
Abstract
Mantle cell lymphoma (MCL) is a currently incurable B-cell malignancy. Lenalidomide (Len) has been demonstrated to be one of the most efficient new treatment options. Because Len and 1α,25-dihydroxyvitamin (VD3) synergize to kill breast cancer cells, we investigated whether VD3 could increase the ability of Len to induce MCL cell death. While MCL cells were weakly sensitive to Len (1 μM), the addition of VD3 at physiological dose (100 nM) strongly increased cell death, accompanied by slowdown in cell cycle progression in MCL cell lines (n=4 out of 6) and primary samples (n=5 out of 7). The Len/VD3 treatment markedly increased the expression of the BH3-only BCL2-interacting killer (Bik) without affecting the expression of other Bcl-2 molecules. Immunoprecipitation assays demonstrated that Bik was free from anti-apoptotic partners, Bcl-2 and Bcl-xL, in treated cells. Moreover, silencing of BIK prevented apoptosis induced by Len/VD3, confirming the direct involvement of Bik in cell death. Bik accumulation induced by Len/VD3 was related to an increase in BIK mRNA levels, which resulted from a demethylation of BIK CpG islands. The sensitivity of MCL cells to Len/VD3 was similar to the response to 5-azacytidine, which also induced demethylation of BIK CpG islands. These preclinical data provide the rationale to investigate the role of VD3 in vivo in the response to Len.
Similar content being viewed by others


Main
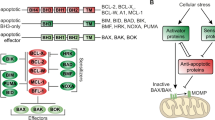
Mantle cell lymphoma (MCL) is an aggressive non-Hodgkin’s lymphoma (NHL) that accounts for ∼5–8% of all NHLs in adults.1, 2 Intensive chemotherapy regimens combined with anti-CD20 antibodies with or without autologous stem cell transplantation have significantly improved patients’ outcomes, but most patients relapse and survive only an average of ∼5 years from the time of diagnosis.3 Moreover, this intensive regimen is not applicable to all patients, especially to elderly patients. As in other type of cancers, there is an obvious need for new therapies in MCL. Immunomodulatory drugs such as lenalidomide (Len) were first introduced to treat multiple myeloma, where Len has proven benefits. More recently, Len has also been successfully used for relapsed and refractory patients with MCL.4, 5, 6, 7, 8, 9 Several phase III trials comparing Len (with or without chemotherapy) versus standard treatment options are ongoing in MCL. Despite a proven efficacy in MCL, ∼60% of MCL patients remain resistant to Len. Len exhibits direct antitumor efficacy and modulates the tumor environment, especially the immune environment, but the mechanisms of resistance to Len remain partially unknown.10, 11 1α,25-dihydroxyvitamin D3 (VD3) has a well-described function as an endocrine steroid hormone that regulates calcium metabolism, but its physiological role is not limited to this function.12 The effects of VD3 in cancer have been under investigation for over a decade.13, 14 In NHL patients, the level of VD3 in the serum was recently evaluated as a prognostic marker, where a deficiency in VD3 predicts worse overall survival.15 In contrast, Rosen et al.12 who performed a large systematic review concluded that there is no evidence that VD3 could reduce the risk of cancer development or mortality.12 While the relationship between VD3 and the incidence of cancer remains unresolved, some authors suggest that VD3 could act as an anticancer agent through its anti-proliferative, pro-differentiation, anti-inflammatory and anti-angiogenic properties by modulating the activity of transduction pathways or altering the transcriptional regulation of genes.12, 16 Indeed, VD3 modulates >900 genes involved in differentiation, cell cycle control, apoptosis, immune response and migration.14, 17 Because VD3 is safe and well tolerated, the combination of VD3 plus anticancer agent(s) is as an attractive treatment option. Recently, the combination of VD3 and Len was demonstrated to synergistically induce cell death through the modulation of Bcl-2 expression in breast cancer cells.18 In the present work, we assessed the efficacy of VD3 combined with a low dose of Len in modulating the apoptosis of MCL cell lines and primary cells.
Results
Addition of VD3 increased Len-induced cell death in MCL cells
We first assessed the sensitivity of six MCL cell lines to Len and VD3 at varying concentrations, respectively, ranging from 0.001 to 10 μM and from 1 to 150 nM. After 6 days of treatment, the JEKO-1, MINO, REC-1 and Z-138 cells were weakly sensitive to Len (black color), whereas the GRANTA-519 and UPN-1 cells were resistant (>80% of viability at 10 μM, gray color; Figure 1a). The GRANTA-519, UPN-1 and REC-1 cells were resistant to VD3 (>80% of viability at 150 nM), whereas the JEKO-1, MINO and Z-138 cells were weakly sensitive with a maximum of loss viability of 32% for Z-138 cells (Figure 1b). In contrast to the sensitivity of MCL cells to Len, which increased linearly with concentration, the sensitivity to VD3 reached a plateau (e.g., 75 nM in Z-138 cells). Because cereblon (CRBN) is the receptor for Len, we investigated whether the resistance of MCL cell lines could be related to lack of CRBN expression.24, 25 All of MCL cell lines expressed CRBN, and there was no correlation between the level of CRBN expression and the sensitivity of MCL cell lines (Supplementary Figure S1A). MCL cell lines expressed VD3 receptor (VDR) and VDR expression was further increased by VD3 in both sensitive and resistant MCL cell lines, indicating that resistance to VD3 was not related to the lack of VDR expression (Supplementary Figures S1B and C).

