Hes1 plasmids were a kind gift from Avinash Bhandoola lab (NIH, USA).
Flow cytometry
In brief, K562 cells were washed twice in × 1 PBS, fixed in 2% PFA for 10 min at room temperature. Then, cells were permeabilized by Saponin followed by blocking with 5% blotto at room temperature for 30 min. Rabbit anti-Hes1 antibody (Abcam, Cambridge, USA) was incubated as per manufacturer’s protocol. Secondary antibody incubation was done at RT for 30 min and washed with PBS. Cells were assessed for Hes1 expression on FACS Calibur flow cytometer (San Jose, USA). The data were analysed using CellQuestPro software (USA) and shown as histograms and dotplots.
CRISPR-mediated miRNA182 knockout
Designing of MIR182 constructs—the CRISPR and CRISPR-associated (CAS) system is RNA-based genome-engineering platform. Recently shown in vitro reconstitution of the Streptococcus pyogenes type II CRISPR system demonstrated that crRNA fused to a normally trans-encoded tracrRNA was sufficient to direct the Cas9 protein to sequence-specifically cleave target DNA sequences matching the crRNA. Here we engineer a single system of this bacterial type II CRISPR system in human cell lines. For designing the guide RNAs (gRNAs), we identified all the NGG sites (represents protospacer adjacent motif sequence) in MIR182 cluster region ±1000 bp of the target site. To direct Cas9 to cleave sequences of interest, we designed crRNA-tracrRNA fusion oligonucleotides, referred to as gRNAs under the human U6 polymerase III promoter. To identify CRISPR target sites around MIR182 cluster that should be cleavable without off-target cuts, we therefore examined all 23 bp sequences of the form 5′-GBBBBBBBBBBBBBBBBBBBNGG-3′, where the B’s represented the bases at the genome location, for which no sequence of the form 5′-NNNNNNNBBBBBBBBBBBBBNGG-3′ existed at any other location in the human genome.59 Identification of CRISPR target sites across MIR182 locus was done using the Zhang lab tool, MIT. A single plasmid system expressing Cas9, eGFP and GuideRNA was synthesised to target MIR182 loci (Supplementary Figure 5b). The CRISPR plasmids induced double-strand breaks in HEK293T cells assessed by surveyor nuclease assay represented as indel percentage (Figure 3b). In HEK293 cells, CRISPR plasmids transfection generated deletion of the MIR182 locus as shown by agarose gel analysis (Supplementary Figure 6a). Each off-target site was assessed for indels. The top five off-targets were chosen based on their quality score and examined for their potential to generate indels Supplementary Figure 8. The off-targets of Cas9+0482 and Cas9+9678 did not reveal indels, whereas Cas9+0425-05 off-target present in the non-coding genomic region induced indels (Figure 3f). A number of 0.5 million K562 cells were nucleofected with CRISPR MIR182 plasmids and cultured for 72 h. Further, expression of plasmids was monitored by eGFP expression (Supplementary Figure 5c). FACS sorting of 0.5% high eGFP-positive cells was performed on FACS ARIA with an anticipation of higher editing efficiency (Supplementary Figure 5d). A total of 30–50 cells were cultured in a 48-well plate for 10 days and analysed for deletion clones by PCR method. A 1560 bp long DNA sequence of the MIR182 locus was amplified using MIR182 genomic locus-specific primers.
Surveyor mutation detection assay
In brief, HEK293T cells were transfected with CAS9-GFP-GuideRNAs targeting MIR182 flanking regions. After 48 h, genomic DNA was extracted from the pool of cells, and analysed for Indels using Surveyor mutation detection kit (Transgenomic, Omaha, USA) as per manufacturer’s protocol. In brief, PCR amplicons from mutant (test) and wild-type (reference) DNA were hybridized by heating and cooling the mixture to form hetero- and homoduplexes. The annealed heteroduplex/homoduplex mixture was treated with Surveyor Nuclease. DNA fragments were analysed by agarose gel electrophoresis. The formation of new cleavage products due to the presence of one or more mismatches was indicated by the presence of additional bands. The relative size of these cleavage products indicated the location of the mismatch or mismatches (Figure 3a). Intensity of individual DNA bands was measured by ImageJ software. Indel percentage was calculated as per reference protocol.38, 59
Off-target analysis
Off-targets were identified by the Zhang lab, MIT tool. The top five off-targets for each guide RNAs were analysed by the surveyor nuclease mutation detection system.
