
Abstract
Peroxisome proliferator-activated receptor α (PPARα) has been reported to induce a potent anti-inflammatory response. Autophagy is a recently recognized rudimentary cellular response to inflammation and injury. The aim of the present study was to test the hypothesis that PPARα activation mediates autophagy to inhibit liver inflammation and protect against acute liver failure (ALF). PPARα expression during ALF and the impact of PPARα activation by Wy-14 643 on the hepatic immune response were studied in a D-galactosamine/lipopolysaccharide-induced mouse model. Autophagy was inhibited by 3-methyladenine or small interfering RNA (siRNA) against Atg7. In both the mouse model and human ALF subjects, PPARα was significantly downregulated in the injured liver. PPARα activation by pretreatment with Wy-14 643 protected against liver injury in mice. The protective effect of PPARα activation relied on the suppression of inflammatory mechanisms through the induction of autophagy. This hypothesis is supported by the following evidence: first, PPARα activation suppressed proinflammatory responses and inhibited phosphorylated NF-κBp65, phosphorylated JNK and phosphorylated ERK pathways in vivo. Second, protection by PPARα activation was due to the induction of autophagy because inhibition of autophagy by 3-methyladenine or Atg7 siRNA reversed liver protection and inflammation. Third, PPARα activation directly induced autophagy in primary macrophages in vitro, which protected cells from a lipopolysaccharide-induced proinflammatory response. Here, for the first time, we have demonstrated that PPARα-mediated induction of autophagy ameliorated liver injury in cases of ALF by attenuating inflammatory responses, indicating a potential therapeutic application for ALF treatment.
Similar content being viewed by others


Main
Acute liver failure (ALF), an inflammation-mediated hepatocellular injury process, is a clinical syndrome that results from hepatocellular apoptosis and hemorrhagic necrosis.1 ALF frequently results from viral hepatitis, ingestion of drugs or toxic substances, or hepatic ischemia–reperfusion injury, among others. The prognosis for ALF is extremely poor, and there is currently no effective therapy for the end stage of the disease other than liver transplantation.2 Although the nature of ALF has been extensively studied, the mechanisms by which organ damage occurs are not completely understood.
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily of ligand-activated transcription factors, with its subfamily consisting of three members: PPARα, PPARβ and PPARγ.3, 4 PPARα has been reported to be involved in a number of cellular processes, including lipid and lipoprotein metabolism,5 apoptosis6 and inflammatory responses.7, 8, 9 Studies have demonstrated that PPARα exhibits potent anti-inflammatory activity through suppressing nuclear factor-κB (NF-κB) and/or modulating the activation (phosphorylation) of signal transducer and activator of transcription1 (STAT1)-related inflammatory signaling in cultured neuronal cells.7, 10 PPARα-null mice exhibit an aggravated reaction to various inflammatory stimuli in the skin, blood vessels, intestine and lung.11, 12, 13, 14 Recently, in a model of lipopolysaccharide (LPS)-induced hepatic inflammatory injury, reduced PPARα expression was shown to be associated with increased tissue bacterial load in sepsis.15 Additionally, a lack of PPARα exacerbates LPS-induced liver toxicity through STAT1 inflammatory signaling and increases oxidative/nitrosative stress.16 However, the functional role of PPARα in the pathogenesis of ALF remains elusive. An ALF model induced by the coinjection of D-galactosamine (D-GalN) and LPS has been widely used to examine the underlying mechanisms of ALF.17, 18 In the present study, we used D-GalN/LPS to induce ALF in mice and to explore the roles of PPARα in the context of ALF.
Macroautophagy (referred to hereafter as autophagy) is a highly evolutionarily conserved process found in virtually all types of eukaryotic cells. Autophagy involves the sequestration of regions of cytosol within double-membrane-bound compartments followed by lysosome-based degradation of the contents. Previous studies have suggested that autophagy represents an adaptive strategy by which cells can remove damaged organelles and enhance survival following bioenergetics-induced stress.19, 20, 21 In addition, accumulating evidence has demonstrated multiple roles of autophagy in the regulation of cell death, differentiation and the anti-microbial response in mammals.20, 21 In recent years, emerging evidence has indicated that the autophagy process may have an essential role for the host during bacterial clearance and may also interact with inflammatory processes, which consequently may impact the outcomes of disease progression.22, 23 There is a complex reciprocal relationship between autophagy pathway/proteins and inflammation.24, 25 Recent observations have revealed a relationship between autophagy and inflammasome-associated proinflammatory cytokine maturation in macrophages.26, 27
Given the above information, we speculated that PPARα activation may serve as a protective function to restrain liver inflammation in cases of ALF. Moreover, we further hypothesized that PPARα activation attenuates inflammatory response by regulating autophagy in ALF. In the present study, PPARα activation protected mice from D-GalN/LPS-induced ALF and significantly downregulated the expression of proinflammatory cytokines. Moreover, PPARα activation resulted in elevated autophagy induced by D-GalN/LPS in mice, and autophagy inhibition by 3-methyladenine (3-MA) or Atg7 siRNA reversed the hepatoprotective effect of PPARα activation and restored the inflammatory response in ALF mice. Hence, by orchestrating autophagy signaling, PPARα is essential for the inflammation mechanism in the ALF immune response cascade.
Results
Intrahepatic expression of PPARα is suppressed in D-GalN/LPS-induced ALF
According to the pathologic characteristics of liver and the levels of serum transaminase, massive hepatic injury was apparent after 4–6 h as determined by hematoxylin and eosin (H&E) staining (Figure 1a), which was in agreement with the increased serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) enzyme levels (Figure 1b); thus, the mice ALF model is successfully formatted at 6 h after D-GalN/LPS injection. Next, we investigated a possible association between PPARα and ALF induced by D-GalN/LPS treatment. Accompanying the liver injury, PPARα mRNA and protein levels gradually declined over the same time period (Figure 1c and Suplementary Figure 1). These results indicated that PPARα is suppressed with the gradual progression of D-GalN/LPS-induced ALF.

