
Abstract
Members of the WD-repeat protein interacting with phosphoinositides (WIPI) family are phosphatidylinositol 3-phosphate (PI3P) effectors that are essential for the formation of autophagosomes. Autophagosomes, unique double-membraned organelles, are characteristic for autophagy, a bulk degradation mechanism with cytoprotective and homeostatic function. Both, WIPI-1 and WIPI-2 are aberrantly expressed in several solid tumors, linking these genes to carcinogenesis. We now found that the expression of WIPI-1 was significantly reduced in a large cohort of 98 primary acute myeloid leukemia (AML) patient samples (complex karyotypes; t(8;21); t(15,17); inv(16)). In contrast, the expression of WIPI-2 was only reduced in acute promyelocytic leukemia (APL), a distinct subtype of AML (t(15,17)). As AML cells are blocked in their differentiation, we tested if the expression levels of WIPI-1 and WIPI-2 increase during all-trans retinoic acid (ATRA)-induced neutrophil differentiation of APL. According to the higher WIPI-1 expression in granulocytes compared with immature blast cells, WIPI-1 but not WIPI-2 expression was significantly induced during neutrophil differentiation of NB4 APL cells. Interestingly, the induction of WIPI-1 expression was dependent on the transcription factor PU.1, a master regulator of myelopoiesis, supporting our notion that WIPI-1 expression is reduced in AML patients lacking proper PU-1 activity. Further, knocking down WIPI-1 in NB4 cells markedly attenuated the autophagic flux and significantly reduced neutrophil differentiation. This result was also achieved by knocking down WIPI-2, suggesting that both WIPI-1 and WIPI-2 are functionally required and not redundant in mediating the PI3P signal at the onset of autophagy in NB4 cells. In line with these data, downregulation of PI3KC3 (hVPS34), which generates PI3P upstream of WIPIs, also inhibited neutrophil differentiation. In conclusion, we demonstrate that both WIPI-1 and WIPI-2 are required for the PI3P-dependent autophagic activity during neutrophil differentiation, and that PU.1-dependent WIPI-1 expression is significantly repressed in primary AML patient samples and that the induction of autophagic flux is associated with neutrophil differentiation of APL cells.
Similar content being viewed by others


Main
Macroautophagy (hereafter referred to as autophagy), or cellular self-digestion, is: (a) involved in the maintenance of cellular homeostasis, (b) responsible for a constitutive turnover of cytoplasmic material and long-lived proteins that are either damaged or functionally redundant, (c) highly conserved, and (d) linked to a variety of diseases including neurodegenenerative disorders and cancer.1, 2, 3 The ubiquitin–proteasome pathway, on the other hand, rather participates in the degradation of short-lived proteins.4 Autophagy mainly consists of four steps and follows a hierarchical ordered recruitment of autophagy related (ATG) proteins to the phagophore assembly site (PAS). Firstly, the initiation step critically involves the ULK1 complex, which regulates the following nucleation step by activating phosphatidylinositol 3-kinase class III (PI3KC3) kinases ultimately resulting in the formation of an autophagosome precursor, called phagophore. Further steps include the activity of two ubiquitin-like conjugation systems, and the product LC3-PE (or LC3-II) which is required for phagophore elongation and closure to generate an autophagosome.5
During the nucleation step, PI3KC3 is acting in concert with Beclin 1, VPS15 and ATG14L to produce PI3P. This PI3P signal is essential for autophagosome formation as evidenced by the fact that the use of PI3K inhibitors (wortmannin, 3-MA, LY29002) at concentrations preferentially blocking PI3KC3-abolished autophagy6, 7, 8 (reviewed in Petiot et al.9). Transduction of the PI3P signal occurs via specific PI3P-binding effectors, including the FYVE-domain protein DFCP110 and the WD-repeat protein interacting with phosphoinositides (WIPI) beta-propeller protein family.11 The human WIPI family consists of four members (WIPI-1 to WIPI-4) with different isoforms.11 To date, human WIPI-1, WIPI-2 and WIPI-4 proteins were functionally associated with the process of autophagy.11, 12, 13, 14 WIPI proteins function as autophagic PI3P effector proteins with autophagosomal membrane binding capacity,15 and thereby translocate from the cytosol to the forming PAS. They subsequently become membrane proteins of autophagosomal membranes, visible by fluorescent microscopy as punctate structures.15
Acute myeloid leukemia (AML) is characterized by a block of myeloid differentiation at different stages and represents the most common acute leukemia in adults.16 Current differentiation and cytotoxic therapies for AML, with the exception of a few subgroups with favorable genetic disease, need to be improved. Recently, we and others published that autophagy is essential for differentiation therapy of APL patients,17, 18, 19, 20, 21 a disease characterized by the translocation t(15;17) resulting in the expression of the oncogenic PML–RARA fusion protein and an accumulation of promyelocytic cells. Nevertheless, data on the molecular mechanisms underlying this myeloid differentiation-associated autophagy are scarce and further investigations of this particular autophagic pathway are warranted.
