
Abstract
By screening a collection of one hundred combinations of thiazolidinone compounds, we identified one combination (M4) that synergistically inhibited the growth of H460 and H460/TaxR cells and tumor growth in H460/TaxR xenograft mice. A whole genome microarray assay showed that genes involved in negative regulation of microtubule polymerization or depolymerization, intracellular protein kinase cascade, positive regulation of histone acetylation, cell cycle arrest and apoptosis were upregulated. Further analysis proved that the four compounds act as either microtubule polymerization inhibitors or histone deacetylase inhibitors. They act synergistically targeting multiple proteins and leading to the regulation of cell cycle checkpoint proteins, including p53, p21, cdc25C and cdc2, the activation of caspases, JNK, p38 cascades and the inactivation of Akt. These events resulted in the G2/M cell cycle arrest and cell apoptosis. These data provide a new strategy for discovering anticancer drugs and drug combinations for drug-resistant cancers.
Similar content being viewed by others
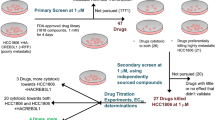

Main
Lung cancer is one of the main causes of death among all diseases. The five-year survival rate of lung cancer is merely 16.3% (http://www.lung.org/lung-disease/lung-cancer/resources/facts-figures/lung-cancer-fact-sheet.html). More than 80% of lung cancer cases are non-small cell lung cancer (NSCLC) cases. Cancer treatments include surgery, radiotherapy and chemotherapy. However, drug resistance and the dose-limiting toxicity are key reasons to account for unsuccessful cancer chemotherapy.1
Discovery of single target-specific drugs has been the focus of cancer drug development. Research in this area has led to the successful drugs such as Gleevec,2 Tarceva and Iressa.3 However, tumorigenesis is a complex process with many genes, proteins, and signaling pathways involved. Thus, single-target drugs have gradually showed some disadvantages, such as lack of efficacy, serious side effects, and development of drug resistance.4, 5, 6 Although drug resistance can be caused by various mechanisms, a critical one is from multidrug resistance (MDR),7 a phenotype of cross-resistance to multiple drugs with diverse chemical structures. A major cause of MDR is the overexpression of the MDR-1 gene that encodes P-glycoprotein (P-gp).8 Many chemotherapy drugs including taxanes and vinca alkaloids are substrates of P-gp. The P-gp prevents the intracellular accumulation of these drugs by increasing their efflux, leading to MDR.9 Efforts have been made to inhibit P-gp in order to reverse MDR.7, 10, 11 However, clinical trials have shown compromised results due to some inevitable side effects.12 Therefore, the discovery of novel compounds or compound combinations that are not substrates of P-gp is a more effective strategy to overcome drug resistance.13
Recently, multi-level and multi-targeting therapies14, 15 have shown potential applications in cancer treatment. Such therapies, including multi-component drugs or multi-targeting drugs, may produce concerted pharmacological intervention of multiple targets and signaling pathways that drive the growth of tumors. For example, drug combination may be a promising strategy for treating multi-factorial diseases such as cancer16 and acquired immunodeficiency syndrome.17 Synergistic action of such drugs may overcome side effects that resulted from high doses of single-target drugs, increase drug selectivity, and offer an opportunity for more precise control of biological systems.18 Drug combinations that simultaneously impact multiple targets are more effective to overcome MDR and lower side-effects19 in cancer cell inhibition20, 21 and tumor shrinkage.22, 23, 24
Previously, we have reported that the thiazolidinone derivatives are useful anticancer agents with P-gp-evading property and minimal side effects.13, 25 Some of these compounds inhibit tubulin polymerization, cause cell cycle arrest and induce apoptosis. They also target various kinases depending on their chemical structures. We assumed that their proper combinations may produce synergistic cancer inhibitory effects. Thus, the purpose of this study is to identify thiazolidinone compound combinations that have synergistic inhibitory effects on P-gp overexpressing NSCLC and to elucidate their possible targets and the affected signaling pathways. (1) To this end, by screening compound combinations prepared from a thiazolidinone compound library13 in a NSCLC cell line H460 and its drug-resistant variant H460/TaxR, a four-compound combination was identified that synergistically inhibit the growth of cancer cells from both lines. (2) We determined the antitumor activity of this combination in drug-resistant H460/TaxR xenograft mice models. The drug combination was highly effective in inhibiting tumor growth and prolonging mice survival. (3) We also investigated the molecular basis of the observed anticancer effects. Our study showed that individual compounds in this combination act as either tubulin polymerization inhibitors or histone deacetylase (HDAC) inhibitors. To the best of our knowledge, this is the first work that reports the synergistic anticancer activity of similarly structured agents by targeting tubulin depolymerization and HDAC simultaneously.
Results
A potent compound combination in cancer cell growth inhibition
Using a cell growth-inhibitory screening against H460 and H460/TaxR cell lines, a potent compound combination M4, containing compounds 27, 107, 167 and 254 was identified (Figures 1a and b). Individual compounds and M4 inhibited cancer cell growth in a dose-dependent manner (Supplementary Figure S1). In addition to its toxicity towards both H460 and H460/TaxR cells, this combination also exhibited a minimal toxicity towards NHFB (Figure 1c) and induced a lower percentage of cell apoptosis in NHFB (Supplementary Figure S1c). To test whether individual compounds or M4 is the substrate or the inhibitor of the P-gp, we investigated the cancer cell growth inhibition in the presence of the P-gp inhibitor and the accumulation of rhodamine 123 (Rho 123) in H460/TaxR cells under the treatment of individual compounds or M4. The results showed that P-gp inhibition did not affect the concentration of drug to cause 50% inhibition of growth (GI50) values of the four compounds or M4 in H460/TaxR cells obviously, whereas the GI50 value of paclitaxel was decreased 18-fold (Table 1). Furthermore, individual compounds or M4 treatment did not increase the accumulation of Rho 123 in H460/TaxR cells (Supplementary Figure S2). These results indicate that individual compounds or M4 could be neither the substrate nor the inhibitor of the P-gp. The GI50 value of M4 was about twofold lower than the lowest GI50 value by any individual compound (Table 1), implying additive or synergistic action by the compound combination.