Genotype for MIR182 deletion
MIR182 genomic site flanking the target sites (1560 bp) was amplified using PCR and was run on a 2% agarose gel containing EtBr. The product length was matched to the expected product and deletion was confirmed by presence of 756, 819 and 1497 bp DNA fragments. Each PCR product was purified using Qiagen PCR purification kit as per manufacturer’s instructions and given for Sanger sequencing analysis. Each chromatogram was read using FinchTV software.
Statistical analysis
Data points are expressed as mean±standard error. Student t-test was performed to find the significance of differences. P values of less than.05 were considered to be significant.
References
Costa G, Kouskoff V, Lacaud G . Origin of blood cells and HSC production in the embryo. Trends Immunol 2012; 33: 215–223.
Crans HN, Sakamoto KM . Transcription factors and translocations in lymphoid and myeloid leukemia. Leukemia 2001; 15: 313–331.
Yang Y, Liu X, Xiao F, Xue S, Xu Q, Yin Y et al. Spred2 modulates the erythroid differentiation induced by imatinib in chronic myeloid leukemia cells. PLoS One 2015; 10: e0117573.
Buckle AM, Mottram R, Pierce A, Lucas GS, Russell N, Miyan JA et al. The effect of Bcr-Abl protein tyrosine kinase on maturation and proliferation of primitive haematopoietic cells. Mol Med 2000; 6: 892–902.
Palma C a, Tonna EJ, Ma DF, Lutherborrow M a . MicroRNA control of myelopoiesis and the differentiation block in acute myeloid leukaemia. J Cell Mol Med 2012; 16: 978–987.
Garzon R, Croce CM . MicroRNAs in normal and malignant hematopoiesis. Curr Opin Hematol 2008; 15: 352–358.
O’Connell RM, Zhao JL, Rao DS . MicroRNA function in myeloid biology. Blood 2011; 118: 2960–2969.
Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci USA 2009; 106: 1814–1819.
Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C et al. TGF-β induces miR-182 to sustain NF-κB activation in glioma subsets. J Clin Invest 2012; 122: 3563–3578.
Yang W-B, Chen P-H, Hsu T, Fu T-F, Su W-C, Liaw H et al. Sp1-mediated microRNA-182 expression regulates lung cancer progression. Oncotarget 2014; 5: 740–753.
Pedranzini L, Mottadelli F, Ronzoni S, Rossella F, Ferracin M, Magnani I et al. Differential cytogenomics and miRNA signature of the acute myeloid leukaemia Kasumi-1 cell line CD34+38- compartment. Leuk Res 2010; 34: 1287–1295.
Guglielmelli P, Tozzi L, Pancrazzi A, Bogani C, Antonioli E, Ponziani V et al. MicroRNA expression profile in granulocytes from primary myelofibrosis patients. Exp Hematol 2007; 35: 1708–1718.
Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S et al. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci USA 2009; 106: 1814–1819.
Wei Q, Lei R, Hu G . Roles of miR-182 in sensory organ development and cancer. Thorac Cancer 2015; 6: 2–9.
Stittrich A, Haftmann C, Sgouroudis E, Kühl AA, Hegazy AN, Panse I et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol 2010; 11: 1057–1062.
Kouri FM, Hurley L a, Day ES, Hua Y, Merkel TJ, Queisser a et al. miR-182 integrates apoptosis, growth and differentiation programs in glioblastoma. Genes Dev 2015; 2: 732–745.
Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood 2006; 108: 28–37.
Calabretta B, Perrotti D . The biology of CML blast crisis. Blood 2004; 103: 4010–4022.
Van Etten R . a. Oncogenic signaling: new insights and controversies from chronic myeloid leukemia. J Exp Med 2007; 204: 461–465.
Era T, Witte ON . Regulated expression of P210 Bcr-Abl during embryonic stem cell differentiation stimulates multipotential progenitor expansion and myeloid cell fate. Proc Natl Acad Sci USA 1999; 97: 1737–1742.