Here, we demonstrate that PI3P-dependent WIPI-mediated autophagy is required for neutrophil differentiation of AML cells. We found that WIPI mRNA expression is significantly repressed in clinical AML samples being associated with an immature myeloid differentiation phenotype. Genetically imparing WIPI or PI3KC3 but not Beclin 1 function resulted in significantly attenuated neutrophil differentiation of APL cells. Our data provide evidence that WIPI-mediated autophagy is critical for cellular myeloid leukemia differentiation and is compromised in AML.
Results
Aberrant WIPI expression in primary AML patient samples and PU.1-dependent induction of WIPI-1 expression during neutrophil differentiation of APL cells
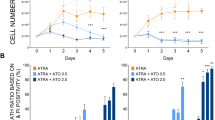
On the basis of the crucial role of WIPI proteins as PI3P effectors in the process of autophagy11, 12, 13, 15 and the initial findings, that all human WIPI genes are aberrantly and differentially expressed in a variety of solid tumors,11 we quantified WIPI-1 and WIPI-2 gene expression in clinical AML patient samples. We found that WIPI-1 mRNA abundance was significantly reduced in our cohort of AML patients (n=98) when compared with the expression of WIPI-1 in healthy granulocytes (n=13; Figure 1a, left panels). In more detail, WIPI-1 message was downregulated in complex karyotype (n=24), t(8,21) (n=20), t(15;17) (n=20) and inv(16) (n=16) AML subtypes but not in normal karyotype AML (n=18; Figure 1a, right panels). In contrast, WIPI-2 mRNA expression was only significantly inhibited in APL patients (t(15;17)), a distinct AML subtype (Supplementary Figure 1a). Moreover, WIPI-1 was threefold higher expressed in healthy granulocytes when compared with normal CD34+ progenitor cells (Figure 1a, left panels), whereas WIPI-2 was equally expressed in both of these cell populations (Supplementary Figure 1a). In summary, we found a positive association between low WIPI-1 expression and an immature myeloid phenotype.

Significantly decreased WIPI-1 mRNA expression in primary AML patient samples and increase in WIPI-1 expression upon ATRA treatment of NB4 APL cells was detected. (a) WIPI-1 mRNA levels of granulocytes from healthy donors and AML patient samples were measured using qPCR. Data represent log2 expression levels and were normalized to the expression levels of the two housekeeping genes HMBS and ABL. For better readability, we multiplied the results by (−1) and excluded Ct values higher than 40 (ΔCt=40−CtWIPI-1−(Mean CtHMBS and CtABL1) × (−1)). (b) mRNA levels of WIPI-1 expression was measured in control, and ATRA-treated NB4, NB4-R2 APL cell lines at day 4 using qPCR. Values were normalized to the housekeeping gene HMBS and given as n-fold mRNA expression relative to the control of day 4. (c, d) Increase in endogenous WIPI-1 puncta was detected using confocal microscopy and was further quantified using ImageJ software. (e) PI3P:WIPI-1 binding assay of cell lysates from NB4 cells treated with 1 μM ATRA for 6 days. Each membrane was loaded with equal total protein lysate. M.W.U, *P<0.05. **P<0.01, ***P<0.001. M.W.U, Mann–Whitney U-test
To functionally evaluate the role of both WIPI-1 and WIPI-2 in ATRA-induced neutrophil differentiation of myeloid leukemia cells, we used NB4 APL cells as a model. NB4 cells are differentiated towards neutrophil-like cells by administration of ATRA. In line with higher WIPI-1 expression in mature neutrophils when compared with AML blast or CD34+ progenitor cells (Figure 1a), ATRA treatment of NB4 cells resulted in a significant three and fourfold upregulation of WIPI-1 mRNA at day 4 and 6, respectively (Figure 1b, upper panel). Importantly, WIPI-1 mRNA levels did not change in the ATRA-resistant NB4-R2 cells (Figure 1b, lower panel) indicating that the upregulation of WIPI-1 expression is not due to a stochastic stress response to ATRA administration. Lastly, WIPI-2 mRNA levels did neither change during neutrophil differentiation of NB4 cells nor in NB4-R2 cells (Supplementary Figure 1b).