The chemical structures of compounds in M4, the cytotoxicity and synergistic effect of M4 on H460 and H460/TaxR cells. (a) The chemical structures of compounds in M4. (b) An overlay of structures of the four compounds in M4. (c) Effect of M4 treatment on cell growth in cancer cells and normal cells. The four compounds 27, 107, 167 and 254 were mixed with equal molar ratio. The cells were treated with different concentrations (μM ) of M4 for 72 h. The cell growth inhibition was measured by SRB assay. Each experiment was repeated three times independently. Data are shown as means±S.D. (d, e) Combination index (CI) for treatment in H460 and H460/TaxR cells, respectively. The CI was determined using the Compusyn software to evaluate the interaction of the four compounds in M4. A solid horizontal line marks CI=1. The CI offers definition for addictive (CI=1), synergism (CI<1), and antagonism (CI>1) in drug combination. The data are the mean values from three independent experiments
In order to evaluate whether the action of M4 was additive or synergistic in H460 and H460/TaxR cells, we analyzed the combination effects using the combination index (CI) method.26 The Fa-CI plot for M4 showed that the CI value was <1 over the entire range, CI values for H460 and H460/TaxR cell lines were 0.18 and 0.24, respectively, at the 50% effective dose, suggesting a strong synergism by M4 (Figures 1d and e). Similar analyses were also carried out for all the two-compound and three-compound combinations by these four compounds in H460/TaxR cells. The GI50 values of all the two-compound and three-compound combinations were less potent than that of M4 (Supplementary Table S1). Antagonistic effect was indicated for almost all the two-compound and three-compound combinations at 50% effective dose for H460/TaxR cells, except an additive effect by 107–167 combination and a moderate synergism by 27–167–254 combination.
Briefly, a potent compound combination M4 was identified to have strong anticancer effects with minimal toxicity to normal cells and no reduction on activity in drug-resistant cells. Four compounds in M4 showed strong synergism in anticancer action by data analysis. The detailed mechanism, however, awaits verifications by various experiments to be discussed later.
Enhanced antitumor effects in the H460/TaxR xenograft mice by M4
To evaluate in vivo effects of M4, we next investigated the effects of individual compound or M4 on the tumor growth in mice with H460/TaxR xenograft tumors. The experiment was terminated and the mice were killed on day 22. The mean volumes of subcutaneous tumors in M4- or compound-treated animals decreased relative to those in vehicle-treated mice as the dose increased. Compound 27 (10.0 mg/kg) and paclitaxel (20 mg/kg) inhibited the tumor growth obviously, but not significant compared with the solvent control (Figure 2a and Supplementary Table S2). Compound 107, 167 or 254 at low dose did not inhibit the tumor growth obviously. However, treatment with compound 27, 107, 167 and 254 alone at higher doses caused tumor growth inhibition, but not significant compared with the solvent control (Figure 2b and Supplementary Table S2). By contrast, in the group treated with M4, the tumor growth was significantly inhibited (P=0.009). On day 22, the mean tumor volume in animals treated with solvent was 1103±111 mm3, whereas the mean tumor volume in animals treated with M4 was 465±74 mm3. Furthermore, M4 significantly increased the survival rate of tumorous mice compared with control or paclitaxel groups (Figure 2c). The body weights of all mice were monitored to evaluate the side effects of M4 and each compound. As shown in Figure 2d, there is no obvious body weight loss in mice throughout the experiment, indicating that M4 did not have evident systemic toxicity at the dose used in this investigation. Lower survival rate and body weight loss in mice in the paclitaxel group reflected the toxicity of the drug towards normal tissues. Taken together, these results suggested that M4 induced an enhanced antitumor effect in H460/TaxR xenograft mice compared with individual compounds with no obvious toxicity.

Antitumor effects of individual compounds and M4 on the human lung cancer H460/TaxR in vivo. The mice received the treatments of individual compounds or M4 every day by subcutaneous injection for 11 days, or the treatment of paclitaxel every other day for three times as positive control. The experiment was terminated and the mice were killed on day 22. (a) Antitumor effects of individual compound at low dose or paclitaxel as positive control. (b) Antitumor effects of individual compounds or M4 at high dose. Each point refers to the mean value of 6–7 tumors. **, the value is significantly different from that of control (P<0.01). (c) Effects of paclitaxel or M4 on mice survival (Kaplan–Meier plot). (d) Effects of paclitaxel or M4 on the body weight of mice
Perturbation of gene expression in H460 and H460/TaxR by M4
To gain further insight into the mechanism of action of M4, the changes in gene expression levels were determined by an Agilent Whole Human genome Oligo Microarray platform (Agilent Technologies, Santa Clara, CA, USA). The microarray analysis showed that only 46 genes were upregulated and 70 genes were downregulated (>twofolds) in H460 cells following treatment with a combined concentration of 1.0 μM of M4 for 4 h (Supplementary Figure S3a), whereas a total of 1408 genes were altered (>twofolds) in H460/TaxR cells under the same treatment (Supplementary Figure S3b). The results showed that more genes were altered in H460/TaxR cells compared with H460 cells (Supplementary Figure S3c). MDR1/ABCB1 gene was strongly altered with a 153-fold increase among other altered genes confirming that P-gp overexpression had a major role. However, our data could not exclude the possibility of the other altered pathways.
The heat map (Figure 3a) showed the altered genes related to the synergistic growth inhibitory effect and the affected biological processes. In H460/TaxR cells, genes involved in negative regulation of microtubule polymerization, such as TBCD, FKBP4, AURKB and KIF18B, and genes involved in regulating JUN kinase activity, such as MAP3K9, MAP4K2, and AXIN1, were all upregulated in cells treated with M4. These suggested a role of M4 in regulating microtubule depolymerization. Genes involved in regulating histone acetylation, such as DMAP1, HCFC1 and PHF15 and genes in response to tumor necrosis factor such as TNFRSF12A, TRAF4, TRAP1, CD40 and MADD were also upregulated, suggesting another role of M4 in inhibiting the HDAC. Moreover, several p53 target genes including AEN, BOK, E2F7 and PHLDA3 were also upregulated, indicating the activation of p53 in cells after treatment. Consistent with this, genes involved in positive regulation of cell apoptosis and cell cycle arrest such as BCL2L13, CDKN1A (p21, Cip1), TNFRSF12A, BRCA2, CDK2, and CDK10 were also upregulated after treatment (Figure 3b).

Micrarray analysis of gene expression changes in H460/TaxR cells treated with M4 for 4 h. (a) The heat map shows the effect of M4 on the representative gene expression in H460/TaxR cells involved in the synergistic anticancer activity of M4. (b) Representation of differentially expressed gene involved in crucial pathways in cells treated with M4, red-genes upregulated, blue-genes downregulated. The number in the bracket is the fold change of the gene expression
Individual compounds and M4 perturbed tubulin polymerization/depolymerization in H460 and H460/TaxR cells
Our previous works27, 28, 29 have shown that some thiazolidinone compounds inhibited tubulin polymerization and induced G2/M arrest in human cancer cells. In this work, we also found that the expression of genes involved in microtubule depolymerization was affected by M4 treatment. Furthermore, genes involved in protein modification, particularly H4 acetylation were also upregulated. Inhibition of HDAC might induce polymerization through tubulin acetylation.