PPARα expression is suppressed during ALF progression. Mice were intraperitoneally injected with D-GalN (700 mg/kg) and LPS (10 μg/kg) at 2, 4 and 6 h (10 mice per group). The mice in the control group (n=8) were injected with PBS only. (a) Representative livers and H&E staining of livers from the control and the 2-, 4- and 6-h groups. (b) Serum AST and ALT enzyme levels from the control and the 2-, 4- and 6-h groups. (c) Gene expression of PPARα was measured by qRT-PCR in the livers of the control and the 2-, 4- and 6-h groups. The average target gene/HPRT ratios for each experimental group were plotted. (d) Protein expression levels of PPARα were measured by western blot assays in the livers of the control and the 2-, 4- and 6-h groups. A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample
PPARα activation protects against D-GalN/LPS-induced ALF, which is associated with suppressed hepatic inflammation
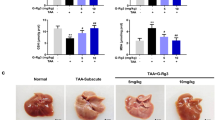
We then evaluated whether PPARα activation could rescue the injury by applying Wy-14 643, a PPARα ligand activator. Pretreatment with Wy-14 643 for 2 h before D-GalN/LPS treatment resulted in complete protection against ALF. In the survival analysis, the mice in the D-GalN/LPS control group began to perish 6 h after D-GalN/LPS injection, and the survival rate of these mice was 30% (3 of 10 mice) at the 48-h time point. By contrast, the survival rate after Wy-14 643 treatment was 70% (7 of 10 mice; Figure 2a). The gross morphology of the liver after Wy-14 643 treatment appeared substantially normal, and the liver architecture was well preserved (Figure 2b). With respect to liver damage, the mice subjected to Wy-14 643 treatment showed significantly lower sALT and sAST levels compared with the ALF group (Figure 2c). These results demonstrated that PPARα is critical for D-GalN/LPS-induced ALF and that its activation protects mice from liver injury.

Wy-14 643 protects against D-GalN/LPS-induced liver injury and suppresses liver inflammation. Wy/D-GalN/LPS-treated mice were administered Wy-14 643 (6 mg/kg) via tail vein injection 2 h before D-GalN/LPS exposure (n=10); D-GalN/LPS-treated mice were pretreated with vehicle (dimethylsulfoxide (DMSO)) 2 h before D-GalN/LPS exposure (n=10). Control mice were pretreated with vehicle (DMSO) 2 h before PBS injection (n=8). The mice were killed 6 h after D-GalN/LPS treatment, and the liver and serum samples were collected (apart from a). (a) The survival rate of mice was measured in the Wy/D-GalN/LPS-treated group and the D-GalN/LPS-treated group (10 mice per group). (b) Representative livers and H&E staining of livers from different groups. (c) Serum AST and ALT enzyme levels from different groups. (d) Gene expression of TNF-α, IL-1β, IL-6, CCL-1, CCL-2, CXCL-1 and CXCL-10 at 6 h after D-GalN/LPS injection and IL-12p40 at 2 h after D-GalN/LPS injection (n=8). The average target gene/HPRT ratios for each experimental group were plotted. (e) Liver MPO levels 6 h after D-GalN/LPS injection. (f) BMDMs were stimulated with LPS for 24 h in the absence or presence of Wy-14 643, and then the BMDM-conditioned media were collected. The primary hepatocytes stimulated with actinomycin D in the absence or presence of BMDM-conditioned media for 12 h. The MTT assay and LDH assays were measured. (g) The levels of phosphorylated MAP kinases, including JNK, ERK, p38 and phosphorylated NF-κBp65, phosphorylated Akt and β-actin, were measured by western blotting. A representative blot from three samples of every group is shown. Densitometry analysis of the proteins was performed for each sample
To determine the impact of PPARα activation on the induction of inflammatory cytokines by D-GalN/LPS-treated ALF, the livers were harvested 6 h after D-GalN/LPS injection. Indeed, PPARα activation by Wy-14 643 attenuated the expression of proinflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6 and IL-12p40. Furthermore, we found that PPARα activation suppressed the expression of several chemokines, including chemokine (C–C motif) ligand 1 (CCL-1), CCL-2, chemokine (C–X–C motif) ligand-1 (CXCL-1) and CXCL-10, which are important for the recruitment of neutrophils (Figure 2d). We also assayed myeloperoxidase (MPO) activities to measure neutrophil infiltration. As shown in Figure 2e, treatment with Wy-14 643 significantly decreased MPO activity-related injury compared with the D-GalN/LPS insult. Because hepatocyte death is the major cause of ALF, we next measured the impact of the conditioned medium from bone marrow-derived macrophages (BMDMs) stimulated with LPS in the absence or presence of Wy-14 643 on hepatocyte death in vitro, with actinomycin D used to mimic the effect of D-GalN. The results showed that conditioned medium of BMDMs stimulated with LPS in the presence of Wy-14 643 led to an increased number of live hepatocytes and lower LDH levels (Figure 2f). These results suggest that the protection of liver injury by PPARα activation is mediated by reducing inflammation and neutrophil infiltration in the liver in cases of ALF.
NF-κB and mitogen-activated protein kinases (MAPKs) are two of the most important transcription factors in the inflammatory pathways that has major roles in ALF.28 Thus, we assessed the impact of PPARα activation on these two pathways. As shown in Figure 2g and Suplementary Figure 2, the phosphorylation levels of NF-κBp65 were increased by D-GalN/LPS insult and suppressed by Wy-14 643 treatment. In a similar manner, activation of JNK and ERK was also attenuated by Wy-14 643 treatment in the ALF mouse model. Furthermore, we found that the phosphorylation levels of Akt is also increased by D-GalN/LPS insult and suppressed by Wy-14 643 treatment. These results demonstrated that PPARα activation-mediated suppression of inflammation is regulated by the NF-κB and MAPK pathways.
PPARα activation protects mice from ALF through autophagy mechanisms
Because a previous study has shown that PPARγ activation induces autophagy in breast cancer cells,29 we evaluated whether PPARα activation promotes autophagy pathways in the context of ALF. Quantitative reverse transcription-PCR (qRT-PCR) results showed that, compared with D-GalN/LPS-treated mice, PPARα activation by Wy-14 643 pretreatment in ALF mice promoted the expression of Atg7 and Atg5 genes, which are important genes relating to the autophagy pathway, but did not influence Beclin-1 (Figure 3a). These alterations were confirmed by western blot analyses (Figure3b and Supplementary Figure 3). Moreover, as shown in Figure 3b and Supplementary Figure 3, PPARα activation by Wy-14 643 pretreatment not only increased the PPARα expression but also promoted lipidated LC3 form (LC3II) conversion and p62 protein degradation in D-GalN/LPS-induced mice. The results showed that PPARα activation promotes autophagy during ALF.