To monitor the autophagic status, we assessed the localization of endogenous WIPI-1 protein in NB4 cells during neutrophil differentiation by immunocytochemistry. Using this imaging method, the presence of fluorescent WIPI-1 puncta reliably reflects the formation of autophagosomal membranes.15 Indeed, a significant increase of WIPI-1 puncta per cell was measured upon ATRA-induced neutrophil differentiation (Figure 1c and d). As WIPI-1 specifically binds to PI3P,15 we asked whether increased WIPI-1 expression upon ATRA treatment (Figure 1b, upper panel) would result in a higher proportion of WIPI-1 protein binding to PI3P. Indeed, a phospholipid–protein overlay assay with native cell extracts revealed that endogenous WIPI-1 protein from ATRA-treated NB4 APL cells bound more prominently to immobilized PI3P when compared with untreated cells (n=3, a representative result is provided in Figure 1e). Satisfactory, this result also confirmed that endogenous WIPI-1 extracted from NB4 APL properly binds to PI3P.
To test if low WIPI-1 expression in immature AML blasts might be attributed to aberrant transcriptional regulation by myeloid transcription factors, we analyzed the 5.8 kb genomic region upstream of the WIPI-1 transcriptional start site for putative binding sites of the key myeloid transcription factor PU.1 using MatInspector. We identified 4 putative PU.1 binding sites (A–D) in the 5′-promoter region of WIPI-1 (Figure 2a). By using chromatin immunoprecipitation and anti-PU.1 antibodies, we found binding of PU.1 to all 4 PU.1 binding sites (A–D) in the 5′-promotor of human WIPI-1 (Figure 2b). In order to show PU.1-dependent regulation of WIPI-1 expression, we either knocked down or overexpressed PU.1 in NB4 APL cells. Knocking down PU.1 impaired induction of WIPI-1 gene expression upon ATRA treatment (Figure 2c), whereas tamoxifen treatment of the inducible PU.1-ER fusion protein led to a significant twofold induction of WIPI-1 expression (Figure 2d).

PU.1-dependent WIPI-1 regulation in NB4 APL cells. (a) Schematic representation of a 5.8-kb human WIPI-1 promoter fragment. Using MatInspector, 4 putative PU.1 binding sites (circles) are found in the human WIPI-1 promoter. (b) In vivo binding of PU.1 to the 4 binding sites was shown by ChIP in NB4 cells using antibodies against PU.1. Antibodies against acetyl-histone H3 and IgG served as positive and negative controls, respectively. GAPDH amplification was shown as a negative control for the different pull-downs. (c) Upper panel: WIPI-1 mRNA expression was measured in NB4 shPU.1 knockdown cells upon ATRA treatment at day 4. Lower panel: PU.1 western blot analysis of NB4 SHC002 control and PU.1 knockdown cells. Total protein expression was used as loading control. (d) NB4 cells, transduced with an inducible PU-1-ER expressing vector were treated with 4-OHT to induce PU.1 translocation to the nucleus. WIPI-1 mRNA levels were assessed by qPCR and normalized to the HMBS housekeeping gene. Results are given as n-fold regulation compared with untreated, control transduced NB4 pBabe cells. M.W.U, ***P<0.001. M.W.U, Mann–Whitney U-test
Together, we found that PU.1 regulates WIPI-1 expression during neutrophil differentiation.
Inhibiting WIPI-1 or WIPI-2 significantly attenuates neutrophil differentiation
We addressed the question whether or not WIPI-1 is required for ATRA-induced neutrophil differentiation by using lentivirally delivered shRNA targeting WIPI-1. Clearly, knocking down WIPI-1 impaired neutrophil differentiation as evidenced by significantly reduced CD11b protein and CEBPE mRNA levels, both bona fide markers for neutrophil differentiation of AML cell lines (Figures 3a–d, top row panels). Interestingly, knocking down WIPI-2 also resulted in impaired neutrophil differentiation (Figures 3a–d, second row panels). These results demonstrate that the neutrophil differentiation depends on WIPI function.