To verify it, we tested their effect on microtubule assembly–disassembly process in a cell-free system. Under normal conditions, α- and β-tubulin subunits dimerize and then self-assemble to form cylindrical microtubules in a time-dependent manner. This process is indicated by a steady increase of fluorescence with time in dimethyl sulfoxide (DMSO)-treated group (Figure 4a). Compound 167 and 254 showed no effect on tubulin polymerization at the concentration tested. The microtubule stabilizing agent paclitaxel enhanced the polymerization of tubulins as the fluorescence of the system increased rapidly, whereas microtubule destabilizing agent, colchicine, completely inhibited tubulin polymerization. Compound 27, 107 or M4 inhibited polymerization of tubulin compared with that of the solvent.

Effect of individual compounds and M4 on tubulin depolymerization of H460 and H460/TaxR cells. (a) The inhibition of tubulin assembly in vitro. The assay was performed in a black 96-well plate. Each well of the assay plate contained 5 μl control buffer or test compounds (5 μM) was pre-warmed at 37 °C for 1 min, then 50 μl tubulin solution was pipetted into each well rapidly, and the polymerization dynamics of tubulin was monitored through measuring the change of fluorescence (excitation/emission 355 nm/460 nm) every 2 min for 42 min at 37 °C on 1420 Multi-label counter (Perkin Elmer). The data are the mean values from three independent experiments. (b–d) Cells were treated with different concentrations (0, 0.25, 0.5 and 1.0 μM) of individual compounds or M4 for 24 h. Paclitaxel, VCR and SAHA were used to treat cells as positive controls. Polymeric (P) and soluble (S) tubulin fraction were isolated as explained in Materials and Methods, and equal amounts of proteins were resolved by SDS–PAGE followed by immunoblotting with anti-α-tubulin antibody. S/P in cells treated with DMSO has been normalized to 1.0
To further confirm that whether the balance of tubulin polymerization and depolymerization was altered by individual compounds or M4, we quantitatively assessed the ratio between soluble and polymeric tubulins in live cells. The soluble and polymeric fractions of tubulins in cells were analyzed by western blot (Figures 4b–d). Paclitaxel, vincristine (VCR) and suberoylanilide hydroxamic acid (SAHA), a typical HDAC inhibitor, were used to treat cells as positive controls. The results showed that paclitaxel and SAHA increased the polymeric fraction of tubulin in H460 cells obviously, and VCR increased the soluble fraction of tubulin. However, all individual compounds increased the polymeric fraction of tubulin in a concentration-dependent manner, whereas M4 increased the soluble fraction of tubulin at 0.25 and 0.5 μM.
In H460/TaxR cells, paclitaxel and VCR have no obvious effects on depolymerization/polymerization of tubulin because of the efflux of the two drugs through P-gp; SAHA increased the polymeric fraction of tubulin. Although at all the concentrations tested, 27, 167 or M4 has no significant effects on the balance of tubulin depolymerization and polymerization; 107 increased the soluble fraction of tubulin at 0.5 μM obviously; 254 decreased the soluble fraction of tubulin at 1.0 μM. All in all, although 27, 107 or M4 exhibited the tubulin depolymerization activity, each compound or M4 might also act as HDAC inhibitors and induce tubulin polymerization through tubulin acetylation.
Each compound and M4 induced G2/M phase arrest and regulated the expression of cell cycle checkpoint proteins in H460 and H460/TaxR cells
The polymerization/depolymerizaiton of tubulin and HDAC activity are both related to the stability of microtubule and thus the cell cycle regulation. We analyzed the effects of individual compounds and M4 on cell cycle. After 12 h of treatment, paclitaxel, VCR, SAHA, and individual compounds or M4 all induced G2/M arrest in H460 cells (Figures 5a and c). In H460/TaxR cells, only SAHA, 27, 254 and M4 induced obvious G2/M arrest.

Cell cycle analysis of H460 and H460/TaxR cells treated with compound alone or M4. (a, c) Cells were treated with 1.0 μM of 27, 107, 167, 254, M4 (total concentration is 1.0 μM), paclitaxel (100 nM), VCR (100 nM) and SAHA (10 μM), respectively, for 12 h. Then the cell cycle distribution was analyzed using the flow cytometer (FACS Calibur, BD, San Jose, CA, USA). (b, d) Western blot analysis of the cell cycle checkpoint proteins in H460 and H460/TaxR cells. The experiment was repeated at least twice
We next examine the effects of individual compounds or M4 on the expression of G2/M checkpoint proteins in cells after treatment for 24 h. Consistent with cell cycle arrest, individual compounds or M4 treatment caused a marked increase of p53, phosphorylated/inactivation of cdc25C(Ser216), and an obvious decrease in the total level of cdc2 in both cell lines. Cyclin B1 expression was slightly increased by 27, 107 and decreased by 254 in H460 cells, whereas in H460/TaxR cells, individual compounds or M4 increased the expression of cyclin B1 except 27. In addition, individual compounds or M4 increased the level of p21 (waf1/cip1) obviously in H460 cells. By contrast, the basal level of p21 (waf1/cip1) is much higher in H460/TaxR cells, and after treatments, the level of p21 decreased but still in a relative high level (Figures 5b and d). Further analysis showed that individual compounds or M4 exhibited different mode on the induction of p53 target genes AEN, BOK, E2F7 and PHLDA3 (Supplementary Figure S4). These results suggest that inhibition of proliferation of H460 and H460/TaxR cells by individual compounds or M4 involve G2/M arrest through modulation of p53, p21 and cell cycle-related protein expression.
Induction of cell apoptosis by M4 through both death receptor and mitochondrial pathways
Inhibiting tubulin and HDAC induces cell apoptosis in cells various pathways, we assessed whether each compound or M4 induced cell apoptosis through the activation of caspase-3/7, caspase-8 and caspase-9. In H460/TaxR cells, individual compounds or M4 treatments activated caspase-8, caspase-9 and caspase-3/7 in a concentration-dependent manner (Figures 6b–d). The evaluation of Annexin V-positive cells showed that paclitaxel, VCR and SAHA did not induce H460/TaxR apoptosis obviously (Supplementary Figure S5) at the concentration tested, whereas individual compounds or M4 enhanced cell apoptosis significantly (Figure 6e and Supplementary Figure S5). Consistently, increased levels of pro-apoptotic proteins Bad and decreased levels of anti-apoptotic protein Bcl-xL were observed, leading to the release of cytochrome c (Cyto c) from the mitochondria to the cytosol (Figure 6f). Furthermore, individual compounds or M4 treatment resulted in a decrease of the mitochondrial membrane potential (ΔΨm) (Figure 6g). Similar events related to the induction of cell apoptosis were detected in H460 cells (Supplementary Figures S6 and S7). These results revealed that individual compounds or M4 induced cell apoptosis through both death receptor and mitochondrial pathways.