The combined Len/VD3 treatment induced apoptosis and inhibited cell cycle progression in MCL cells. (a–c) Dose–response of cells to Len and VD3. Cells (2 × 105 cells/ml) were incubated with or without increasing concentrations of Len (a), VD3 (b) or combined Len/VD3 (c) for 6 days, and viability was assessed using MTT assay. The data represent the mean±S.E. of three independent experiments. (d) Len/VD3 induced apoptosis and inhibited cell cycle progression. Cells (2 × 105 cells/ml) were incubated with or without 1 μM Len, 100 nM VD3 or Len/VD3 (1 μM and 100 nM) for 6 days and were then stained with Annexin V or PI. A representative experiment out of three is shown. (e) The Len/VD3 treatment induced apoptosis in primary MCL cells. Primary cells (1 × 106 cells/ml) from seven independent patients were incubated with or without 10 μM Len, 100 nM VD3 or Len/VD3 for 6 days and were stained with Annexin V. *P<0.05
We next investigated the efficacy of the combined 1 μM Len and 100 nM VD3 treatment for 6 days. The addition of VD3 did not overcome the resistance of GRANTA-519 and UPN-1 to Len (Figure 1c). However, the Len/VD3 treatment decreased the viability to 68±5%, 56±8%, 52±6% and 14±7%, respectively, with a combination index of 0.88, 0.52, 0.62 and 0.52 in the MINO, JEKO-1, REC-1 and Z-138 cells, respectively. To determine whether the loss of viability was related or not to the induction of apoptosis, we performed Annexin V staining and cell cycle analysis in JEKO-1, Z-138 and GRANTA-519. In the presence of Len/VD3, 46±11% of JEKO-1 cells and 84±9% of Z-138 cells were Annexin V positive and 19±7% of JEKO-1 cells and 39±9% of Z-138 cells contained subG1 DNA content (Figure 1d). Moreover, the Len/VD3 treatment decreased the proportion of cells in the S and G2 phases in the sensitive cell lines. In contrast, Len/VD3 treatment did not induce any significant Annexin V staining or modifications in the cell cycle in the resistant GRANTA-519 cells.
We investigated the effect of Len/VD3 in primary cells from patients with MCL (Figure 1e and Table 1). Seven peripheral blood samples collected from patients at the time of diagnosis were purified using anti-CD19 immunomagnetic beads. The CD19+ MCL cells were incubated for 6 days with 10 μM Len and with or without 100 nM VD3. Cell death induced by Len (median value 9%) was enhanced by the addition of VD3, with a median value of 21% (P=0.016, Wilcoxon matched-pairs signed rank test). As observed in cell lines, the combined Len/VD3 treatment increased the percentage of cell death in all samples sensitive to Len but not in those resistant to Len (samples 1 and 2, Table 1).
Combined Len/VD3 treatment induced intrinsic pathway of apoptosis and accumulation of BH3-only BIK protein
Len/VD3 treatment induced cleavage of caspase 9 in sensitive JEKO-1 cells but not in resistant GRANTA-519 cells (Figure 2a), suggesting the involvement of the intrinsic pathway of apoptosis, which was confirmed by the induction of mitochondrial depolarization, as assessed using JC-1 staining (Figure 2b). To confirm the role of the intrinsic pathway, we performed Bax silencing experiments (Figure 2c). Indeed, the transfection with siBAX prevented by 78±21% the Len/VD3-induced decrease in cell number (P<0.01, paired t-test): the mean cell number (millions cells/ml) was 3.3±0.2 and 1.5±0.4 in untreated and Len/VD3-treated siCt cells, respectively, versus 3.3±0.3 and 2.9±0.3 in untreated and Len/VD3-treated siBAX cells, respectively (Figure 2d).

The combined Len/VD3 treatment activated caspase 9, induced mitochondrial depolarization and involved Bax. (a) The Len/VD3 treatment induced caspase 9 activation. MCL cells (2 × 105 cells/ml) were incubated for 4 days with or without 1 μM Len, 100 nM VD3 or Len/VD3. Cells were then lysed and activation of caspase 9 was assessed by western blotting. A representative experiment out of three is shown. (b) The Len/VD3 treatment induced mitochondrial depolarization. Z-138 cells (2 × 105 cells/ml) were incubated for 4 days with or without 1 μM Len and 100 nM VD3, and then stained with JC-1. A representative experiment out of three is shown. (c–e) Silencing of BAX prevented cell death induced by Len/VD3 combination. JEKO-1 cells (5 × 105/ml) were seeded for 48 h with or without the Len/VD3 combination (1 μM Len and 100 nM VD3) before transfection with sicontrol (siCt) or siBAX RNA. Then, transfected cells were reseeded (5 × 105/ml) for additional 3 days with or without the Len/VD3 combination. (c) Western blotting analysis of Bax expression. Bax expression was assessed in siCt- and siBAX-transfected JEKO-1 cells treated or not with Len/VD3 combination. (d) Len/VD3 induced a Bax-dependent decrease in cellularity. The cellular density was measured by a direct counting. The data represent five independent experiments. (e) Len/VD3 induced a Bax-dependent cell death. Cells were stained with Annexin V and fluorescence was analyzed on a FACSCalibur. The data represent five independent experiments. *P<0.05, **P<0.01
Similarly, the transfection with siBAX reduced by 68±4% the Len/VD3-induced cell death (P=0.01), but it did not significantly induced cell death in untreated cells (P=0.25): the mean cell death was 11±2% and 28±6% in untreated and Len/VD3-treated siCt cells, respectively, versus 12±3% and 18±5% in untreated and Len/VD3-treated siBAX cells, respectively (Figure 2e).