Impaired neutrophil differentiation in NB4 WIPI-1, WIPI-2, PI3KC3 but not in BECN1 knockdown cells. (a) SHC002, shWIPI-1, shWIPI-2, shPI3KC3 and shBECN1 expressing NB4 cells were differentiated for 4 days and knockdown efficiency was measured by qPCR. (b) Neutrophil differentiation was assessed by measuring CD11b surface expression with FACS analysis, a representative CD11b histogram is shown. (c) Bar graphs of the median fluorescence intensity of three independent experiments are shown. (d) CEBPE mRNA expression was measured by qPCR in SHC002, shWIPI-1, shWIPI-2, shPI3KC3 and shBECN1 expressing NB4 cells upon 1 μM ATRA treatment after 4 days. (e) NBT assay was performed at day 4 in SHC, shWIPI-1, shWIPI-2, shPI3KC3 and shBECN1 NB4 APL cells and NBT-positive cells were quantified from two independent experiments done in duplicate. (f) Relative transcript abundance for WIPI-1 and WIPI-2 was calculated as described in.12 M.W.U, *P<0.05. **P<0.01, ***P<0.001. M.W.U, Mann–Whitney U-test
To further address the PI3P-dependent initiation of autophagy during APL differentiation, we investigated if lentivirally delivered shRNA targeting of PI3KC3 or its regulatory subunit Beclin 1 (BECN1) interferes with ATRA-induced neutrophil differentiation. Knocking down PI3KC3 resulted in drastically impaired neutrophil differentiation (Figures 3a–d, third row panels). Accordingly, inhibiting PI3P production using the small compound PI3K inhibitor LY294002 that inhibits the formation of WIPI-1 puncta,13 also reduced the abundance of CD11b, marking NB4 differentiation (Supplementary Figure 2). However, inhibiting Beclin 1 did not affect ATRA-induced neutrophil differentiation as similar CD11b and CEBPE levels in NB4 control and Beclin 1-knockdown cells were found (Figures 3a–d, fourth row panels). This indicates that PI3P-dependent WIPI function at autophagy initiation is required for neutrophil differentiation of NB4 cells but that the regulatory subunit of the PI3KC3 complex Beclin 1 might not be involved in PI3P production in this cell type.
Importantly, we also tested if knocking down WIPI-1, WIPI-2, PI3KC3 or Beclin 1 affected neutrophil function using a nitroblue tetrazolium (NBT) assay that measures the production of reactive oxygen species (Figure 3e). Seventy-two percent of control cells transduced with non-targeting shRNAs reduced colorless NBT into intracellular dark deposits after 96 h of ATRA treatment (Figure 3e). Only 39% of WIPI-1 and 50% of WIPI-2 APL knockdown cells stained NBT positive (Figure 3e, quantification on the right). Similarly, knocking down PI3KC3 attenuated neutrophil function, whereas inhibiting Beclin 1 did not influence NBT reduction upon neutrophil differentiation (Figure 3e). This result further underlines that WIPI-1, WIPI-2 and PI3KC3 but not Beclin 1 participate in ATRA-induced neutrophil differentiation and function of myeloid leukemia cells.
Furthermore, direct comparison of relative WIPI-1 and WIPI-2 transcript levels revealed that WIPI-2 (90–99%) is expressed more in undifferentiated and differentiated NB4 APL cells than WIPI-1 (1–10%), which suggests a rate-limiting role of WIPI-1 during ATRA-induced neutrophil differentiation of NB4 cells (Figure 3f).
To control whether Beclin 1-knockdown cells are autophagy-competent, we measured LC3 puncta formation upon both short-term starvation and ATRA treatment.22, 23 As expected, knocking down Beclin 1 inhibited starvation-induced autophagy (Supplementary Figure 3a, black bars); however, knocking down Beclin 1 did not affect ATRA-induced autophagy (Supplementary Figure 3a, white bars). Moreover, we employed a biochemical standard method to determine the turnover of long-lived proteins by autophagy and measured the proteolysis of radiolabeled proteins in Beclin 1-knockdown cells.23 The proteolysis in Beclin 1-knockdown cells under basal conditions was not significantly decreased upon bafilomycin A1 treatment whereas ATRA-treated samples showed significantly reduced proteolysis upon combined treatment with bafilomycin A1 suggesting that basal but not ATRA-induced autophagy requires Beclin 1. Consistent with the role of Beclin 1 in starvation-induced autophagy, Beclin 1-knockdown cells did not show increased proteolysis during starvation compared to control cells (Supplementary Figure 3b), supporting a function for Beclin 1 in canonical autophagy found during starvation or during basal conditions but not during ATRA treatment. Of note, Beclin 1-independent entries to autophagic sequestration have been reported.5
WIPI-1- and WIPI-2-dependent autophagy during neutrophil differentiation