Cell apoptosis analysis on the human lung caner cell H460/TaxR treated with compound alone or M4 for 24 h. (a) The morphology of cells treated with 1.0 μM individual compounds or M4. (b–d) Caspase-8, caspase-9 or caspase-3/7 activity in H460/TaxR cells treated with 0.5 or 1.0 μM individual compounds or M4 for 24 h. Cells were treated for 24 h in 96-well plate, and the caspase substrates were added to each well. Samples were read using a Victor2 multilabel counter (PerkinElmer) to detect the light produced through the luciferase reaction for caspase 8 or 9 activity or to detect the fluorescence at 485 nm (excitation)/535 nm (emission) for caspase3/7 activity. (e) The percentage of cells undergoing apoptosis after treatment with 1.0 and 10.0 μM individual compounds or M4 for 24 h. Cells were double stained with Annexin V and 7-AAD, and were analyzed by flow cytometry. The values presented are the means of three independent experiments±S.D. *, **, *** the value is significantly different from that of control (P<0.05, P<0.01, P<0.001, respectively); #, ##, ###the value is significantly different from that of M4 treatment at the same concentrations (P<0.05, P<0.01, P<0.001, respectively). (f) Cells were treated with individual compounds or M4 for 24 h, and the cell lysates were immunoblotted for the expression of apoptosis-related proteins. In addition, after treatment, the cells were harvested and separated into cytosolic and mitochondrial fractions, the expression of Cyt C in the cytosol were analyzed by western blot. (g) The disruption of mitochondrial membrane potential of individual compounds or M4 was measured by flow cytometry using the Guava EasyCyte MitoPotential kit
Each compound and M4 inhibited HDAC activity
On the basis of these events induced by individual compounds or M4 in H460 and H460/TaxR cells, we next further determined the HDAC inhibitor activities of individual compounds or M4 in H460/TaxR cells. Inhibition of HDAC by individual compounds or M4 is usually indicated by histone H4 acetylation, tubulin acetylation, and Akt phosphorylation state. Western blot analysis showed hyperacetylation of α-tubulin and histone H4 after treating H460/TaxR cells with individual compounds or M4 for 24 h (Figure 7a). Such treatments resulted in a decrease in Akt activation (phosphorylation). In particular, M4 showed a prolonged inhibition of Akt signaling with no subsequent reactivation in 24 h, although Akt reactivation was observed after treatments with individual compounds.

Individual compounds and M4 mediate the protein acetylation, Akt dephosphorylation and MAPK in H460/TaxR cells. (a) Time-dependent effect of compound alone or M4 on protein acetylation, the Akt and the MAPK signaling pathway. The cells were treated with 27 (1.0 μM), 107 (2.0 μM), 167 (2.0 μM), 254 (1.0 μM) or M4 (1.0 μM) for indicated periods, and the cell lysates were immunoblotted. (b) The scheme of the signal pathway involved in the anticancer activity of compound alone or M4. (c) A working model showing how the four compounds in M4 produce synergistic anticancer effect
Previous reports have demonstrated that tubulin polymerization inhibitors and HDAC inhibitors both affect the mitogen-activated protein kinase (MAPK) signaling pathway.25, 30 We also observed the upregulation of genes that are involved in positive regulation of JUN kinase activity in microarray experiments. Therefore, we next examined the effects of individual compounds or M4 on activation of MAPK-related kinase signaling. Western blot analysis showed that individual compounds or M4 led to increases in phosphorylation of JNK, total JNK, and phosphorylation of p38 and total p38 within 24 h. In contrast, the phosphorylation of ERK1/2 and overall levels of ERK1/2 remained unaffected probably owing to the opposing effect between anti-tubulin agent and HDAC inhibitor on this protein. We also observed the activation of Erk5 within 8 h of treatment and its subsequent inactivation. These findings indicated that the activation of JNK, p38 and the inactivation of Akt had key roles in the synergic anticancer activity of M4 (Figures 7b and c).
Discussion
P-gp-independent antitumor activity of M4
Drug resistance and the dose-limiting toxicity are main road blockers in current cancer chemotherapy. Drug combination chemotherapy may generate synergistic cancer cells killing in drug-resistant cancer cells.31 Thiazolidinone compounds have been found to escape P-gp efflux in paclitaxel-resistant cancer cells.13, 25 In this work, we identified a combination M4 containing 27, 107, 167 and 254 compounds, producing synergistic inhibitory effects in human NSCLC H460 cells and drug-resistant H460/TaxR cells by screening 100 compound combinations. Rho 123 efflux assay and P-gp activity inhibition indicated individual compounds or M4 was neither the substrate nor the inhibitor of P-gp (Supplementary Figure S2 and Supplementary Table S1). Further analysis showed that individual compounds or M4 induced G2/M arrest and cell apoptosis in both H460 and H460/TaxR cells. M4 treatment resulted in an increased antitumor activity in H460/TaxR xenograft without systemic toxicity in vivo (Figure 2). In contrast, even using a very low-dose, paclitaxel still showed evident side effects (Figure 2).
Microtubule depolymerization and the inhibition of HDAC account for the synergistic anticancer activity of M4
In order to reveal the mechanism of the synergism, we investigated molecular basis underlying the observed synergistic anticancer activity of M4. The microarray analysis showed that genes involved in negative regulation of microtubule assembly, cytoskeleton organization, positive regulation of JUN kinase activity and histone acetylation were upregulated by M4 (Figure 3), indicating that M4 might act as a multi-targeting inhibitor combination. Consistently, we observed the regulation of cell cycle checkpoint proteins, including p53, p21, cdc25C (Ser216) and cdc2 (Tyr15), the hyperacetylation of histone H4 and nonhistone α-tubulin, the dephosphorylation of Akt, the activation of caspases, the release of Cyto C from the mitochondria and the decrease of ΔΨm (Figure 6 and Supplementary Figure S6). These results demonstrated that by targeting different cellular targets, compounds in M4 acted synergistically to inhibit cancer cell growth and tumor growth in mice.