PPARα activation protects mice from ALF through autophagy mechanisms. SiAtg7/Wy/D-GalN/LPS-treated mice were pretreated with Atg7 siRNA (50 μM/kg) for 48 h via tail vein injection and then administered Wy-14 643 (6 mg/kg) 2 h before D-GalN/LPS exposure (n=8); control siRNA/Wy/D-GalN/LPS-treated mice were pretreated with control siRNA (50 μM/kg) for 48 h via tail vein injection, and then administered Wy-14 643 (6 mg/kg) 2 h before D-GalN/LPS exposure (n=9); 3-MA/Wy/D-GalN/LPS-treated mice were coadministered 3-MA (10 mg/kg) and Wy-14 643 at 2 h before D-GalN/LPS exposure (n=7). Mice were killed 6 h after D-GalN/LPS treatment, and liver and serum samples were collected (apart from (d). (a) Gene expression levels of autophagy-related proteins, including Atg7, Atg5 and Beclin-1, were measured by qRT-PCR in livers from control mice, D-GalN/LPS-treated mice and Wy/D-GalN/LPS-treated mice. The average target gene/HPRT ratios for each experimental group were plotted. (b) Protein expression levels of autophagy-related proteins, including LC3B, Atg7, Atg5, Beclin-1, p62 and PPARα, were measured by western blotting in livers from control mice, D-GalN/LPS-treated mice and Wy/D-GalN/LPS-treated mice. A representative blot for three samples from each group is shown. Densitometry analysis of the proteins was performed for each sample. (c) Protein expression levels of Atg7 and β-actin were measured by western blotting. A representative blot for two samples from every group is shown. (d) Serum AST and ALT enzyme levels from different groups. (e) The survival rate of mice was measured in different groups (10 mice per group). (f) Representative livers and H&E staining of livers ( × 200) from different groups. (g) Gene expression levels of PPARα were measured by qRT-PCR in control mice (n=8), D-GalN/LPS-treated mice (n=10), siAtg7/D-GalN/LPS-treated mice (n=8), 3-MA/D-GalN/LPS-treated mice (n=9) and Wy-14 643/D-GalN/LPS-treated mice (n=8). (h) Protein expression levels of LC3B, p62 and β-actin were measured by western blotting in livers from D-GalN/LPS-treated mice (n=6), Wy/D-GalN/LPS-treated mice (n=8) and CQ/Wy/D-GalN/LPS-treated mice (n=8). A representative blot for two samples from each group is shown. Densitometry analysis of the proteins was performed for each sample
Next, we sought to confirm that PPARα activation leads to the induction of autophagy to protect the liver from injury. First, we applied a specific inhibitor, 3-MA, to block autophagy. Next, we used siRNA to knockdown Atg7. The inhibition of liver Atg7-specific siRNA in vivo was confirmed by the reduced Atg7 levels in the mice (Figure 3c); furthermore, the Atg7 downregulation and 3-MA treatment also decreased the lipidation of LC3I to LC3II (Suplementary Figure 4). The results showed that hepatic protection by Wy-14 643 in ALF was completely negated by the inhibition of autophagy, which was evident by the decreased survival (Figure 3e), the significantly higher sALT and sAST levels (Figure 3d), the relatively worse gross morphology and the relatively less preserved liver architecture by histology (Figure 3f). We also verified whether pharmacologic treatment with 3-MA and Atg7 siRNA affects PPARα expression. The results showed that there were no differences in PPARα gene expression levels between D-GalN/LPS-treated mice and 3-MA- or Atg7 siRNA-treated mice, but the gene expression of PPARα is increased in Wy-14 643-treated mice (Figure 3g). Autophagic flux in D-GalN/LPS-treated mice was monitored after treatment with Wy-14 643 in the presence or absence of chloroquine (CQ). As shown in Figure 3h and Suplementary Figure 5, Wy-14 643 pretreatment induced the lipidation of LC3I to LC3II and the degradation of p62 in D-GalN/LPS-induced mice; the CQ pretreatment increased LC3II and p62 accumulation compared with mice treated with Wy-14 643 and D-GalN/LPS. Thus, these results demonstrated that hepatoprotective mechanisms of PPARα activation depend on autophagy pathways.

PPARα promotes autophagy to suppress liver inflammation in ALF. (a) Gene expression levels of TNF-α, IL-1β and IL-6 at 6 h after D-GalN/LPS injection in livers from the above groups (the same as Figure 3) and IL-12p40 at 2 h after D-GalN/LPS injection in livers from the above groups (n=8). The average target gene/HPRT ratios for each experimental group were plotted. (b) Liver MPO levels at 6 h after D-GalN/LPS injection in livers from the above groups (the same as Figure 3). (c) Gene expression levels of CCL-1, CCL-2, CXCL-1 and CXCL-10 at 6 h after D-GalN/LPS injection in livers from the above groups (the same as Figure 3). The average target gene/HPRT ratios for each experimental group were plotted

PPARα activation regulates the primary macrophage TLR4 response in vitro. BMDMs were stimulated with LPS for 1 h (for IL-12p40) or 6 h (for TNF-α, IL-1β, IL-6, inducible nitric oxide synthase (iNOS), CCL-1, CCL-2, CXCL-1 and CXCL-10) in the absence or presence of Wy-14 643 (10, 25 or 50 μM). (a) Gene induction of TNF-α, IL-1β, IL-6, IL-12p40 and iNOS was measured by qRT-PCR. The average target gene/HPRT ratios for each experimental group were plotted. (b) Gene induction of CCL-1, CCL-2, CXCL-1 and CXCL-10 was measured by qRT-PCR. The average target gene/HPRT ratios for each experimental group were plotted. (c) Protein expression levels of PPARα were measured by western blotting in BMDMs stimulated by LPS in the absence or presence of Wy-14 643 (50 μM). Densitometry analysis of the proteins was performed
PPARα activation promotes autophagy to suppress the inflammatory response in vivo
Given the ability of PPARα activation to suppress liver inflammation and promote autophagy, we sought to determine whether an autophagy pathway is required for PPARα-mediated suppression of the inflammatory response. In the context of PPARα activation in the ALF mice, 3-MA- or Atg7-specific siRNA restored the gene expression of TNF-α, IL-1β, IL-6 and IL-12p40 (Figure 4a). Furthermore, autophagy inhibition increased neutrophil infiltration in mouse livers (Figure 4b) and upregulated the gene expression of chemokines (Figure 4c). These results demonstrated that the autophagy pathways induced by PPARα activation may contribute to the suppression of liver inflammation in the context of ALF.