Next, we asked if the involvement of the PI3P effector proteins WIPI-1 and WIPI-2 in neutrophil differentiation can be attributed, at least in part, to their role in autophagy. To address this question, we analyzed the autophagic flux in NB4 WIPI-1 and WIPI-2 knockdown cells by LC3-II western blotting in the presence or absence of the lysosomal inhibitor bafilomycin A1. We found markedly decreased amounts of endogenous, lipidated LC3-II protein after 4 days of ATRA treatment in combination with bafilomycin A1 treatment indicating reduced autophagic levels in these knockdown cells compared with control cells (Figure 4a). Further, we expressed EGFP-Cherry-tagged LC3 in NB4 WIPI-1 as well as WIPI-2 knockdown cells. Intracellular localization of EGFP-Cherry-LC3 visualizes autophagosomal membranes following LC3 lipidation.24 In general, upon both autophagy inductions (e.g., starvation) and inhibition (e.g., bafilomycin A1) an increase in LC3 puncta formation is observed. Using EGFP-Cherry-LC3 however, induction and inhibition of autophagy can be distinguished. As the EGFP signal is more sensitive to the acidic and/or proteolytic conditions of the lysosomal lumen compared with the Cherry signal, an increase in red LC3 puncta (EGFP−/Cherry+) per cell reflects active autophagic flux, and a decrease in colocalizing EGFP+/Cherry+ signals (yellow puncta) reflects the inhibition of autophagy. Using control shRNAs, we detected an accumulation of EGFP−/Cherry+ LC3 puncta (red) at day 4 of ATRA treatment when compared with untreated cells indicating increased autophagic flux. An accumulation of yellow LC3 puncta was detected after blocking autophagosomal degradation (bafilomycin A1, Baf A1) during ATRA treatment (Figure 4b). In contrast, when either WIPI-1 or WIPI-2 were knocked down by lentiviral shRNA targeting, the acculumation of EGFP−/Cherry+ LC3 (red) puncta upon ATRA treatment was attenuated as well as of EGFP+/Cherry+ (yellow) LC3 puncta formation upon ATRA and bafilomycin A1 co-treatment (Figure 4b). Moreover, the turnover of long-lived proteins was measured in WIPI-1 and WIPI-2 knockdown cells. We found that the proteolysis of long-lived 14C-labeled proteins was significantly increased after 4 days of ATRA treatment in NB4 SHC002 control cells. In NB4 WIPI-1 and WIPI-2 knockdown cells, however, this increase was significantly abolished (Figure 4c and Supplementary Figure 4a). Furthermore, inhibiting the WIPI-1 upstream regulator PU.1 resulted in significantly reduced long-lived protein degradation as well (Figure 4d and supplementary Figure 4b). As knocking down PU.1 attenuates ATRA-mediated differentiation, we employed ATRA-resistant NB4-R2 cells to test if decreased autophagy seen in PU.1-knockdown cells is mainly due to the attenuated differentiation potential in these cells. Autophagic flux as determined by LC3-II protein abundance in the presence of the lysosomal inhibitor bafilomycin A1 was inhibited in ATRA-resistant NB4-R2 cells (Supplementary Figure 4c). These data clearly show that induction of autophagic flux is associated with neutrophil differentiation of APL cells.

Attenuated differentiation of WIPI-1 and WIPI-2 knockdown APL cells is in part due to impaired autophagy. (a) Autophagic flux was measured in SHC002 and shWIPI-1 or shWIPI-2 expressing NB4 cells upon 1 μM ATRA treatment at day 4 in the presence or absence of bafilomycin A1 by measuring endogenous LC3-II protein levels on western blot and four independently performed experiments were quantified. GAPDH was used as loading control. (b) Stable EGFP-Cherry-LC3 expressing NB4 cells were transduced either with SHC002, shWIPI-1 or shWIPI-2 and cells were treated with 1 μM ATRA for 4 days. Bafilomycin A1 was added after 3 days. Representative pictures of EGFP-Cherry-LC3 puncta from three independently performed experiments are shown. (c) The rate of long-lived protein degradation was measured in control and ATRA-treated SHC002 control cells or WIPI-1, WIPI-2 knockdown cells, in the presence or absence of the lysosomal inhibitor bafilomycin A1.21 The degradation rate for long-lived proteins was calculated as the percentage of radioactivity in the TCA-soluble fraction relative to the total radioactivity in nonsoluble fractions. Further, values were subtracted from the corresponding sample treated with bafilomycin A1. (d) Long-lived protein degradation in NB4 SHC002 control and PU.1 knockdown cells. Analysis as in c. M.W.U, *P<0.05. **P<0.01. M.W.U, Mann–Whitney U-test
In order to further determine autophagic levels during ATRA-induced differentiation in Beclin 1-knockdown cells, we perfomed long-lived protein degradation and EGFP-Cherry-LC3 tandem assays in the respective knockdown cells at day 4. Inhibiting Beclin 1 did not significantly decrease proteolysis of long-lived proteins and no differences for EGFP-Cherry-LC3 puncta formation were observed in Beclin 1 knockdown compared with control cells after 4 days of ATRA treatment (Supplementary Figure 4d and e). On the other hand, NB4 PI3KC3 knockdown cells showed a clear trend (P=0.07) towards a reduction of autophagic flux during APL differentiation when compared with control cells (Supplementary Figure 4e). To exclude that increased cell death causes the differences in the flux assays, we performed Annexin V and propidium iodide (PI) staining. We observed similar increased cell death levels in Beclin 1, WIPI-1 and PI3KC3 knockdown as compared with control cells (Supplementary Figure 4f). Thus, we exclude that differences in cell death among the different autophagy knockdown cells are responsible for the different autophagic flux following ATRA treatment in Beclin 1 versus the other knockdown cells.