To further characterize the Len/VD3-induced apoptosis, we then assessed changes in the expression of pro- and anti-apoptotic proteins by western blotting. Len/VD3 weakly increased the expression of the BH3-only Noxa in the sensitive JEKO-1 and Z-138 cell lines, and Puma proteins in JEKO-1 cells. By contrast, Len/VD3 markedly increased that of Bik in the four sensitive cell lines (JEKO-1, Z-138, MINO and REC-1) and not in the two resistant (GRANTA-519 and UPN-1) cell lines (Figure 3). Furthermore, the expression of the other BH3-only proteins (i.e., Bid, Bad, Bim, Bax and Bak) and of the anti-apoptotic proteins (i.e., Bcl-2, Bcl-xL and Mcl-1) was not affected by the treatment.

The combined Len/VD3 treatment induced BIK expression in sensitive cells. MCL cells (2 × 105 cells/ml) were incubated for 4 days with or without 1 μM Len and 100 nM VD3. Cells were then lysed and expression of the indicated proteins was assessed (western blotting)
Bik is known to interact with anti-apoptotic proteins, mainly Bcl-2 and Bcl-xL.19, 22, 26 We performed an IP assay to evaluate the binding of Bik in control and Len/VD3-treated JEKO-1 cells. As shown in Figure 4a, the Bik IP assay confirmed that Bik was bound to Bcl-2 but not to Bcl-xL or Mcl-1. In Len/VD3-treated cells, Bik was strongly associated with Bcl-2, weakly with Bcl-xL and not at all with Mcl-1, and a proportion was found free from Bcl-2 and Bcl-xL (Figure 4b).

A pool of Len/VD3-induced Bik protein was free from Bcl-2 and Bcl-xL. (a) IP assay of Bik in JEKO-1 cells. The Bik IP assay was performed in lysate of cells (107 cells) cultured for 4 days with or without 1 μM Len and 100 nM VD3. The expression of Bik, Bcl-2, Bcl-xL and Mcl-1 was analyzed in pellet (IP) and supernatant (OUT). (b) Reciprocal Bcl-2 and Bcl-xL IP assays. IP assays were performed by the simultaneous addition of both anti-Bcl-2 and anti-Bcl-xL antibodies to the lysates. The protein expression was analyzed in the total lysate (IN), pellet (IP) and supernatant (OUT) fractions
Silencing of BIK inhibited the apoptosis induced by Len/VD3
To directly investigate the implication of Bik in Len/VD3-induced apoptosis, we transiently transfected JEKO-1 and MINO cells with siBIK RNA to prevent an increase in Bik expression. To this end, cell transfection was performed at day 2 after the addition of Len/VD3. At day 5, the induction of Bik expression in Len/VD3-treated cells was reduced by 80±5% in JEKO-1 cells and 90±10% in MINO cells in the presence of siBIK RNA (Figure 5a).

The silencing of BIK decreased cell death induced by Len/VD3. (a–d) MCL cells (5 × 105/ml) were seeded for 48 h with or without Len/VD3 prior or not to transfection with siControl (siCt) or siBIK RNA, and the cells were reseeded (5 × 105 cells/ml) for an additional 3 days with or without Len/VD3. (a) Western blotting analysis of Bik expression. Bik expression was assessed in siCt and siBIK MCL cells that were or were not treated with combined Len/VD3. A representative experiment out of three is shown. (b) The silencing of BIK prevented the Len/VD3-induced cell growth inhibition. The cellular density was determined by direct counting. The data represent four independent experiments. (c) The silencing of BIK decreased Len/VD3-induced death. The cells were stained with Annexin V. The data represent four independent experiments. (d) The silencing of BIK decreased Len/VD3-induced subG1 peak. The cells were stained with PI. The data represent four independent experiments. (e–g): JEKO-1 cells (5 × 105/ml) were seeded for 48 h with or without Len/VD3 prior or not to transfection with siCt or siNOXA or siPUMA RNA, and the cells were reseeded (5 × 105 cells/ml) for an additional 3 days with or without Len/VD3. The data represent three independent experiments. (e) Western blotting analysis of Puma and Noxa expression in JEKO-1 cells. A representative experiment out of three is shown. (f) SiNOXA and siPUMA RNA did not prevent Len/VD3-induced inhibition of growth. The cellular density was determined by direct counting. The data represent three independent experiments. (g) SiNOXA and siPUMA RNA did not inhibit Len/VD3-induced cell death. Cells were stained with Annexin V. The data represent three independent experiments. *P<0.05, **P<0.01. NS, not significant
The transfection with siBIK prevented by 88±13% and 72±25% the Len/VD3-induced decrease in cell number in JEKO-1 and MINO cells, respectively, (P<0.01 and <0.01, respectively, paired t-test): the mean Len/VD3-induced decrease in cell number was 60±9% and 55±7% in siCt JEKO-1 and MINO cells, respectively, versus 6±8% and 16±15% in siBIK JEKO-1 and MINO cells, respectively (Figure 5b).