Hasserjian RP, Boecklin F, Parker S, Chase A, Dhar S, Zaiac M et al. ST1571 (imatinib mesylate) reduces bone marrow cellularity and normalizes morphologic features irrespective of cytogenetic response. Am J Clin Pathol 2002; 117: 360–367.
Müller MC, Cortes JE, Kim DW, Druker BJ, Erben P, Pasquini R et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: Analysis of responses according to preexisting BCR-ABL mutations. Blood 2009; 114: 4944–4953.
Brendel C, Scharenberg C, Dohse M, Robey RW, Bates SE, Shukla S et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 2007; 21: 1267–1275.
Chu S, Xu H, Shah NP, Snyder DS, Forman SJ, Sawyers CL et al. Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood 2005; 105: 2093–2098.
Graham SM, Jørgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002; 99: 319–325.
Zhao JL, Ma C, O’Connell RM, Mehta A, Diloreto R, Heath JR et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell 2014; 14: 445–459.
Li J . Quiescence regulators for hematopoietic stem cell. Exp Hematol 2011; 39: 511–520.
Doré LC, Crispino JD . Transcription factor networks in erythroid cell and megakaryocyte development. Blood 2011; 118: 231–239.
Akashi K, Traver D, Miyamoto T, Weissman IL . A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000; 404: 193–197.
Marley SB, Deininger MW, Davidson RJ, Goldman JM, Gordon MY . The tyrosine kinase inhibitor STI571, like interferon-alpha, preferentially reduces the capacity for amplification of granulocyte-macrophage progenitors from patients with chronic myeloid leukemia 197. Exp Hematol 2000; 28: 551–557.
Amin HM, Yang Y, Shen Y, Estey EH, Giles FJ, Pierce SA et al. Having a higher blast percentage in circulation than bone marrow: clinical implications in myelodysplastic syndrome and acute lymphoid and myeloid leukemias. Leukemia 2005; 19: 1567–1572.
States U . Erythroid variant of chronic myelogenous leukemia. Leukemia 2008; 1: 21–24.
Leary JF, Ohlsson-Wilhelm BM, Giuliano R, LaBella S, Farley B, Rowley PT . Multipotent human hematopoietic cell line K562: lineage-specific constitutive and inducible antigens. Leuk Res 1987; 11: 807–815.
Lee SM, Bae JH, Kim MJ, Lee HS, Lee MK, Chung BS et al. Bcr-Abl-independent imatinib-resistant K562 cells show aberrant protein acetylation and increased sensitivity to histone deacetylase inhibitors. J Pharmacol Exp Ther 2007; 322: 1084–1092.
Vaz C, Ahmad HM, Sharma P, Gupta R, Kumar L, Kulshreshtha R et al. Analysis of microRNA transcriptome by deep sequencing of small RNA libraries of peripheral blood. BMC Genomics 2010; 11: 288.
Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008; 134: 521–533.
Montagner S, Dehó L, Monticelli S . MicroRNAs in hematopoietic development. BMC Immunol 2014; 15: 14.
Mandal PK, Ferreira LMR, Collins R, Meissner TB, Boutwell CL, Friesen M et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014; 15: 643–652.
Ruijtenberg S, van den Heuvel S . Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016; 15: 196–212.
Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010; 463: 676–680.
Shet AS, Jahagirdar BN, Verfaillie CM . Chronic myelogenous leukemia: mechanisms underlying disease progression. Leukemia 2002: 1402–1411.
Ning F, Wang F, Li M, Yu Z-S, Hao Y, Chen S . MicroRNA-182 modulates chemosensitivity of human non-small cell lung cancer to cisplatin by targeting PDCD4. Diagn Pathol 2014; 9: 143.
Qin J, Luo M, Qian H, Chen W . Upregulated miR-182 increases drug resistance in cisplatin-treated HCC cell by regulating TP53INP1. Gene 2014; 538: 342–347.
Wang YQ, De Guo R, Guo RM, Sheng W, Yin LR . MicroRNA-182 promotes cell growth, invasion, and chemoresistance by targeting programmed cell death 4 (PDCD4) in human ovarian carcinomas. J Cell Biochem 2013; 114: 1464–1473.
Puissant A, Dufies M, Fenouille N, Sahra Ben I, Jacquel A, Robert G et al. Imatinib triggers mesenchymal-like conversion of CML cells associated with increased aggressiveness. J Mol Cell Biol 2012; 4: 1–14.