PPARα activation regulates the primary macrophage TLR4 response in vitro
To further confirm our in vivo experimental findings, we investigated the cellular mechanism of the effects of PPARα activation on macrophages in response to toll-like receptor 4 (TLR4) stimulation by LPS. BMDMs were stimulated by LPS in either the absence or the presence of Wy-14 643 (10, 25 and 50 μM). Remarkably, the expression levels of cytokines (Figure 5a) and chemokines (Figure 5b) by LPS-stimulated macrophages were attenuated by Wy-14 643 treatment in a dose-dependent manner. We also measured the PPARα expression levels in BMDMs stimulated by LPS in the absence or presence of Wy-14 643 by western blot analysis. As shown in Figure 5c, compared with only LPS treatment, PPARα expression levels were higher in BMDMs stimulated by LPS in the presence of Wy-14 643. These results indicated that PPARα activation indeed suppresses the LPS-triggered expression of proinflammatory cytokines and chemokines in vitro.
PPARα activation suppresses proinflammatory response by promoting autophagy in vitro
We then sought to confirm the role of autophagy in vitro in the PPARα-activated macrophage response. Western blot results showed that Wy-14 643 treatment promoted the expression level of LC3II/I, Atg7, Beclin-1 and Atg5 proteins; meanwhile, Wy-14 643 treatment also increased the degradation of p62 (Figure 6a). Moreover, we transfected the GFP-LC3 plasmid into BMDMs to observe the formation of autophagosomes. As shown in Figure 6b, the GFP-LC3 signal was weak in the control cells, but was bright and punctate after Wy-14 643 treatment, in a dose-dependent manner. To confirm that autophagy induction by Wy-14 643 was exclusively due to PPARα activation and not to a nonspecific effect of this agonist, we examined the effect of Wy-14 643 treatment on autophagy-related gene expression using an RNA interference approach to downregulate PPARα expression in macrophages. As shown in Figure 6c, PPARα siRNA reduced the Wy-14 643 treatment-induced upregulation of Atg7, Atg5 and Beclin-1 genes. Furthermore, we tested the effect of Atg7 siRNA or 3-MA treatment in DMEM on autophagy. As shown in Figure 6d, compared with LPS-treated cells, Wy-14 643 treatment increased the ratio of LC3II/I, the expression levels of Atg7 and PPARα, and also increased the degradation of p62; moreover, compared with Wy-14 643-treated cells, Atg7 siRNA or 3-MA treatment decreased the ratio of LC3II/I, the expression levels of Atg7, decreased the degradation of p62 and almost no change of PPARα.

PPARα activation suppresses proinflammatory response by promoting autophagy in vitro. (a) BMDMs were stimulated with or without Wy-14 643 (10, 25 or 50 μM). Total cell lysates were analyzed for LC3B, p62, Atg7, Beclin-1 and Atg5, as well as total β-actin protein levels by western blotting. Densitometry analysis of the proteins was performed for each sample. Data are shown as mean±S.E.M (n=3). *P<0.05 or #P<0.01, compared with dimethylsulfoxide (DMSO)-treated BMDMs. (b) Transfected GFP-LC3 plasmid for 12 h, BMDMs were preincubated with Wy-14 643 (10, 25 or 50 μm) for 24 h to observe the formation of autophagosomes. The percentage of cells with GFP-LC3 puncta in the groups receiving different concentrations. GFP-positive cells were defined as cells that display bright, punctate staining. Approximately 50 cells were counted, and the experiment was repeated at least three times. (c) After transfection of PPARα siRNA or control siRNA (3 mg/ml) for 36 h, BMDMs were incubated with or without Wy-14 643 (50 μM) for 6 h. Gene induction of Atg7, Atg5 and Beclin-1 was measured by qRT-PCR. The average target gene/HPRT ratios for each experimental group were plotted. (d) After transfection of Atg7 siRNA (3 mg/ml) for 36 h or preincubation with 3-MA (10 mM/l) for 2 h, BMDMs were incubated with or without Wy-14 643 (50 μM) for 2 h and then incubated in LPS (20 ng/ml) for 6 h. Western blot analysis shows the LC3BII/I ratio, Atg7, PPARα and p62 degradation. β-Actin is shown as a loading control; densitometry analysis of the proteins was performed for each sample; data are shown as mean±S.E.M (n=3). (e) Wy-14 643 activates autophagic flux in the primary hepatocytes of mice. Hepatocytes were treated with 50 μM Wy-14 643 in the absence or presence of CQ (10 μM) for 24 h. Western blot analysis shows the LC3BII/I ratio and p62 degradation. β-Actin is shown as a loading control; densitometry analysis of the proteins was performed for each sample; data are shown as mean±S.E.M (n=3). (f and g) After transfection of Atg7 siRNA (3 mg/ml) for 36 h or preincubated 3-MA (10 mM/l) for 2 h, BMDMs were incubated with or without Wy-14 643 (50 μM) for 2 h and then incubated with LPS (20 ng/ml) for 6 h. Cytokine and chemokine gene induction was measured by qRT-PCR. The average target gene/HPRT ratios for each experimental group were plotted
Autophagic flux in primary mouse hepatocytes was monitored after treatment with Wy-14 643 in the presence or absence of CQ. The influence of Wy-14 643, with or without CQ, on LC3I/II levels and p62 was assessed by western blotting. As shown in Figure 6e, Wy-14 643 induced the lipidation of LC3I to LC3II and the degradation of p62. The addition of CQ further increased LC3II conversion, and also decreased the degradation of p62 compared with cells treated with Wy-14 643 only. Taken together, analysis of the level of LC3-II/I and p62/β-actin ratio showed that PPARα activation can directly induce autophagic flux in BMDMs.