Similarly, the transfection with siBAX reduced by 38±19% and 37±10% the Len/VD3-induced cell death in JEKO-1 and MINO cells, respectively, (P<0.01 and P=0.02, respectively), but it did not significantly induce cell death in untreated cells (P=1 and 0.3, respectively): the mean cell death was 12±2% and 27±5% in untreated and Len/VD3-treated siCt JEKO-1 cells, respectively, versus 12±1% and 22±5% in untreated and Len/VD3-treated siBIK JEKO-1 cells, respectively, and 10±2% and 27±6% in cells untreated and Len/VD3-treated siCt MINO cells, respectively, versus 11±3% and 21±5% in siBIK MINO cells, respectively (Figure 5c). Similarly, siBIK RNA significantly inhibited the appearance of the subG1 peak in both JEKO-1 (P=0.01) and MINO (P=0.03) cells treated with Len/VD3 (63±9% and 73±12% of inhibition, respectively; Figure 5d). Because expression of Puma and Noxa was slightly induced by Len/VD3 in JEKO-1 cells, we assessed their role using siRNA experiments (Figure 5e). The transfection of siNOXA or siPUMA neither impaired cell number nor induced apoptosis (Figures 5f and g). The silencing of Puma or Noxa did not prevent the Len/VD3-induced decrease in cell number, which was 48±5%, 41±6% and 48±7% in siCt, siNOXA and siPUMA cells, respectively (Figure 5f). Similarly, the silencing of Puma or Noxa did not prevent the Len/VD3-induced cell death: the mean cell death was 21±2%, 21±1% and 22±2% in siCt, siNOXA and siPUMA cells, respectively (Figure 5g). Altogether, these data demonstrated that Bik mediated the Len/VD3-induced apoptosis.
Len/VD3 treatment increased the proportion of unmethylated BIK CpG islands and mimicked 5-aza
The transcription of the BIK gene is positively regulated by the transcription factor TEF and negatively regulated by the methylation of CpG islands.22, 23, 27, 28, 29 As shown in Figure 6a, BIK mRNA expression was increased by 13±1.3-fold (P=0.003) in the sensitive JEKO-1 cells and by 1.3±0.2-fold (P=0.1) in the resistant GRANTA-519 cells. Because expression of TEF remained unchanged (Supplementary Figure S2), we assessed whether the increase in BIK expression was related to a modulation of BIK CpG islands methylation. We determined the level of methylated over unmethylated BIK CpG islands located within intron 1 by methylation-specific PCR, as previously described by Hatzimichael et al.23 (Supplementary Figure S3). Although the methylation level of the BIK CpG islands was variable both within cell lines and patients’ samples, it appeared that BIK CpG islands were mainly methylated with a methylated over unmethylated ratio ranging from 2.3 to 8.2 in cell lines and from 1.4 to 8.9 in primary samples (Figure 6b). We then assessed the proportion of methylated over unmethylated BIK CpG islands in cells treated with Len/VD3 or the methylation inhibitor 5-aza. As shown in Figure 6c, both Len/VD3 and 5-aza treatments decreased the level of methylated BIK CpG islands and increased that of unmethylated BIK CpG islands. The median methylated over unmethylated ratio in JEKO-1, Z-138 and MINO cells decreased 3.3-fold (range 2–5) and 4-fold (range 3–5) with Len/VD3 and 5-aza, respectively, while it remained unchanged in the resistant GRANTA-519 cells.

The Len/VD3 treatment increased BIK mRNA expression via the demethylation of CpG islands. (a) The Len/VD3 treatment increased BIK mRNA expression. JEKO-1 cells were treated for 4 days with 1 μM Len and 100 nM VD3. BIK mRNA expression was quantified by qRT-PCR assay. The data represent the mean±S.E. of three independent experiments. (b) BIK CpG islands were constitutively methylated in MCL cell lines and patient samples (P1–P4). Methylation-specific PCR was performed on genomic DNA as described within the Material and Methods section and in Supplementary Figure S3. (c) The Len/VD3 treatment induced the demethylation of BIK CpG islands. MCL cells were treated for 4 days with 1 μM Len and 100 nM VD3 or for 3 days with 1 μM 5-aza. Methylation-specific PCR was performed on genomic DNA. (d) 5-aza increased expression of BIK mRNA. MCL cells were treated for 3 days with 1 μM 5-aza. qRT-PCR assay was performed as described within the legend of Figure 6a. The data represent the mean±S.E. of three independent experiments. (e) 5-aza induced expression of Bik. MCL cells (2 × 105/ml) were incubated for 3 days with or without 1 μM 5-aza. Cells were then lysed and expression of the indicated proteins was assessed by western blotting. (f) 5-aza induced cell death in MCL cell lines sensitive to Len/VD3 treatment. Cells (2 × 105/ml) were treated for 3 days with 1 μM 5-aza and cell viability was assessed using MTT assay. The data represent the mean±S.E. of three independent experiments. *P<0.05, **P<0.01
The 5-aza-induced decrease in methylated over unmethylated ratio was associated with an increase in BIK expression: the fold increase was 13±2 and 0.9±0.4 in JEKO-1 and GRANTA-519 cells, P=0.001 and 0.1, respectively, Figure 6d. The increase in BIK expression was confirmed at the protein level and except Noxa, expression of which was increased in JEKO-1 and Z-138 cells, expression of the other Bcl-2 family proteins was not significantly modified in both cell lines; Figure 6e. Interestingly, 5-aza decreased the viability in MCL cell lines sensitive to Len/VD3 (JEKO-1, MINO, Z-138 and REC-1) but not in resistant cell lines (GRANTA-519 and UPN-1), as shown in Figure 6f.