Xu J, Lamouille S, Derynck R . TGF-β-induced epithelial to mesenchymal transition. Cell Res 2009; 19: 156–172.
Tang X, Zheng D, Hu P, Zeng Z, Li M, Tucker L et al. Glycogen synthase kinase 3 beta inhibits microRNA-183-96-182 cluster via the β-Catenin/TCF/LEF-1 pathway in gastric cancer cells. Nucleic Acids Res 2014; 42: 2988–2998.
Hirata H, Ueno K, Shahryari V, Tanaka Y, Tabatabai ZL, Hinoda Y et al. Oncogenic miRNA-182-5p targets Smad4 and RECK in human bladder cancer. PLoS One 2012; 7: 1–8.
Yang H, Hui H, Wang Q, Li H, Zhao K, Zhou Y et al. Wogonin induces cell cycle arrest and erythroid differentiation in imatinib-resistant K562 cells and primary CML cells. Oncotarget 2014; 5: 8188–8201.
Yeh C-H, Moles R, Nicot C . Clinical significance of microRNAs in chronic and acute human leukemia. Mol Cancer 2016; 15: 37.
Stittrich A-B, Haftmann C, Sgouroudis E, Andrea Kühl A, Nabil Hegazy A, Panse I et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol 2010; 11: 0–5.
Wang C, Ren R, Hu H, Tan C, Han M, Wang X et al. MiR-182 is up-regulated and targeting Cebpa in hepatocellular carcinoma. Chin J Cancer Res 2014; 26: 17–29.
Dambal S, Shah M, Mihelich B, Nonn L . The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res 2015; 43: 7173–7188.
Kato T, Sakata-Yanagimoto M, Nishikii H, Ueno M, Miyake Y, Yokoyama Y et al. Hes1 suppresses acute myeloid leukemia development through FLT3 repression. Leukemia 2014: 1–10.
Nakahara F, Sakata-Yanagimoto M, Komeno Y, Kato N, Uchida T, Haraguchi K et al. Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood 2010; 115: 2872–2881.
Grebe D, Pluskalová M, Marinov I, Klamová H . Imatinib mesylate affects tyrosine kinase activity in both leukemic and normal primary mononuclear blood cells. J Appl Biomed 2006; 4: 95–104.
Sarma NJ, Takeda A, Yaseen NR . Colony forming cell (CFC) assay for human hematopoietic cells. J Vis Exp 2010: 3–7.
Hogge BDE, Lansdorp P, Reid D, Eaves C . Enhanced detection, maintenance, and differentiation of primitive human hematopoietic cells in cultures containing murine fibroblasts engineered to produce human steel factor, interleukin-3, and granulocyte colony-stimulating factor. Blood 88: 3765–3773.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE et al. RNA-guided human genome engineering via Cas9_Sup. Science 2013; 339: 823–826.
Acknowledgements
We thank Soumen Chakraborty for providing the K562 and KCL22 cell lines. We thank Jyotsna Dhawan and Khairul Ramlee for valuable suggestions. We also thank central imaging and flow cytometry (CIFF) facility for FACS analysis. This work was supported by DBT, CSIR and NCBS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U Simon
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Arya, D., Sachithanandan, S., Ross, C. et al. MiRNA182 regulates percentage of myeloid and erythroid cells in chronic myeloid leukemia. Cell Death Dis 8, e2547 (2018). https://doi.org/10.1038/cddis.2016.471
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2016.471
This article is cited by
-
Novel evidence that the ABO blood group shapes erythropoiesis and results in higher hematocrit for blood group B carriers
Leukemia (2023)
-
Engineered model of t(7;12)(q36;p13) AML recapitulates patient-specific features and gene expression profiles
Oncogenesis (2022)
-
Hes1 Controls Proliferation and Apoptosis in Chronic Lymphoblastic Leukemia Cells by Modulating PTEN Expression
Molecular Biotechnology (2022)
-
Non-coding RNAs in cancer: platforms and strategies for investigating the genomic “dark matter”
Journal of Experimental & Clinical Cancer Research (2020)
-
Construction and comprehensive analysis of a ceRNA network to reveal potential prognostic biomarkers for hepatocellular carcinoma
Cancer Cell International (2019)