To further examine the notion of the PPARα-mediated autophagy in the macrophages activated by TLR4 signaling, we added 3-MA in Wy-14 643-treated macrophage cultures or transfected siRNA Atg7 into macrophages before Wy-14 643 treatment. Indeed, the LPS-induced proinflammatory cytokine and chemokine levels were restored after inhibition of autophagy (Figures 6f and g). Thus, we concluded that PPARα activation promotes autophagy to regulate macrophage TLR4 response by downregulating the gene expression of proinflammatory markers and chemokines.
Decreased expression of PPARα in the liver of ALF patients with HBV infection
To determine whether PPARα participates in the progression of ALF in patients with hepatitis B virus (HBV) infection, we used liver tissues to measure the changes in PPARα among normal subjects, chronic hepatitis B (CHB) patients and ALF patients with HBV infection. Relative to the normal controls, the qRT-PCR results showed that PPARα gene expression levels were not different in the cases of CHB, but were significantly reduced in the cases of ALF (Figure 7a). Similar results were obtained by western blot analyses (Figure 7b) and immunofluorescence staining in liver tissue (Figure 7c). These results indicated that PPARα expression is also depressed in patients with ALF induced by HBV infection.

PPARα expression is decreased in the liver of ALF patients with HBV infection. (a) Gene expression of PPARα was measured by qRT-PCR in the livers of normal subjects (n=8), CHB patients (n=12) and ALF patients (n=12). The average target gene/HPRT ratios for each experimental group were plotted. (b) Protein expression levels of PPARα were measured by western blotting in the livers of normal subjects (n=8), CHB patients (n=12) and ALF patients (n=12). A representative blot from two samples of every group is shown. Densitometry analysis of the proteins was performed for each sample. (c) Immunofluorescence staining for PPARα (red) in the liver of normal subjects, CHB patients and ALF patients with HBV infection. Representative images of each experiment are shown. Original magnification, × 400. (d) In the D-GalN/LPS-induced ALF mice, PPARα was suppressed, which promotes downregulation of autophagy and increases the expression of proinflammatory cytokines and chemokines. These events lead to incremental neutrophil infiltration and inflammation in the liver and ultimately induce the development of ALF. Red arrows indicate PPARα agonist; Wy-14 643 induced changes in ALF mice relative to their DMSO-injected counterparts
Discussion
Although PPARα activation exerts strong anti-inflammatory properties in various animal models of liver injury,30, 31, 32, 33, 34 its role in the context of ALF has not yet been explored. The main findings of the present study were that PPARα activation promotes autophagy, which may depress liver inflammation to inhibit hepatocyte apoptosis and, consequently, result in liver protection (as depicted in Figure 7d).
Increasing reports in the literature indicate that the systemic inflammatory response, whether through hepatic inflammation and/or sepsis, has a major role in determining the outcome of ALF patients.35, 36 Kupffer cells (macrophages in the liver) not only contribute to the early phase of the disease but also cause sustained inflammation.37, 38 Moreover, immune-mediated liver injury is a consequence of the recruitment of effector leukocytes to the liver where they mediate tissue damage.39 Leukocyte migration from the vascular lumen into the surrounding extravascular tissue makes a significant contribution to liver injury in ALF.28, 40 Studies have shown that neutrophil accumulation in a liver with ALF can be triggered by a variety of proinflammatory mediators and chemokines.41, 42 Our results showed that PPARα activation significantly reduced the D-GalN/LPS-induced acute liver damage by reducing the expression of proinflammatory cytokines and chemokines, regulating the activities of the NF-κB and MAPK pathways. Therefore, we speculate that one of the protective mechanisms of PPARα activation is the reduced activation of the NF-κB and MAPK pathways in the liver in cases of ALF. In one regard, this mechanism can repress the cascade activation of inflammatory cytokines and directly control liver inflammation. By contrast, PPARα activation significantly decreased the expression of chemokines, which resulted in mitigating neutrophil migration and infiltration in the liver, further indirectly alleviating inflammation. Taken together, our results confirmed that PPARα activation can effectively treat liver injury due to ALF. This finding implicates PPARα as an important target for the effective intervention of ALF.
What is the underlying mechanism mediating the effect of PPARα activation on liver inflammation? The regulation of autophagy via PPARα signaling is a novel finding. Autophagy is an intracellular degradation process through which long-lived cytosolic proteins and organelles are degraded by lysosomes and recycled. Autophagic flux can be monitored by combining the measurement of LC3II and p62 levels. An increase of LC3II conversion with the degradation of p62 indicates the induction of autophagic flux, whereas the increase of LC3II conversion without the degradation of p62 indicates the inhibition of autophagic flux.42 Our results demonstrated that PPARα activation can upregulate the autophagy-related genes Atg5 and Atg7; more importantly, PPARα activation increase LC3II conversion and p62 degradation in vivo and in vitro. In conclusion, our study revealed that PPARα activation promotes autophagic flux in the progression of D-GalN/LPS-induced ALF.
Because autophagy affects the effector cells of innate and adaptive immunity, which mediate the inflammatory response, its activity in these cells influences the anti-microbial response, and the development of an effective cognate immune defense. Loss or decreased autophagy may lead to necrotic death that can initiate an inflammatory reaction in phagocytes through their surface and cytosolic receptors; thus, autophagy can shape inflammatory responses.43 Our preliminary research found that autophagy is promoted in the early phase of D-GalN/LPS-induced ALF and inhibited in the later phase of D-GalN/LPS-induced ALF, and it has a protective role in ALF (Ren Feng et al., unpublished paper). We further explored the role of autophagy in the protection of PPARα activation-treated ALF. We found that inhibition of autophagy reversed the protective effect of PPARα activation on ALF, restored the upregulation of gene expression of the inflammatory cytokines and chemokines and reversed the decreased infiltration and migration of neutrophils in the liver. We demonstrated a new pathogenic mechanism of ALF that PPARα activation attenuates the inflammatory response to protect against D-GalN/LPS-induced ALF in mice by promoting autophagy.