In addition, we observed the induction of p53 target genes resulting from the microtubule disruption and DNA damage.32 It is noteworthy that the expression of p53 target genes including AEN, BOK, E2F7 and PHLDA3 are differently induced with individual compounds or M4 treatment (Supplementary Figure S4). Among these p53 target genes that have been regulated, AEN, BOK and E2F7 are all essential mediators of p53-dependent apoptosis induced by DNA damage.33, 34, 35 PHLDA3 encodes a PH domain-only protein, which is a p53-regulated repressor of Akt.36 These results indicated that p53 activation has key roles in the synergistic anticancer effects between HDAC inhibitor and microtubule-depolymerizing agents as reported previously.37
The synergistic anticancer activity of HDAC inhibitors in combination with conventional tubulin inhibitors
In the past years, HDAC inhibitors have been developed as a promising new class of drugs for cancers. Some compounds exhibit strong antitumor activities in vivo with low toxicity in preclinical studies.38 The proposed anticancer mechanisms of HDAC inhibitors includes the activation of death receptor and/or the mitochondrial pathway of apoptosis, the generation of reactive oxygen species (ROS), the inhibition of angiogenesis, the induction of autophagy,39, 40 and DNA damage.41
It has been demonstrated that the histone hyperacetylation induced by HDAC inhibitors causes structural alterations in chromatin, which may expose portions of DNA that are normally protected by heterochromatin to DNA-damaging agents, such as UV, cytotoxic drugs or ROS.41, 42 In addition, HDAC inhibitors regulate protein function through acetylating nonhistone proteins such as tubulin and Hsp90.43, 44 Based on this, the combination of HDAC inhibitors with conventional anticancer drugs achieved synergy in cancer treatment.45, 46, 47, 48, 49, 50, 51 Since acetylation of tubulin by HDAC stabilizes the microtubule, preclinical studies demonstrated that HDAC inhibitors combined with the microtubule stabilizing agents produced synergistic anticancer effects. However, little is known on how HDAC inhibitors and microtubule-depolymerizing agents act to achieve synergy.37, 46
Here, for the first time, we demonstrate that a combination of tubulin polymerization inhibitors and HDAC inhibitors achieve a significant enhancement in anticancer activity in vitro and in vivo. We have also improved our understanding on how the anticancer activities of individual compounds help to reach the synergistic anticancer activity of M4. In conclusion, we propose that: (1) screening compound combinations is an efficient strategy to discover new anticancer drug combinations; (2) combination of HDAC inhibitors with tubulin polymerization inhibitors is an effective method for cancer therapy.
Materials and Methods
Reagents
The thiazolidinone derivatives used for compound combination screening in this study were reported previously.13 All compounds were dissolved in DMSO and stored at 4 °C as stocks. Each compound was diluted before use. Compound combinations were prepared by mixing the four compounds with equal molar ratio. RPMI 1640, fetal bovine serum, penicillin, streptomycin, and all other tissue culture reagents were obtained from Life Technologies (Grand island, NY, USA). The Guava Nexin Assay reagent and Guava EasyCyte MitoPotential kit were obtained from Millipore (Billerica, MA, USA). Caspase-Glo 8 assay kit, Caspase-Glo 9 assay kit and Apo-ONE Homogeneous Caspase-3/7 Assay Kit were from Promega (Promega Biotech CO., Ltd, Madison, WI, USA). The Mitochondria isolation kit was obtained from Pierce Biotechnology (Rockford, IL, USA); Sulforhodamine B (SRB) and other chemicals reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The paclitaxel-sensitive lung cancer cell H460 and P-gp-overexpressing paclitaxel-resistant cell H460/TaxR were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 mg/ml streptomycin, as previously reported.27 NHFBs were maintained in Dulbecco’s modified Eagle’s medium with the same supplements. All cells were maintained in the presence of 5% CO2 at 37 °C.
Cytotoxicity assay
The inhibitory effects of thiazolidinone combinations on cell growth were determined by the SRB assay.52 In brief, cells were seeded in 96-well flat-bottom plates in a volume of 100 μl at 4 × 103 cells/well and incubated for 24 h at 37 °C. The cells were treated with compound combinations on the next day at the concentrations of 1.0 μM for 72 h for the first round screening. Compound combinations showing obvious inhibition on H460/TaxR cells is selected for the second round screening. In the second round screening, both H460 and H460/TaxR cells were treated with the combinations at different concentrations for 72 h. Cell growth was calculated using the following formula: percentage growth inhibition=100−(mean OD sample−mean OD on day 0)/(mean OD neg control−mean OD on day 0) × 100. Final concentration of DMSO in treated cells was <0.1%.
Analysis of combined effects of the four compounds in M4
To assess the combined effect of the four compounds in the selected combination (M4) containing 27, 107, 167 and 254, both H460 and H460/TaxR cells were treated with M4 or compound alone at doses that corresponded to 0.2, 0.4, 0.8, 0.9, 1.0, 1.2, and 1.5 times GI50 values for 72 h and measured for growth inhibition by SRB assay. Cell growth was calculated using the following formula: percentage fractional affected (fa)=100−(mean OD sample−mean OD on day 0)/(mean OD neg control−mean OD on day 0) × 100. Each experiment was performed at least three times. In order to provide more insight into the interactions of the four components in M4, the cancer cell growth-inhibitory activities of two-compound combinations and three-compound combinations were also carried out at the same time. The median effect analysis developed by T-C Chou was used to identify the interaction between the four compounds in their anticancer activity.26, 53 Data were expressed as CIs, which offers definition for addictive (CI=1), synergism (CI<1) and antagonism (CI>1) in drug combination.54 The CI values were calculated based on the equation stated below. (D)1/(Dx)1+(D)2/(Dx)2+(D)3/(Dx)3+(D)4/(Dx)4=CI (Dx)1 is the dose of compound 1 alone that inhibits x%. Likewise, (Dx)2, (Dx)3, (Dx)4 are the doses of compound 2, 3, 4 alone that inhibits x%. (D)1 is the portion of compound 1 in combination also inhibits x%. Again, (D)2, (D)3, (D)4 is the portion of compound 2, 3, 4 in combination inhibits x%.
Rho 123 accumulation assay
To evaluate if individual compounds or M4 inhibits the P-gp function of H460/TaxR cells, 2 × 104 cells/well were seeded into a 24-well plate. After the cell grew up to 70% percent, 2.5 μM Rho 123, together with different compounds (DMSO, Reversan, compounds 27, 107, 167, 254, M4) at 2.5 μM were added and incubated for 4 h. Then the cell were collected, washed and resuspended in 100 μl phosphate-buffered saline (PBS). The Rho 123 accumulation in cells was determined by the Guava EasyCyt flow cytometer system.