In summary, blocking WIPI-1 or WIPI-2 and PI3KC3 but not Beclin 1 significantly impairs ATRA-induced neutrophil differentiation at least in part due to impaired autophagy.
Discussion
Deregulation of human WIPI expression was so far only described in a variety of solid cancers, including kidney, pancreatic, skin, uterine and ovarian cancer.11 We now add AML to the list of cancers with aberrant low WIPI expression. We found that WIPI-1, but not WIPI-2 with the exception of APL, is significantly downregulated in AML patients when compared with mature, healthy neutrophils. Hence, decreased WIPI-1 levels might contribute to the immature myeloid phenotype of AML blasts. Supporting this observation, WIPI-1 but not WIPI-2 levels were significantly upregulated during neutrophil differentiation of APL cells. However, the inhibition of WIPI-1 as well as of WIPI-2 significantly impaired neutrophil differentiation. This might be explained by the much higher relative WIPI-2 levels compared with WIPI-1 in NB4 APL cells that are already sufficient to allow differentiation. High WIPI-2 expression in these cells may indicate additional functions of WIPI-2 also in non-differentiating conditions.12 Moreover, our results indicate that WIPI-1 and WIPI-2 are functionally not redundant during ATRA-induced granulocytic differentiation, thereby supporting the initial observation that all WIPI family members function as PI3P effectors but have different roles in autophagy and cancer.11, 25
The analysis of the transcriptional regulation of ATG genes is still in its infancy. Several studies reported that the p53 family of tumor suppressors or E2F transcription factors regulate expression of several ATG genes.26, 27 Recently, GATA-1 was identified as the first hematopoietic transcription factor regulating the LC3/ATG8 family of autophagy genes that mediate mitochondrial clearance during erythropoiesis.28 Here, we identify WIPI-1 as a new target of PU.1, an essential transcription factor for neutrophil differentiation that is functionally impaired in AML. Further supporting PU.1-mediated transcriptional regulation of WIPI-1, Wang et al.29 described PU.1 binding to a site 20 kb upstream of WIPI-1 transcriptional start in the course of a global survey of PU.1 binding sites in U937 AML cells. As GATA-1, a functional PU.1 agonist during erythroid differentiation, interacts with PU.1, it is likely that GATA-1 and PU.1 cooperate in controlling ATG gene expression in hemtatopoietic cells.30, 31
As data from Trocoli et al.19 suggest a Beclin 1-independent autophagy mechanism during ATRA-induced granulocytic differentiation, we asked whether autophagy required for ATRA-induced granulocytic differentiation depends on canonical PI3P generation as well as their PI3P effector proteins. We have shown that PI3P effector proteins (WIPIs) contribute to ATRA-induced granulocytic differentiation. In addition, we showed that inhibition of WIPI-1 and WIPI-2 reduced autophagy flux during ATRA-induced neutrophil differentiation suggesting that these proteins support neutrophil differentiation, at least in part, by activating autophagy. Moreover, WIPI-1 puncta are increased upon ATRA administration indicating that WIPI-1 binds to autophagosomal membranes as described for canonical autophagy11, 32 and not just increases LC3 lipidation as described for resveratrol-mediated non-canonical autophagy.13 Further, we earlier demonstrated that differentiation-associated autophagy depends on ATG5 puncta formation21 and now we show that WIPI-1/WIPI-2 function upstream of the ATG5-LC3 axis as described by others.13, 33 If WIPI-2 acts upstream of WIPI-1 or vice versa as described,12 it is still under discussion and should rather be investigated in starvation-induced autophagy. In conclusion, we identify both PI3P effector proteins, WIPI-1 and WIPI-2 as positive regulators of neutrophil differentiation in APL cells.
Canonical autophagy is mainly characterized by the generation of the Beclin 1/PI3KC3 complexes and requires the generation of the phosphatidolinositol 3-phosphat (PI3P) signaling lipid34 (reviewed in Burman et al.35). We clearly demonstrate that neutrophil differentiation of APL cells depends on PI3P using pharmacological (LY294002) and genetic inhibition of PI3KC3. Both PI3P inhibition methods resulted in the same phenotype regarding differentiation and autophagy, which is consistent with previous published data.36 This further underlies our hypothesis that PI3P, mainly but not exclusively originating from PI3 kinases class III,37 is crucial for ATRA-induced neutrophil differentiation in an autophagy-dependent manner.