A previous study demonstrated that reductions in PPARα levels resulted in a significant inhibition of HBV replication in HepG2 cells.44 However, the role of PPARα expression in the progression of ALF patients with HBV infection remains unclear. Our studies further explored this role and showed that PPARα levels are not influenced in CHB patients compared with normal subjects, whereas it is downregulated in ALF patients with HBV infection. It can be speculated that PPARα may have an important role in the pathogenesis of HBV infection-induced ALF, especially in the regulation of liver inflammation; however, the mechanisms may differ between mouse ALF induced by D-GalN/LPS and human ALF induced by HBV. Based on our findings, we speculate that PPARα levels can be considered an early warning sign of ALF with HBV infection. Additional in-depth research for these questions is needed.
A limitation of our study is that we could not assess exactly how PPARα regulates the autophagy pathway. We have found that PPARα activation significantly inhibits the phosphorylation levels of Akt in D-GalN/LPS-induced ALF, and previous studies have shown that activation of the PI3K-Akt pathway significantly inhibits the autophagic pathway.45, 46 Therefore, we hypothesize that PPARα activation may regulate autophagy through the PI3K-Akt pathway signaling pathway in the progression of ALF. At present, this hypothesis is under further exploration.
In summary, this study adds to the general understanding of mechanisms of ALF and provides new insight into the importance of PPARα in regulating liver inflammation, partially through an autophagy pathway. Therefore, PPARα activation represents a potent strategy to ameliorate ALF pathology. Further preclinical studies on PPARα agonists are warranted for the development of a clinically applicable therapeutic strategy against ALF.
Materials and Methods
Animal experiments
Male wild-type (C57BL/6) mice (aged 8–12 weeks) were purchased from Capital Medical University (Beijing, China) and housed in the Capital Medical University animal facility under specific pathogen-free condition and received humane care according to Capital Medical University Animal Care Committee guidelines.
The mice were intraperitoneally injected with D-GalN (700 mg/kg; Sigma, St. Louis, MO, USA) and LPS (10 μg/kg; InvivoGen, San Diego, CA, USA) to induce ALF or with saline in the control animals. The PPARα activator Wy-14 643 (6 mg/kg; Sigma) was administered via injection into the tail vein 2 h before D-GalN/LPS exposure. Suppression of autophagy was achieved by tail vein injection of 3-MA (10 mg/kg; Sigma) or siRNA for Atg7 (50 μM/kg). CQ (C6628; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in normal saline solution. Mice were pre-treated with CQ (50 mg/kg, intraperitoneally) 2 h before D-GalN/LPS exposure. The mice were killed at various time points after D-GalN/LPS treatment, and liver and serum samples were collected for future analysis.
Human specimens
Normal liver samples were collected from eight patients undergoing hepatic resection for liver transplantation. CHB samples were obtained from the livers of 12 patients undergoing liver puncture biopsy. ALF samples were obtained from the livers of 12 patients with HBV infection undergoing liver transplantation. This study meets the ethical guidelines of the 1975 Declaration of Helsinki, and the study protocol was approved by the Medical Ethics Committee of Beijing YouAn Hospital. Informed consent was obtained from all patients. The clinical characteristics and details of the patients included in the study are shown in Table 1.
Hepatocellular damage assay, liver histology and MPO assay
Serum aminotransferase, liver histology by H&E staining and MPO assays were performed as described previously.47, 48
Quantitative real-time PCR analysis and primer sequences
Total RNA was isolated from hepatic samples using Trizol reagent according to the manufacturer’s protocol. A total of 2.5 μg of RNA was reverse transcribed into cDNA using SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). Quantitative PCR was performed using the DNA Engine with Chromo 4 Detector (MJ Research, Waltham, MA, USA). The following were added to a final reaction volume of 20 μl: 1 × SuperMix (Platinum SYBR Green qPCR Kit; Invitrogen); cDNA (2 μl); and 0.5 μM of each primer. The amplification conditions were as follows: 50 °C for 2 min and 95 °C for 5 min, followed by 50 cycles of 95 °C for 15 s and 60 °C for 30 s. The primers used to amplify the specific mouse gene fragments are listed in Table 2.
Western blot analyses
Protein was extracted from liver tissue in RIPA buffer together with phosphatase and protease inhibitors. Proteins in SDS-loading buffer were subjected to SDS-12% polyacrylamide gel electrophoresis and transferred to a PVDF membrane (Bio-Rad, Hercules, CA, USA). Antibodies against PPARα (Abcam, Cambridge, MA, USA), phosphorylated ERK, phosphorylated JNK, phosphorylated p38, phosphorylated NF-κBp65, LC3B, Atg7, Atg5, Beclin-1, Lamp1 and β-actin (Cell Signaling Technology Inc., Santa Cruz, CA, USA) were used for western blot analysis. The membranes were probed with primary antibodies (1:500–1000) in 10 ml of blocking buffer overnight at 4 °C. After washing, the membranes were further probed with the appropriate horseradish peroxidase-conjugated secondary antibody (1:2000) in 10 ml of blocking buffer for 1 h at room temperature. SuperSignal West Pico chemiluminescent substrates (Thermo Fisher Scientific, Rockford, IL, USA) were used for chemiluminescence development.
Atg7 siRNA treatment in vivo
Autophagy was inhibited through the siRNA against Atg7 (50 μM/kg; Jima, Suzhou, China); its sequence is 5′-GCAUCAUCUUCGAAGUGAATT-3′. Atg7 knockdown was achieved by siRNA using an Entranster in vivo transfection reagent (Engreen Biosystem Co., Beijing, China) via hydrodynamic tail vein injection in mice. Scrambled siRNA (50 μM/kg) was used as a control. The processes were performed following the manufacturer’s instructions.
BMDM cell culture and treatment
Murine BMDMs, differentiated from bone marrow cells, were prepared by culturing cells in DMEM containing 10% fetal bovine serum, 1% penicillin/streptomycin and 20% L929 conditioned medium for 6 days. LPS (20 ng/ml) was used to activate cells, Wy-14 643 (50 μM) was used to activate PPARα, siRNA PPARα (5′-GAGAUCGGCCUGGCCUUCUAAACAU-3′) was used to inhibit PPARα and 3-MA (10 mM) or Atg7 siRNA (3 mg/ml) was used to inhibit autophagy in the macrophages. Transient transfection was performed with a GFP-LC3 plasmid or Atg7 siRNA using Fugene HD (Roche, Shanghai, China) according to the manufacturer’s instructions.