Human lung tumor xenograft experiment
Five-week-old female athymic nude mice were purchased from Vital River Research Animal Services (Beijing, China) and housed under pathogen-free conditions. This study was carried out following the recommendations from Guide for the Care and Use of laboratory Animals by the National Institutes of Health. The protocol was approved by the Committee on the Ethics on Animal Experimentation of Shandong University. H460/TaxR cells (5 × 106 cells in 0.1 ml PBS) were injected subcutaneously into the dorsal flank of each mouse. When the tumors reached volumes of ∼80 mm3, the mice showing tumors were randomly assigned to 11 groups (n=6). The mice were treated every day for 11 days with subcutaneous administration of (1) 100 μl of solvent (PBS/cremophor/DMSO/ethanol, 7.5:1.5:0.5:0.5); (2) compound 27 at low dose (27L, 10 mg/kg/day); (3) compound 27 at high dose (27H, 42.2 mg/kg/day); (4) compound 107 at low dose (107L, 10.5 mg/kg/day); (5) compound 107 at high dose (107H, 42.2 mg/kg/day); (6) compound 167 at low dose (167L, 11.6 mg/kg/day); (7) compound 167 at high dose (167H, 42.2 mg/kg/day); (8) compound 254 at low dose (254L, 10.1 mg/kg/ day); (9) compound 254 at high dose (254H, 42.2 mg/kg/day); (10) M4 (42.2 mg/kg/day) containing the four compounds with equal molar ratio, that is, 27 (10 mg/kg)+107 (10.5 mg/kg)+167 (11.6 mg/kg)+254 (10.1 mg/kg). Another group was treated with standard anticancer agent paclitaxel (20 mg/kg/day) every other day for three times as positive control. To check the toxicity of these treatments, the animals were weighed every other day. The tumor dimensions were measured with a linear caliper every other day. Tumor volume was determined by the formula L × W2 × 0.5, in which L is the largest diameter of tumor and W is the smallest diameter of the tumor. The experiment was terminated and the mice were sacrificed when the tumors in solvent-treated group grew to 1200 mm3. The mice organs and blood samples were collected and used for hematoxylin–eosin staining and serum chemistry assay to monitor the side effect of the treatment, respectively.
Genome-wide transcriptional profiling with agilent microarray
Gene array was carried out to determine the genes that may have been regulated and involved in the anticancer activity of M4. Briefly, cells were seeded in 100 mm dishes (1.5 × 106/dish) and incubated for 24 h at 37 °C. Cells were treated with vehicle or 1.0 μM M4 for 4 h. After treatment, the cells were washed three times with PBS and suspended in 1.0 ml Trizol reagent (Life Technologies, Inc., Carlsbad, CA, USA). The suspended cells were frozen at −80 °C. For microarray analysis, cells in Trizol were shipped on dry ice to KangChen Bio-Tech (Shanghai, China) for analysis via Agilent Whole Human genome Oligo Microarray platform. The RNA preparation and microarray hybridization were performed according to the manufacturer’s instructions. Briefly, total RNA from each sample was amplified and transcribed into fluorescent cRNA using the manufacturer’s instructions (Agilent’s Quick Amp Labeling protocol, version 5.7, Agilent Technologies). The labeled cRNAs were hybridized onto the Whole Human Genome Oligo Microarray (4 × 44 K, Agilent Technologies). After having washed the slides, the arrays were scanned by the Agilent Scanner G2505C. Agilent Feature Extraction software (version 11.0.1.1) was used to analyze the acquired array images. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v11.5.1 software package (Agilent Technologies). After quantile normalization of the raw data, at least 1 out of 4 genes samples have flags in detected (‘all targets value’) were chosen for further data analysis. Differentially expressed genes were identified through fold change filtering. Pathway analysis and GO analysis were applied to determine the roles of these differentially expressed genes had in these biological pathways or GO terms. Finally, hierarchical clustering was performed to show the distinguishable gene expression profiling between samples.
Reverse transcription (RT)–PCR
Cells were treated with medium containing 0.1% DMSO, 1.0 μM 27, 1.0 μM 107, 1.0 μM 167, 1.0 μM 254 or 1.0 μM M4 for 4 h. Total RNA was extracted using Trizol reagent (Life Technologies, Inc.) according to the manufacturer’s instructions. cDNA synthesis was performed with the RT reagent kit obtained from TaKaRa (Dalian, China) according to the manufacturer’s protocol and amplified by PCR. The following primers were obtained from BioAsia (Shanghai, China) and used for the respective genes: AEN: 5′-CGCAAGGCTGTCCCCTTCCA-3′ and 5′-CTGCGGGCCTCCTGCTGTTC-3′; AXIN1: 5′-ACCGAAAGTACATTCTTGATAAC-3′ and 5′-TCCATACCTGAACTCTCTGC-3′; BOK: 5′-TGTGCCTATTAGGGCTCCGT-3′, and 5′-GCTCCAGGAAAGATGCCTCA-3′; E2F7: 5′ GGAAAGGCAACAGCAAACTCT 3′, and 5′-GGGACACTGAAAGACGGCA-3′ PHLDA3: 5′-CGTTGTTCCCTCTGGCTTGT-3′, and 5′-CTGATTCCTGAGGCGTTGGC-3′. Glyceraldehyde 3-phosphate dehydrogenase cDNA was amplified using the primers from Qiangen (QT01192646) as a control. PCR products were resolved on a 1.2% agarose gel and DNA bands were visualized by ethidium bromide staining. The experiments were repeated at least three times.
Tubulin polymerization assay
Tubulin polymerization was determined with the polymerization assay kit (fluorescence based) according to the manufacturer’s instructions (Cytoskeleton, Inc., Denver, CO, USA). The assay was performed in a black 96-well plates. Each well of the plate contained 5.0 μl control buffer (containing 0.1% DMSO) or 5 μM test compounds (containing 0.1% DMSO) was pre-warmed at 37 °C for 1 min, then 50 μl tubulin solution was pipetted into each well rapidly, and the polymerization dynamics of tubulin was monitored through measuring the change of fluorescence using an excitation wavelength of 355 nm and an emission wavelength of 460 nm every 2 min for 42 min at 37 °C by 1420 Multi-label Counter (PerkinElmer, Boston, MA, USA).
Quantitative assessment of soluble and polymeric tubulins in live cells
Cells were plated in six-well dishes, and allowed to attach for 24 h. The cells were treated with the compound alone or M4 at various concentrations for 24 h, paclitaxel (100 nM), VCR (100 nM) and SAHA (10 μM) were used to treat cells as positive controls. The soluble and polymeric fractions of cell tubulin was prepared according to the protocol reported previously.55 Western blot analysis was performed using rabbit monoclonal anti-α-tubulin antibody (1 : 1000). Intensity of the blot was determined using the Image J software (NIH, Bethesda, MD, USA).
Cell cycle analysis
Cells were seeded in six-well plate at a concentration of 2 × 105 cells/well. Compound alone or M4 at 1.0 μM was added to each well on the next day for 12 h. Paclitaxel (100 nM), VCR (100 nM) and SAHA (10 μM) were used to treat cells as positive controls. To analyze the intracellular DNA content, cells were harvested, washed twice in PBS, and fixed in 70% ethanol at −20 °C overnight. The cells were then suspended in PBS containing 50 μg/ml propidium iodide (Sigma-Aldrich) and incubated at 4 °C for at least 3 h. Cell-cycle analysis was performed using the flow cytometer (FACS Calibur, BD, San Jose, CA, USA).