Discussion
Recently, novel targeted therapies have been investigated in MCL. Among these therapies, Len used alone or in combination has proven clinical efficacy and has emerged as a new promising therapy.30 Indeed, an upcoming phase III clinical trial conducted by the European MCL network will address the question of Len maintenance in elderly patients.31 Despite its proven efficacy in various hematological malignancies, questions regarding both Len’s mechanisms of action and the mechanisms underlying the sensitivity/resistance of tumor cells to Len remain. In the present work, we demonstrated that VD3 enhanced the sensitivity of MCL cells to Len and found that the pro-apoptotic protein Bik functions as a mediator of Len/VD3-induced cell death. Although the addition of VD3 to Len significantly increased cell death, it did not overcome resistance to Len in cell lines or primary cells. The resistance to Len or to Len/VD3 treatment was not owing to a lack of CRBN or VDR expression because all cell lines expressed both receptors, as shown in Supplementary Figure S1.24, 25 We can also exclude the involvement of a defect in the VD3 pathway because VD3 treatment, in both sensitive and resistant cell lines, induced one of its main primary target genes, the VDR. It is interesting to note that GRANTA-519 is EBV-positive, similar to the JVM2 cell line, which was also resistant to both Len and Len/VD3 treatment (data not shown). Thus, among EBV-negative MCL cell lines, only UPN-1 appeared to be resistant to Len and Len/VD3, and UPN-1 is known to contain an Rb deletion, which could contribute to the observed Len resistance. Our experiments demonstrated that the combined Len/VD3 treatment induced a slowdown in cell cycle progression, as indicated by a reduction of phases S and G2M, and an increase in the G1 phase, and this effect was confirmed by a marked decrease in the cell concentration and by the induction of apoptosis. While the expression of the anti-apoptotic proteins Bcl-2, Bcl-xL and Mcl-1 was not altered, that of BH3-only proteins was increased with Len/VD3 treatment. The expression of Noxa or Puma was slightly modulated by Len/VD3 in some cell lines, while that of Bik was markedly increased in all responsive cell lines. As expected, the marked Bik increase was directly involved in apoptosis because Bik silencing significantly reduced apoptosis. By contrast, Len/VD3 did not induce any increase in Bcl-2 or Bcl-xL expression, and IP experiments showed that a pool of Bik was free from its partners Bcl-2 and Bcl-xL. The involvement of Bax in Bik-mediated killing of cells, which has been well established, was further confirmed by the results of its direct silencing.32 However, the lack of both constitutive Bim expression and impact of Puma silencing could suggest a direct activation of Bax. Indeed, endothelial reticulum containing the whole Bik protein was shown to promote Bax membrane insertion and cytochrome c release, which was inhibited by Bcl-2.33, 34 Although Bik was considered as a sensitizer only, the recent study of Du et al.35 demonstrated that BH3 domains other than Bim and Bid, including Bik, can directly activate Bax/Bak in Bim and Bid double knockout MEF cells. Of note, Len/VD3 significantly increased Bik expression without modifying that of the other pro or anti-apoptotic molecules. Thus, in the context of MCL cells, which do not express Bim and express a low level of Bid, the marked Len/VD3-induced increase in Bik expression could directly mediate Bax activation or deplace Bax from Bcl-2/Bax complexes. We showed that the increase in Bik protein expression was related to an increase in BIK mRNA, which was not related to an increase in TEF expression.22 Of note, all of the cell lines expressed TEF, although all of them did not express Bik, suggesting that another mechanism of regulating BIK expression is present in MCL cells. The BIK gene is silenced by methylation in several cancer types, and its upregulation by methylation inhibitors such as 5-aza could contribute to the pro-apoptotic activity of these inhibitors.27, 29 Indeed, we demonstrated that Len/VD3, as well as 5-aza, increased the proportion of unmethylated BIK CpG islands in the sensitive JEKO-1, MINO and Z-138 cell lines but not in the resistant GRANTA-519 cells. Among all of the BH3-only genes, Bik exhibited the greatest change in expression upon the inhibition of methylation in the two cell lines tested. Noxa and Puma expression was also increased but, in contrast to Bik and Noxa, increase in Puma expression was not found in both cell lines. Bim is rarely expressed in MCL cell lines and we did not find that the Len/VD3 treatment or 5-aza induced its expression, confirming that the lack of Bim expression is mostly governed by a chromosomal deletion.36, 37, 38 Interestingly, the sensitivity of MCL cell lines to Len/VD3 and 5-aza was similar and both treatments increased the expression of Bik. Thus, at least in MCL cells, Bik appears to be the BH3-only protein that is commonly induced or increased via demethylation. Similar results were reported in renal carcinoma cells, in which inhibition of methylation significantly induced a strong increase in BIK expression but not in other Bcl-2 family molecules.29 However, the precise molecular mechanism leading to BIK demethylation after exposure to the Len/VD3 treatment remains to be elucidated. DNA methylation and histone modifications are the two arms of epigenetic regulation, and inhibitors of DNA methylation or histone deacetylases (HDAC) are useful in cancer therapy.39, 40 Len and VD3 have a wide range of effects on cancer cells, including modifications of gene expression, particularly of genes involved in transcription.14, 41, 42 VD3 is known to induce the expression of histone demethylase genes such as JMJD3.43 However, because an HDAC inhibitor failed to induce cell death or Bik induction in MCL cell lines (data not shown), histone deacetylation in unlikely to be involved in Len/VD3 activity.
We assessed whether expression of DNA methyl transferases (DNMT) was modulated upon Len/VD3 treatment. DNMT1 is involved in methylation maintenance, whereas DNMT3a and 3b are involved in de novo methylation.44 Len/VD3 decreased DNMT3b expression, but not that of DNMT1 or DNMT3a, in both responsive and resistant cell lines (data not shown). Thus, decrease in DNMT3b expression might be necessary for the unmethylation of BIK CpG islands induced by Len/VD3 in sensitive cell lines, but it does not remain sufficient, as it also occurred in resistant cells. Lack of methylation could be also related to an active demethylation instead of an inhibition of methylation. Indeed, several pathways involving base excision repair have been described but the mechanisms remain poorly understood.44 The precise mechanism leading to inhibition of methylation or to active demethylation of BIK CpG islands remains to be deciphered.