For macrophage-conditioned media, BMDMs were seeded in six-well plates at a density of 4 × 106 cells per well in 3 ml of complete medium and incubated for 12 h. BMDMs were stimulated with LPS (20 ng/ml) in the absence or presence of Wy-14 643 (50 μM) for 24 h. The BMDM-conditioned media were collected, clarified by centrifugation at 400 × g and stored at −20 °C until use.
Isolation and treatment of primary mouse hepatocytes
The mouse livers were perfused with Hank’s solution containing collagenase at 7 weeks of age, and viable hepatocytes were isolated by Percoll isodensity centrifugation as described.49 The primary hepatocytes stimulated with actinomycin D (1 μg/ml; Sigma) in the absence or presence of BMDM-conditioned media for 12 h. The MTT assay (Amersco, Solon, OH, USA) was used as a qualitative index of cell proliferation, and apoptosis was evaluated at 12 h by LDH assays (Biochain Institute, Hayward, CA, USA). The processing was conducted according to the manufacturer’s instructions.
Immunofluorescence staining
Paraffin sections were treated with xylene for 10 min three times. The sections were hydrated through a graded alcohol series and then rinsed three times with distilled water. The slides were incubated for 20 min in 10% goat serum in PBS and then a PPARα rabbit polyclonal antibody (Abcam) overnight at 4 °C. The slides were incubated with Alexa Fluor 568 goat anti-rabbit IgG (1:200; Invitrogen, Grand Island, NY, USA) for 45 min. After three washes with PBS, the nuclei were stained with 4',6-diamidino-2-phenylindole (1 μg/ml; Shizebio, Shanghai, China) for 10 min. The images were examined on a Nikon Eclipse E800 fluorescent microscope (Nikon Corp., Tokyo, Japan).
Statistical analyses
The results are shown as the mean±S.E.M. The statistical analyses were performed using an unpaired Student’s t-test, and P<0.05 (two tailed) was considered significant.
Abbreviations
- ALF:
-
acute liver failure
- PPARα:
-
peroxisome proliferator-activated receptor α
- D-GalN:
-
D-galactosamine
- LPS:
-
lipopolysaccharide
- NF-κB:
-
nuclear factor-κB
- MAPK:
-
mitogen-activated protein kinase
- STAT1:
-
signal transducer and activator of transcription 1
- TNF-α:
-
tumor necrosis factor-α
- ALT:
-
alanine aminotransferase
- AST:
-
aspartate aminotransferase
- MPO:
-
myeloperoxidase
- CHB:
-
chronic hepatitis B
- TLR4:
-
toll-like receptor 4
- BMDM:
-
bone marrow-derived macrophage
- 3-MA:
-
3-methyladenine
- Atg:
-
autophagy gene
- CCL:
-
chemokine (C–C motif) ligand
- CXCL:
-
chemokine (C–X–C motif) ligand
- IL-1β:
-
interleukin-1β
- iNOS:
-
inducible nitric oxide synthase
- CQ:
-
chloroquine
- qRT-PCR:
-
quantitative reverse transcription PCR
- HBV:
-
hepatitis B virus
References
Hoofnagle JH, Carithers RJ, Shapiro C, Ascher N . Acute hepatic failure: summary of a workshop. Hepatology 1995; 21: 240–252.
Riordan SM, Williams R . Mechanisms of hepatocyte injury, multiorgan failure, and prognostic criteria in acute liver failure. Semin Liver Dis 2003; 23: 203–215.
Desvergne B, Wahli W . Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 1999; 20: 649–688.
Kota BP, Huang TH, Roufogalis BD . An overview on biological mechanisms of PPARs. Pharmacol Res 2005; 51: 85–94.
Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC . Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998; 98: 2088–2093.
Chinetti G, Griglio S, Antonucci M, Pineda TI, Delerive P, Majd Z et al. Activation of peroxisome proliferator activated receptors α and γ induces apoptosis of human monocyte derived macrophages. J Biol Chem 1998; 273: 25573–25580.
Delerive P, De BK, Besnard S, Vanden BW, Peters JM, Gonzalez FJ et al. PPARα negatively regulates the vascular inflammatory gene response by negative cross talk with transcription factors NF-κB and AP-1. J Biol Chem 1999; 274: 32048–32054.
Devchand PR, Keller H, Peters JM, Vasquez M, Gonzalez FJ, Wahli W . The PPARα-leukotriene B pathway to inflammation control. Nature 1996; 384: 39–43.
Staels B, Koenig W, Habib A, Merval R, Lebret M, Pineda-Torra I et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature 1998; 393: 790–793.
Lee JH, Joe EH, Jou I . PPAR-alpha activators suppress STAT1 inflammatory signaling in lipopolysaccharide-activated rat glia. NeuroReport 2005; 16: 829–833.
Delayre OC, Becker J, Guenon I, Lagente V, Auwerx J, Frossard N et al. PPARalpha downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res 2005; 6: 91–100.
Di PR, Esposito E, Mazzon E, Genovese T, Muia C, Crisafulli C et al. Absence of peroxisome proliferators-activated receptors (PPAR)alpha enhanced the multiple organ failure induced by zymosan. Shock 2006; 26: 477–484.
Sheu MY, Fowler AJ, Kao J, Schmuth M, Schoonjans K, Auwerx J et al. Topical peroxisome proliferator activated receptor-alpha activators reduce inflammation in irritant and allergic contact dermatitis models. J Invest Dermatol 2002; 118: 94–101.
Seong HY, Mohamed AA, Byoung JS . Activation of PPARa by Wy-14,643 ameliorates systemic lipopolysaccharide-induced acute lung injury. Biochem Biophys Res Commun 2013; 9: 0006-291X(13)00870-X.
Stephen WS, Charles CC, Basilia Z, Hector RW . Reduced peroxisome proliferator-activated receptor α expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock 2012; 37: 164–169.
Seong HY, Ogyi P, Lauren EH, Mohamed AA, Kwan HM, Byoung JS . Lack of PPARα exacerbates lipopolysaccharide-induced liver toxicity through STAT1 inflammatory signaling and increased oxidative/nitrosative stress. Toxicol Lett 2011; 202: 23–29.