Caspases activity assay
Caspase-Glo 8 assay kit, Caspase-Glo 9 assay kit and Apo-ONE homogeneous Caspase-3/7 assay kit were obtained from Promega Biotech (Madison, WI, USA ) and used to analyze the caspase 8, caspase 9 and caspase 3/7 activities, respectively. 1.2 × 104 cells per well were plated in 96-well tissue culture plates and incubated overnight. Cells were then treated with 0.5 or 1.0 μM compound alone or M4 for 24 h. The caspase substrates were added to each well. Contents were mixed by gentle shaking at 300 × g for 2 min, and the plates were incubated at room temperature in the dark for 4 h. Samples were read using a Victor2 multilabel counter (PerkinElmer) to detect the light produced through the luciferase reaction for caspase 8 or 9 activity or to detect the fluorescence at 485 nm (excitation)/535 nm (emission) for caspase3/7 activity. The experiments were repeated three times.
Cell apoptosis assay
The percentage of cells undergoing apoptosis was determined using the Guava Nexin assay (Millipore). Briefly, cells were seeded in six-well plate at a concentration of 2 × 105 cells/well. 1.0 μM or 10 μM compound alone or M4 was added to each well the next day. Paclitaxel (100 nM), VCR (100 nM) and SAHA (10 μM) were used to treat cells as positive controls. After 24 h of treatment, cells were collected (keeping all floating cells) and double stained with Annexin V/7-AAD according to the manufacturer’s instructions (Guava Nexin method). Cells were analyzed by the Guava easyCyt flow cytometer system. All experiments were repeated three times.
Mitochondrion membrane potential (ΔΨm) analysis
Changes of mitochondrion membrane potential in H460 and H460/TaxR cells were measured using flow cytometry (Guava EasyCyte 5, Millipore) after the 24 h treatment. Briefly, cells were cultured in six-well plates and treated with 27 (1.0 μM), 107 (2.0 μM), 167 (2.0 μM), 254 (1.0 μM) or M4 (1.0 μM), respectively, for 24 h. Then, cells were detached by cell dispersal reagent (Guava ViaCount CDRTM, Millipore), and collected by centrifugation for 5 min, 300 × g. The pellets were resuspended in 1.0 ml cell culture medium, then, 4 μl of 50 × JC-1 staining solution was added to 200 μl cells. The cells were incubated in the incubator (37 °C, 5% CO2) for 30 min and measured immediately using flow cytometry. As the positive control, cells were treated with 50 μM CCCP (carbonyl cyanide 3-chlorophenylhydrazone), mitochondrial membrane disrupter, for 30 min at 37 °C before harvesting cells.
Western blot analysis
Western blot analysis was performed with primary antibodies against Akt, phospho-Akt (Ser473), SAPK/JNK, phospho-SAPK/JNK (p-JNK, Thr183/Tyr185), p38, phospho-p38 (p-p38, Thr180/Tyr182), p42 MAP Kinase, phospho-p42/44 (p-p42/44, Thr202/Tyr204), phospho-Erk5 (Thr218/Tyr220), α-Tubulin, p21 Waf1/Cip1, p53, cyclin B1, phospho-cdc2 (Tyr15), cdc2, phosphor-cdc25C (Ser216), cdc25C, Cytochrome C, Bcl-XL, and pro-apoptosis Bcl-2 family antibody kit from Cell Signaling Technology (Beverly, MA, USA); acetyl-tubulin (Ac-Tubulin, T7451) and β-actin from SIGMA; and acetyl-histone H4(Lys16) (Ac-H4) from ImmunoWay Biotechnology Company (Newark, DE, USA). Cells were seeded in 100 mm dishes (1.2 × 106/ dish) and incubated for 24 h at 37 °C. Cells were treated with vehicle, 27 (1.0 μM), 107 (2.0 μM), 167 (2.0 μM), 254 (1.0 μM) or M4 (1.0 μM), respectively, for indicated periods. After treatment, cells were lysed in the extraction buffer (FNN0011, Life Technologies, Inc.) supplemented with protease inhibitor (P2714, Sigma) and 1 mM PMSF. Protein concentrations were determined using a bicinchoninic acid protein assay kit (Bio-Rad Laboratories, Richmond, CA, USA). Equal amounts (15–30 μg) of proteins were subjected to electrophoresis on 10% (w/v) polyacrylamide gels and then electrophoretically transferred to polyvinylidene difluoride membranes (Millipore). The membranes were incubated with primary antibody followed by peroxidase-linked secondary antibody. An enhanced chemiluminescence western blotting system (Millipore) was used to detect the immunoreactive bands. Intensity of the blot was determined using the Image J software.
Statistical analysis
The data were analyzed by the computer software Compusyn (ComboSyn, Inc., NJ, USA) or the SigmaPlot 10.0 program (Systat Software, Inc., San Jose, CA, USA). Student’s t-test was used to determine the statistical significance between control and test groups. P<0.05 was regarded as significant difference.
Abbreviations
- AIDS:
-
acquired immunodeficiency syndrome
- BCA:
-
bicinchoninic acid
- CI:
-
combination index
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- DMSO:
-
dimethyl sulfoxide
- FBS:
-
fetal bovine serum
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- GI50:
-
the concentration of drug to cause 50% inhibition of growth
- HDAC:
-
histone deacetylase
- MAPK:
-
mitogen-activated protein kinase
- MDR:
-
multidrug resistance
- NHFBs:
-
Normal human fibroblasts
- NSCLC:
-
non-small cell lung cancer
- PBS:
-
phosphate-buffered saline
- PVDF:
-
polyvinylidene difluoride
- Rho 123:
-
rhodamine 123
- ROS:
-
reactive oxygen species
References
Scagliotti G, Novello S, Selvaggi G . Multidrug resistance in non-small-cell lung cancer. Ann Oncol 1999; 10: S83–S86.
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001; 344: 1031–1037.
Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13306–13311.
Sams-Dodd F . Target-based drug discovery: is something wrong? Drug Discov Today 2005; 10: 139–147.
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73.
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 2007; 104: 20932–20937.
Teodori E, Dei S, Martelli C, Scapecchi S, Gualtieri F . The functions and structure of ABC transporters: implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr Drug Targets 2006; 7: 893.
Kartner N, Riordan JR, Ling V . Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science 1983; 221: 1285–1288.
Gottesman MM, Fojo T, Bates SE . Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002; 2: 48–58.
Georges E, Sharom J . Multidrug Resistance and Chemosensitization: Therapeutic Implications for. Adv Pharmacol 1990; 21: 185.
Ford J . Experimental reversal of P-glycoprotein-mediated multidrug resistance by pharmacological chemosensitisers. Eur J Cancer 1996; 32: 991.
van Zuylen L, Nooter K, Sparreboom A, Verweij J . Development of multidrug-resistance convertors: sense or nonsense? Invest New Drugs 2000; 18: 205–220.