In the primary MCL samples, Len/VD3 treatment was efficient, but the magnitude of cell death induced was lower in comparison with that of the cell lines. This weaker efficacy could be related to the very low proliferation index of circulating MCL, which proliferate in the lymph nodes but not in peripheral blood. The demethylation of BIK CpG islands induced by Len/VD3 occurred over several days and impacted proliferation, suggesting that Len/VD3 treatment inhibited methylation after replication. Because we failed to induce proliferation of peripheral MCL cells upon exposure to CD40L, TLR agonists and interleukines, and could not obtain samples from lymph nodes, this hypothesis could not be assessed yet.
In conclusion, these data demonstrate for the first time that combined Len/VD3 treatment induces cell death in MCL cells via the induction of Bik expression owing to the demethylation of its promoter, a mechanism that is similar to that of 5-aza. The potential correlation between VD3 levels and the response to Len, and whether VD3 supplementation could improve the response rate of MCL patients treated with Len remain to be defined in vivo. Our findings support the investigation of VD3 level in patients with MCL and generate interest in retrospective or prospective studies, investigating the relationship between the response to Len and serum VD3 levels.
Materials and Methods
MCL cells and cell lines
The MCL cell lines JEKO-1, MINO, REC-1 and GRANTA-519 were purchased from DSMZ (Braunschweig, Germany), Z-138 was purchased from ATCC (Manassas, VA, USA) and UPN-1 was kindly provided by Dr. V Ribrag (Institut Gustave Roussy, Villejuif, France). Each MCL cell line was identified by a complete phenotypic analysis and a HLA Class I typing. MCL cell lines were maintained in RPMI-1640 medium supplemented with 10% FCS and 2 mM glutamine. Blood samples from patients with MCL at the time of diagnosis were collected after informed consent was obtained at the Department of Hematology at University Hospital of Nantes.19 Peripheral MCL cells from the blood were purified after Ficoll–Hypaque separation using immunomagnetic anti-CD19 beads when MCL infiltration was <90% (Class, Paris, France).
Reagents
The active form of VD3, 1,25-dihydroxyvitamin D3 and 5-aza-2′-deoxycytidine (5-aza) were purchased from Sigma-Aldrich (Lyon, France). Len was provided by Celgene Corporation (San Diego, CA, USA).
Viability and cell cycle assays
Cell viability was evaluated by MTT assay. Cells (2 × 104 cells in 100 μl) were seeded for 6 days in 96-well plates and treated with 100 nM VD3 and 1 μM Len either alone or in combination. Cell death was assessed using an Annexin V staining kit (Beckman Coulter, Marseille, France). The cell cycle stage was assessed using propidium iodide (PI) staining (Beckman Coulter). Flow cytometric analysis was performed on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA) using Cell Quest software.
Western blotting
The analysis of protein expression was conducted by western blotting, as previously described.20 The following primary antibodies were used: Mcl-1 (S19, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Bik (FL160, Santa Cruz Biotechnology), Puma (Calbiochem, Merck, Darmstack, Germany), Bcl-xL, Bad, Bid, Bak (BD Biosciences, Le Pont de Claix, France), Bcl-2, caspase 9, (Cell Signalling, Saint Quentin en Yvelines, France), actin (Millipore Bioscience Research Reagents, Merck, Saint-Quentin en Yvelines, France), Noxa (Alexis, Paris, France) and Bax (Enzo Life Sciences, Villeurbanne, France). The following secondary antibodies were used: rabbit anti-goat (Jakson ImmunoResearch, West Baltimore Pike, PA, USA), anti-mouse/anti-rabbit (Roche, Boulogne-Billancourt, France) and goat anti-rabbit (Jakson ImmunoResearch).
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was estimated using the potential-sensitive fluorescent probe JC-1 (Life Technologies, Saint-Aubin, France). Cells were incubated in Hank’s Balanced Salt Solution (Gibco Life Technologies, Saint-Aubin, France) with JC-1 at 5 μg/ml for 30 min at 37 °C. Fluorescence was analyzed on a FACSCalibur.
RNA extraction, reverse transcription and PCR assays
Extraction, reverse transcription of RNA and quantitative PCR was performed as previously described by using Taqman (Applied Biosystems) BIK (Hs00154189_m1) and RPL37a (Hs01102345_m1, housekeeping gene) probes.21 RT-PCR of TEF (thyrotroph embryonic factor) expression was performed as previously described.22
siRNA transient transfections
Control nontargeted small interfering siRNA (siControl, siCt), siBAX, siBIK, siNOXA and siPUMA were purchased from Thermo Scientific (Courtaboeuf, France). MCL cell lines were electroporated using a Nucleofector system (Amaxa, Lonza, Basel, Switzerland). Cells (5 × 105 cells/ml) were treated for 48 h with 1 μM Len and 100 nM VD3, resuspended in the selected Nucleofector solution (R for JEKO-1 and T for MINO), electroporated in the presence of 10 μmol/l siRNA (A23 Nucleofector program for JEKO-1 and T01 for MINO) and reseeded in culture for the remaining days in the presence of 1 μM Len and 100 nM VD3.