Beclin 1, although enzymatically inert, supports the autophagic process by regulating PI3KC3-dependent generation of PI3P.34 Interestingly, Beclin 1 neither impaired neutrophil differentiation nor autophagy flux for short-term treatment as assessed by measuring endogenous LC3 puncta. On the basis of our additional PI3KC3, WIPI-1 and WIPI-2 knockdown data showing impaired neutrophil differentiation due to attenuated autophagy, it is tempting to speculate that the initiation of ATRA-induced granulocytic differentiation of APL cells follows a non-canonical, Beclin 1-independent, autophagy pathway. In addition, it has been reported that ATRA in combination with everolimus could enhance growth arrest and differentiation in NB4 and HL60 cells further illustrating that autophagy contributes to ATRA-induced differentiation.38
In summary, our data clearly support an important role for autophagy in APL differentiation, which is dependent on the PI3P-WIPI-1/2-LC3 axis. Lastly, the newly discovered PI3P-WIPI-1/2-LC3 pathway operative in neutrophil differentiation of APL cells should be taken into consideration when designing therapeutic strategies that target autophagy in cancer treatments.
Material and Methods
Primary patient samples
A cohort of 98 AML patient samples were enrolled on HOVON/SAKK (Dutch-Belgian Hematology-Oncology/ Swiss Group for Clinical Cancer Research Cooperative group) protocols -04, -04A, -29, and -42.39, 40, 41, 42, 43 All patients provided written informed consent in accordance with the Declaration of Helsinki. Bone marrow aspirates or peripheral blood samples were taken at diagnosis. Blasts and mononuclear cells were purified by Ficoll-Hypaque (Nygaard, Oslo, Norway) centrifugation and cryopreserved. The AML samples contained 80–100% blast cells after thawing, regardless of the blast count at diagnosis. Mutational analyses and classification were performed as described previously.44 Please see Supplementary Table 1 for patient characteristics.
Cell lines and culture conditions
The human acute promyelocytic leukemic (APL) cell lines NB4 and its ATRA-resistant NB4-R2 subclone45 were kindly provided by B.E.Torbett. Cell lines were cultured in RPMI-1640 medium (Sigma-Aldrich, Buchs, Switzerland) supplemented with 10% fetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin in humidified atmosphere containing 5% CO2 at 37 °C. Differentiation of NB4 cells was induced with 1 μM ATRA for 4 to 6 days. Successful neutrophil differentiation was assessed by FACS analysis of CD11b protein or quantitative RT-PCR (qPCR) for CEBPE mRNA expression. Functional differentiation was tested by the NBT-reduction assay46 performed as follows: cells were washed with PBS and treated with NBT/PMA for 20 min at 37 °C, 5% CO2. After incubation, cells were transferred to coated glass slides by cytospin and counterstained with Safranin O. NBT-positive cells were counted. At least 200 cells were scored for each determination from two independent experiments done in duplicate. Data was expressed as the percentage of NBT-positive cell number relative to viable cell number. Cell death in SHC002 or shWIPI-1, shPI3KC3 and shBECN1-transfected cells upon ATRA treatment was assessed using AnnexinV/PI staining and flow cytometry. The isolation of primary neutrophils was performed using polymorphprep (AXIS-SHIELD, Axon Lab, Baden, Switzerland). Cells were layered carefully over Polymorphprep and centrifuged for 40 min at 400 g at room temperature. Primary neutrophils were harvested and RNA was isolated as described below.
TaqMan low-density array and real-time qRT-PCR (qPCR)
RNA extraction, RT-PCR, low-density array measurements as well as data analysis were performed as described.47 Gene Expression Assays for WIPI-1, WIPI-2, PI3KC3, BECN1 and CEBPE used in a 96-well format on the ABI 7500 Sequence detection system were Hs00215872_m1, Hs00255379_m1, Hs00176908_m1, Hs00186838_m1 and Hs00357657_m1, respectively (Applied Biosystems, Rotkreuz, Switzerland). HMBS primer and probes have been described previously.48
Autophagy assays
ATRA-induced autophagy was blocked using bafilomycin A1 (BML-CM110, Enzo Life Sciences, Lausen, Switzerland) 100 nM or 200 nM. LY294002 (L9908, Sigma-Aldrich) was used at a concentration of 10 μM. Starvation (EBSS, Sigma-Aldrich (E2888)) was induced for 5 h before analysis. Protein degradation assay was performed as follows. Cells were treated with or without ATRA and bafilomycin A1 for 4 days. 0.3 μCi 14C-Valine (L-(U-14-C)Valine, Code CFB.75, Amersham) per ml/well was added after 2 days of ATRA treatment. Addition of bafilomycin A1 was performed 24 h before analysis. At day 4, assay was performed as described in.49 Short-term assays were performed as follows: cells were incubated for 2 days with 0.3 μCi 14C-Valine per ml/well. At day 2, assay was performed as described in.49
Western blotting
Whole cell extracts were prepared using RIPA or alternatively in lysis buffer containing aminocaproic acid, 50 mM Bis-Tris, 0.5 mM EDTA, 1% Triton-X100, supplemented with proteinase inhibitor cocktail (complete, Roche Diagnostics, Rotkreuz, Switzerland). Eighty to 120 μg of total protein was separated on a 12% SDS-polyacrylamide gel and transferred to membrane. Blots were incubated with primary antibody in TBS 0.05% Tween-20/ 3% BSA overnight at 4 °C, washed and incubated with IRDye- coupled secondary anti-mouse or anti-rabbit antibodies for 1 h at room temperature protected from light. Primary antibodies used were anti-LC3B (NB600-1384; Novus Biologicals (LuBioScience), Luzern, Switzerland), anti-WIPI-1 antiserum11 and anti-GAPDH (MAB374; Millipore, Zug, Switzerland). Secondary antibodies were Infra-RedDye 680LT-conjugated Goat (polyclonal) Anti-Mouse IgG (H+L), (LICOR) (1 : 10 000) and Infra-RedDye 800CW-conjugated Goat (polyclonal) Anti-Rabbit IgG (H+L) (LI-COR). Data were analyzed using the LI-COR Odyssey Infra-Red Imager (Odyssey, Bad Homburg, Germany).