Zhou H, Wu S, Zhai S, Liu A, Sun Y, Li R et al. Design, synthesis, cytoselective toxicity, structure–activity relationships, and pharmacophore of thiazolidinone derivatives targeting drug-resistant lung cancer cells. J Med Chem 2008; 51: 1242–1251.
Borisy AA, Elliott PJ, Hurst NW, Lee MS, Lehár J, Price ER et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA 2003; 100: 7977–7982.
Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK . Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol 2006; 2: 458–466.
Mayer LD, Harasym TO, Tardi PG, Harasym NL, Shew CR, Johnstone SA et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther 2006; 5: 1854–1863.
Hopkins AL . Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 2008; 4: 682–690.
Lehár J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 2009; 27: 659–666.
DiDiodato G, Sharom FJ . Interaction of combinations of drugs, chemosensitizers, and peptides with the P-glycoprotein multidrug transporter. Biochem Pharmacol 1997; 53: 1789–1797.
Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA Jr, Chou TC . Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res 1999; 59: 6178–6184.
Dent P, Curiel DT, Fisher PB, Grant S . Synergistic combinations of signaling pathway inhibitors: mechanisms for improved cancer therapy. Drug Resist Updat 2009; 12: 65–73.
Raben D, Helfrich B, Chan DC, Ciardiello F, Zhao LM, Franklin W et al. The effects of cetuximab alone and in combination with radiation and/or chemotherapy in lung cancer. Clin Cancer Res 2005; 11: 795–805.
Chou TC . Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58: 621–681.
Regales L, Gong Y, Shen R, De Stanchina E, Vivanco I, Goel A et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 2009; 119: 3000.
Teraishi F, Wu S, Sasaki J, Zhang L, Zhu H-B, Davis JJ et al. P-glycoprotein-independent apoptosis induction by a novel synthetic compound, MMPT [5-[(4-methylphenyl) methylene]-2-(phenylamino)-4 (5H)-thiazolone]. J Pharmacol Exp Ther 2005; 314: 355–362.
Chou TC, Talalay P . Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55.
Teraishi F, Wu S, Sasaki J, Zhang L, Davis J, Guo W et al. JNK1-dependent antimitotic activity of thiazolidin compounds in human non-small-cell lung and colon cancer cells. Cell Mol Life Sci 2005; 62: 2382–2389.
Li L, Zhang Q, Liu A, Li X, Zhou H, Liu Y et al. Proteome interrogation using nanoprobes to identify targets of a cancer-killing molecule. J Am Chem Soc 2011; 133: 6886–6889.
Zhang Q, Zhai S, Li L, Li X, Zhou H, Liu A et al. Antitumor selectivity of a novel Tubulin and HSP90 dual-targeting inhibitor in non-small cell lung cancer models. Biochem Pharmacol 2013; 86: 351–360.
Chen C-S, Weng S-C, Tseng P-H, Lin H-P, Chen C-S . Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem 2005; 280: 38879–38887.
Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V . Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm 2012; 10: 337–352.
Stewart ZA, Tang LJ, Pietenpol JA . Increased p53 phosphorylation after microtubule disruption is mediated in a microtubule inhibitor-and cell-specific manner. Oncogene 2001; 20: 113–124.
Kawase T, Ichikawa H, Ohta T, Nozaki N, Tashiro F, Ohki R et al. p53 target gene AEN is a nuclear exonuclease required for p53-dependent apoptosis. Oncogene 2008; 27: 3797–3810.
Yakovlev AG, Di Giovanni S, Wang G, Liu W, Stoica B, Faden AI . BOK and NOXA are essential mediators of p53-dependent apoptosis. J Biol Chem 2004; 279: 28367–28374.
Carvajal LA, Hamard P-J, Tonnessen C, Manfredi JJ . E2F7, a novel target, is upregulated by p53 and mediates DNA damage-dependent transcriptional repression. Genes Dev 2012; 26: 1533–1545.
Kawase T, Ohki R, Shibata T, Tsutsumi S, Kamimura N, Inazawa J et al. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell 2009; 136: 535–550.
Zuco V, De Cesare M, Cincinelli R, Nannei R, Pisano C, Zaffaroni N et al. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLoS One 2011; 6: e29085.
Dokmanovic M, Clarke C, Marks PA . Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 2007; 5: 981–989.
Rosato RR, Almenara JA, Dai Y, Grant S . Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol Cancer Ther 2003; 2: 1273–1284.
Carew JS, Giles FJ, Nawrocki ST . Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett 2008; 269: 7–17.
Lee J-H, Choy M, Ngo L, Foster S, Marks PA . Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci USA 2010; 107: 14639–14644.
Xu W, Parmigiani R, Marks P . Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26: 5541–5552.
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A et al. HDAC6 is a microtubule-associated deacetylase. Nature 2002; 417: 455–458.
Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90 a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 2005; 280: 26729–26734.
Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F . Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res 2003; 63: 7291–7300.
Dowdy SC, Jiang S, Zhou XC, Hou X, Jin F, Podratz KC et al. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol Cancer Ther 2006; 5: 2767–2776.
Arnold NB, Arkus N, Gunn J, Korc M . The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clin Cancer Res 2007; 13: 18–26.
Jane EP, Premkumar DR, Addo-Yobo SO, Pollack IF . Abrogation of mitogen-activated protein kinase and Akt signaling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human glioma cells. J Pharmacol Exp Ther 2009; 331: 327–337.
Yu C, Friday BB, Lai J-P, McCollum A, Atadja P, Roberts LR et al. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res 2007; 13: 1140–1148.
Rahmani M, Reese E, Dai Y, Bauer C, Payne SG, Dent P et al. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res 2005; 65: 2422–2432.
Giommarelli C, Zuco V, Favini E, Pisano C, Dal Piaz F, De Tommasi N et al. The enhancement of antiproliferative and proapoptotic activity of HDAC inhibitors by curcumin is mediated by Hsp90 inhibition. Cell Mol Life Sci 2010; 67: 995–1004.
Vichai V, Kirtikara K . Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006; 1: 1112–1116.
Chou TC, Motzer RJ, Tong Y, Bosl GJ . Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to clinical protocol design. J Natl Cancer Inst 1994; 86: 1517–1524.
Chou TC . Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70: 440–446.
Cassimeris LU, Wadsworth P, Salmon E . Dynamics of microtubule depolymerization in monocytes. J Cell Biol 1986; 102: 2023–2032.
Acknowledgements
This work is supported by Grants from the National Natural Science Foundation of China (20902054, 90913006 and 21307077), the Independent Innovation Foundation of Shandong University (2010TS093).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhang, C., Zhai, S., Li, X. et al. Synergistic action by multi-targeting compounds produces a potent compound combination for human NSCLC both in vitro and in vivo. Cell Death Dis 5, e1138 (2014). https://doi.org/10.1038/cddis.2014.76
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.76