Immunoprecipitation
Immunoprecipitation (IP) assays were conducted in treated and untreated cells (control). Cells were lysed for 40 min on ice in 10 mmol/l Tris-HCl, pH=7.6, 150 mmol/l NaCl, 5 mmol/l EDTA, 1 mmol/l phenylmethylsulfonylfluoride, 2 mg/ml aprotinin and 1% digitonin. The lysates were immunoprecipitated with a mix of proteins A and G.
Methylation-specific PCR
Methylation of the BIK CPG islands was assessed through methylation-specific PCR, as previously described by Hatzimichael et al.23 In brief, genomic DNA was treated with bisulfite (EZ DNA Methylation Kit, ZYMO Research, Proteigene, Saint Marcel, France) and 35 amplification cycles were performed in 25 μl with methylated forward 5′-GGGAGTCGTGTTTAGGTTTTATC-3′ and reverse 5′-GAACAAAAAAAATACGTTTCGAA-3′ primers or with unmethylated forward 5′-GGGGAGTTGTGTTTAGGTTTTATT-3′ and reverse 5′-CAAACAAAAAAAATACATTTCAAA-3′ primers.
Statistical analyses
Statistical analyses were performed using a paired Student’s t-test or a Wilcoxon matched-pairs signed rank test.
Abbreviations
- BIK:
-
BCL2-interacting killer
- CRBN:
-
cereblon
- Len:
-
lenalidomide
- VD3:
-
1α,25-dihydroxyvitamin D3
- IP:
-
immunoprecipitation
- NHL:
-
non-Hodgkin’s lymphoma
- MCL:
-
mantle cell lymphoma
- VDR:
-
VD3 receptor
- 5-aza:
-
5-aza-2′-deoxycytidine
References
Andersen NS, Jensen MK, de Nully Brown P, Geisler CH . A Danish population-based analysis of 105 mantle cell lymphoma patients: incidences, clinical features, response, survival and prognostic factors. Eur J Cancer 2002; 38: 401–408.
Zhou Y, Wang H, Fang W, Romaguer JE, Zhang Y, Delasalle KB et al. Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 2008; 113: 791–798.
Harel S, Delarue R, Ribrag V, Dreyling M, Hermine O . Treatment of younger patients with mantle cell lymphoma. Semin Hematol 2011; 48: 194–207.
Eve HE, Carey S, Richardson SJ, Heise CC, Mamidipudi V, Shi T et al. Single-agent lenalidomide in relapsed/refractory mantle cell lymphoma: results from a UK phase II study suggest activity and possible gender differences. Br J Haematol 2012; 159: 154–163.
Pan B, Lentzsch S . The application and biology of immunomodulatory drugs (IMiDs) in cancer. Pharmacol Ther 2012; 136: 56–68.
Wiernik PH, Lossos IS, Tuscano JM, Justice G, Vose JM, Cole CE et al. Lenalidomide monotherapy in relapsed or refractory aggressive non-Hodgkin's lymphoma. J Clin Oncol 2008; 26: 4952–4957.
Dawar R, Hernandez-Ilizaliturri F . The emerging role of lenalidomide in the management of mantle cell lymphoma (MCL). Best Pract Res Clin Haematol 2012; 25: 185–190.
Witzig TE, Vose JM, Zinzani PL, Reeder CB, Buckstein R, Polikoff JA et al. An international phase II trial of single-agent lenalidomide for relapsed or refractory aggressive B-cell non-Hodgkin's lymphoma. Ann Oncol 2011; 22: 1622–1627.
Goy A, Sinha R, Williams ME, Kalayoglu Besisik S, Drach J, Ramchandren R et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 2013; 31: 3688–3695.
Chanan-Khan AA, Chitta K, Ersing N, Paulus A, Masood A, Sher T et al. Biological effects and clinical significance of lenalidomide-induced tumour flare reaction in patients with chronic lymphocytic leukaemia: in vivo evidence of immune activation and antitumour response. Br J Haematol 2011; 155: 457–467.
Gorgun G, Calabrese E, Soydan E, Hideshima T, Perrone G, Bandi M et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010; 116: 3227–3237.
Rosen CJ, Adams JS, Bikle DD, Black DM, Demay MB, Manson JE et al. The nonskeletal effects of vitamin D: an Endocrine Society scientific statement. Endocr Rev 2012; 33: 456–492.
Manson JE, Mayne ST, Clinton SK . Vitamin D and prevention of cancer–ready for prime time? N Engl J Med 2011; 364: 1385–1387.
Grant WB . Relation between prediagnostic serum 25-hydroxyvitamin D level and incidence of breast, colorectal, and other cancers. J Photochem Photobiol B 2010; 101: 130–136.
Drake MT, Maurer MJ, Link BK, Habermann TM, Ansell SM, Micallef IN et al. Vitamin D insufficiency and prognosis in non-Hodgkin's lymphoma. J Clin Oncol 2010; 28: 4191–4198.
Deeb KK, Trump DL, Johnson CS . Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer 2007; 7: 684–700.
Nemazannikova N, Antonas K, Dass CR . Vitamin D: Metabolism, molecular mechanisms, and mutations to malignancies. Mol Carcinog 2013; 53: 421–431.
Brosseau C, Colston K, Dalgleish AG, Galustian C . The immunomodulatory drug lenalidomide restores a vitamin D sensitive phenotype to the vitamin D resistant breast cancer cell line MDA-MB-231 through inhibition of BCL-2: potential for breast cancer therapeutics. Apoptosis 2012; 17: 164–173.
Touzeau C, Dousset C, Bodet L, Gomez-Bougie P, Bonnaud S, Moreau A et al. ABT-737 induces apoptosis in mantle cell lymphoma cells with a Bcl-2high/Mcl-1low profile and synergizes with other antineoplastic agents. Clin Cancer Res 2011; 17: 5973–5981.