Mignon A, Rouquet N, Fabre M, Martin S, Pagès JC, Dhainaut JF et al. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med 1999; 159: 1308–1315.
Nakama T, Hirono S, Moriuchi A, Hasuike S, Nagata K, Hori T et al. Etoposide prevents apoptosis inmouse liver with D-GalN/LPS-induced fulminant hepatic failure resulting inreduction of lethality. Hepatology 2001; 33: 1441–1450.
Ohsumi Y . Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2: 211–216.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451: 1069–1075.
Levine B, Deretic V . Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7: 767–777.
Beth L, Noboru M, Herbert WV . Autophagy in immunity and inflammation. Nature 2011; 469: 323–335.
Alexander JSC, Stefan WR . Autophagy in inflammatory diseases. Int J Cell Biol 2011; 2011: 732798.
Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V . Toll-like receptors control autophagy. EMBO J 2008; 27: 1110–1121.
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT . Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27: 135–144.
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T et al. loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008; 456: 264–268.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12: 222–230.
Gyongyi S, Pranoti M . Angela D. Innate immune response and hepatic inflammation. Semin Liver Dis 2007; 27: 339–350.
Jie Z, Zh Wei, Bing L, Mathew CC, Diana WM, Min W et al. PPARγ activation induces autophagy in breast cancer cells. Int J Biochem Cell Biol 2009; 41: 2334–2342.
Delayre OC, Becker J, Guenon I, Lagente V, Auwerx J, Frossard N et al. PPARα downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res 2005; 6: 91–100.
Nakajima T, Kamijo Y, Tanaka N, Sugiyama E, Tanaka E, Kiyosawa K et al. Peroxisome proliferator-activated receptor α protects against alcohol-induced liver damage. Hepatology 2004; 40: 972–980.
Nan YM, Kong LB, Ren WG, Wang RQ, Du JH, Li WC et al. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol mediated liver fibrosis in mice. Lipids Health Dis 2013; 12: 11–20.
Seong HY, Ogyi P, Lauren EH, Mohamed AA, Kwan HM, Byoung JS . Lack of PPARα exacerbates lipopolysaccharide-induced liver toxicity through STAT1 inflammatory signaling and increased oxidative/nitrosative stress. Toxicol Lett 2011; 202: 23–29.
Emilia I, Geoff F, Pauline H, Graham R, Isabelle L . Administration of the potent PPARα Agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 2004; 39: 1286–1296.
Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R . The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32: 734–739.
Jalan R, Olde Damink SW, Hayes PC, Deutz NE, Lee A . Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol 2004; 41: 613–620.
Su GL . Lipopolysaccharides in liver injury: molicular mechanisms of kupffer cell activation. Am J Physiol Gastrointest Liver Physiol 2002; 283: G256–G265.
Matsuno K, Nomiyama H, Yoneyama H, Uwatoku R . Kupffer cell-mediated recruitment of dendritic cells to the liver crucial for a host defense. Dev Immunol 2002; 9: 143–149.
Klugewitz K, Adams DH, Emoto M, Eulenburg K, Hamann A . The composition of intrahepatic lymphocytes: shaped by selective recruitment? Trends Immunol 2004; 25: 590–594.
Bertus E, Simon CA, Stephen JW, Andrew PH, David HA . Immune-mediated liver injury. Semin Liver Dis 2007; 27: 351–366.
Wagner JG, Roth RA . Neutrophil migration during endotoxemia. J Leukoc Biol 1999; 66: 10–24.
Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo A, Adeli K et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445–544.
Fésüs L, Demény MÁ, Petrovski G . Autophagy shapes inflammation. Antioxid Redox Signal 2011; 14: 2233–2243.
Hu W, Wang X, Ding X, Li Y, Zhang X, Xie P et al. MicroRNA-141 represses HBV replication by targeting PPARα. PLoS One 2012; 7: e34165.
Wu YT, Tan HL, Huang Q, Ong CN, Shen HM . Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy 2009; 5: 824–834.
Sovan S, Janet ED, Marie F, Moises GA, Zeyn GT, Maria JS et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90: 1383–1435.
Liyan C, Feng R, Haiyan Z, Tao W, Zhengfu P, Li Z et al. Inhibition of GSK3β ameliorates D-GalN/LPS-induced liver injury by reducing ERS-triggered apoptosis. PLoS One 2012; 7: e45202.
Feng R, Zhongping D, Xiuda S, Shen X, Gao F, Bai L et al. The inhibition of GSK3β ameliorates liver ischemia reperfusion injury via an IL-10-mediated immune modulatory mechanism. Hepatology 2011; 54: 687–696.
Klaunig JE, Goldblatt PJ, Hinton DE, Lipsky MM, Chacko J, Trump BF . Mouse liver cell culture. I. Hepatocyte isolation. In Vitro 1981; 17: 913–925.
Acknowledgements
This work was financially supported by China National Key Project of the Twelfth Five-year Plan (2012ZX10002004-006, 2012ZX10004904-003-001 and 2013ZX10002002-006-001), the National Natural Science Foundation of China (81270532, 81372094 and 81300349), the Beijing Excellent Talents Training Funding (2011D003034000022), the Technology Foundation for Selected Overseas Chinese Scholar, Ministry of Personnel of Beijing (2012) and Applied Research for the Clinical Characteristics of Capital (Z121107001012167).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Jiao, M., Ren, F., Zhou, L. et al. Peroxisome proliferator-activated receptor α activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis 5, e1397 (2014). https://doi.org/10.1038/cddis.2014.361
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.361
This article is cited by
-
Regulation of genes involved in the metabolic adaptation of murine microglial cells in response to elevated HIF-1α mediated activation
Immunogenetics (2024)
-
Cannabinoids induce functional Tregs by promoting tolerogenic DCs via autophagy and metabolic reprograming
Mucosal Immunology (2022)
-
The Zinc Ionophore Clioquinol Reduces Parkinson’s Disease Patient-Derived Brain Extracts-Induced Neurodegeneration
Molecular Neurobiology (2022)
-
Withaferin A alleviates fulminant hepatitis by targeting macrophage and NLRP3
Cell Death & Disease (2021)
-
High-content screening of Thai medicinal plants reveals Boesenbergia rotunda extract and its component Panduratin A as anti-SARS-CoV-2 agents
Scientific Reports (2020)