Surget S, Chiron D, Gomez-Bougie P, Descamps G, Menoret E, Bataille R et al. Cell death via DR5, but not DR4, is regulated by p53 in myeloma cells. Cancer Res 2012; 72: 4562–4573.
Maiga S, Gomez-Bougie P, Bonnaud S, Gratas C, Moreau P, Le Gouill S et al. Paradoxical effect of lenalidomide on cytokine/growth factor profiles in multiple myeloma. Br J Cancer 2013; 108: 1801–1806.
Bodet L, Menoret E, Descamps G, Pellat-Deceunynck C, Bataille R, Le Gouill S et al. BH3-only protein Bik is involved in both apoptosis induction and sensitivity to oxidative stress in multiple myeloma. Br J Cancer 2010; 103: 1808–1814.
Hatzimichael E, Dasoula A, Kounnis V, Benetatos L, Lo Nigro C, Lattanzio L et al. Bcl2-interacting killer CpG methylation in multiple myeloma: a potential predictor of relapsed/refractory disease with therapeutic implications. Leuk Lymphoma 2012; 53: 1709–1713.
Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012; 26: 2326–2335.
Zhu YX, Braggio E, Shi CX, Bruins LA, Schmidt JE, Van Wier S et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011; 118: 4771–4779.
Gillissen B, Essmann F, Hemmati PG, Richter A, Oztop I, Chinnadurai G et al. Mcl-1 determines the Bax dependency of Nbk/Bik-induced apoptosis. J Cell Biol 2007; 179: 701–715.
Kim TY, Zhong S, Fields CR, Kim JH, Robertson KD . Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res 2006; 66: 7490–7501.
Ritchie A, Gutierrez O, Fernandez-Luna JL . PAR bZIP-bik is a novel transcriptional pathway that mediates oxidative stress-induced apoptosis in fibroblasts. Cell Death Differ 2009; 16: 838–846.
Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S et al. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ 2006; 13: 619–627.
Wang M, Fayad L, Wagner-Bartak N, Zhang L, Hagemeister F, Neelapu SS et al. Lenalidomide in combination with rituximab for patients with relapsed or refractory mantle-cell lymphoma: a phase 1/2 clinical trial. Lancet Oncol 2012; 13: 716–723.
Dreyling M, Kluin-Nelemans HC, Bea S, Klapper W, Vogt N, Delfau-Larue MH et al. Update on the molecular pathogenesis and clinical treatment of mantle cell lymphoma: report of the 11th annual conference of the European Mantle Cell Lymphoma Network. Leuk Lymphoma 2012; 54: 699–707.
Gillissen B, Essmann F, Graupner V, Starck L, Radetzki S, Dorken B et al. Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J 2003; 22: 3580–3590.
Germain M, Mathai JP, Shore GC. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J Biol Chem 2002; 277: 18053–18060.
Mathai JP, Germain M, Shore GC, BH3-only BIK . regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem 2005; 280: 23829–23836.
Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T . BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem 2011; 286: 491–501.
Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood 2007; 109: 271–280.
Tagawa H, Karnan S, Suzuki R, Matsuo K, Zhang X, Ota A et al. Genome-wide array-based CGH for mantle cell lymphoma: identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005; 24: 1348–1358.
Perez-Galan P, Dreyling M, Wiestner A . Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood 2011; 117: 26–38.
Khan O, La Thangue NB . HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 2012; 90: 85–94.
Ren J, Singh BN, Huang Q, Li Z, Gao Y, Mishra P et al. DNA hypermethylation as a chemotherapy target. Cell Signal 2011; 23: 1082–1093.
Kim K, An S, Cha HJ, Choi YM, Choi SJ, An IS et al. Lenalidomide induces apoptosis and alters gene expression in non-small cell lung cancer cells. Oncol Lett 2013; 5: 588–592.
Hickey CJ, Schwind S, Radomska HS, Dorrance AM, Santhanam R, Mishra A et al. Lenalidomide-mediated enhanced translation of C/EBPalpha-p30 protein up-regulates expression of the antileukemic microRNA-181a in acute myeloid leukemia. Blood 2013; 121: 159–169.
Pereira F, Barbachano A, Singh PK, Campbell MJ, Munoz A, Larriba MJ . Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle 2012; 11: 1081–1089.
Chen ZX, Riggs AD . DNA methylation and demethylation in mammals. J Biol Chem 2011; 286: 18347–18353.
Acknowledgements
This work was supported by grants from Ligue Régionale Contre le Cancer (2013), Région des Pays de la Loire and INCa (PAIR program). We thank Celgene for providing us with lenalidomide.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Author contributions
CB participated in the design of the study, performed experiments and participated in the writing of the paper. CD and SM performed experiments. CT participated in the design of the study and collected patients’ samples. PM and MA participated in the design of the study and in the writing of the paper. SLG and CPD designed the study and wrote the paper.
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Brosseau, C., Dousset, C., Touzeau, C. et al. Combination of lenalidomide with vitamin D3 induces apoptosis in mantle cell lymphoma via demethylation of BIK. Cell Death Dis 5, e1389 (2014). https://doi.org/10.1038/cddis.2014.346
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.346
This article is cited by
-
Prognostic value of 25-hydroxy vitamin D in extranodal NK/T cell lymphoma
Annals of Hematology (2021)
-
Characterization of an alternative BAK-binding site for BH3 peptides
Nature Communications (2020)

{kind=link}
{kind=link}
{kind=link}