Phospholipid–protein overlay assay
Aminocaproic acid, 50 mM Bis-Tris, 0.5 mM EDTA, 1% Triton-X 100 supplemented with proteinase inhibitor cocktail (complete, Roche Diagnostics)) supplemented with TBS+0.5% Tween in 3% BSA were used to overlay membrane-immobilized phospholipid membranes (Echelon Biosciences Inc., Salt Lake City, UT, USA; P-6001). LICOR detection of bound WIPI-1 protein using anti-WIPI-1 antiserum was performed as described above.
ChIP assay
Chromatin immunoprecipitation assay (ChIP) was performed as described.40 Genomic regions containing putative PU.1 binding sites were amplified by PCR using the following primers: site A; forward 5′-TGGTTGGGTGCCATGAATGACCA-3′ and reverse 5′-TGCACAGGGCATTCACAGGGG-3′, site B; forward 5′-AGCAACCCAATGGGCTTCCCCT-3′ and reverse 5′-GGAAGGGGGAGGGGCAGGGA-3′, site C; forward 5′-ACTGGACAGCAATGCCTGAAAACA-3′ and reverse 5′-TGGCCAGGGTGTGCAGAACAAA-3′, site D; forward 5′-GCTGCGGATCGGATAGGTCTTGT-3′ and reverse 5′-TGACGGAGCTGGAAGGTGGGG-3′. In addition, an unrelated sequence in the GAPDH gene was used as a negative control.50
Generation of stable knockdown cells and EGFP-Cherry-LC3 expressing cells
pLKO.1 lentiviral vectors expressing small hairpin (sh)RNAs targeting WIPI-1, WIPI-2, PI3KC3 and BECN1 or a non-targeting shRNA control (SHCOO2) were purchased from Sigma-Aldrich. A lentiviral vector expressing EGFP-Cherry-LC3 was provided by Dr. MS Soengas. Lentiviral production and transduction of NB4 cells was done as described.51 EGFP-Cherry-LC3 NB4 cells with WIPI-1, WIPI-2 and BECN1 knockdown were generated by serial transduction with both lentiviral vectors described above. Transduced cell populations were selected for 4 days using 1.5 μg/ml puromycin. qPCR was performed to evaluate knockdown efficiency in NB4 WIPI-1, WIPI-2 and BECN1 knockdown cells, whereas western blotting was employed for demonstration for beclin 1 inhibition. To generate PU.1-inducible cells, the pCl10A1 retroviral packaging plasmid and the pBabe-PUER-puro (kindly provided by Dr. H Yoshida) were co-transfected into 293T cells. Cell culture medium was removed after 1 day and cells were incubated for 2 min with PBS/15% glycerol. PBS/15% glycerol was replaced by fresh medium. Viral supernatants were harvested 48 h later. NB4 cells were transduced for 24 h in the presence of 8 μg/ml polybrene. Transduced NB4 cell populations were selected with 1.5 μg/ml puromycin for 4 days.
Fluorescence microscopy
EGFP-Cherry-LC3 expressing NB4 cells were fixed with 2% paraformaldehyde washed with PBS (0.5% of Glycin). Subsequently, cells were placed on a poly-L-lysine coated cover glass and mounted in Fluorescence Mounting Medium (S3032; DAKO, Baar, Switzerland). Images were taken on a confocal microscope at × 60 magnification for puncta assay and on a fluorescence microscope for the NBT assay.
Immunofluorescence microscopy
Cells were fixed with 2% paraformaldehyde washed with PBS (0.5% of Glycin). Subsequently, cells were placed on a poly-L-lysine-coated cover glass permeabilized with triton. Alternatively, cells were fixed with ice-cold methanol for 4 min after cytospun and then washed with PBS (0.5% of Glycin). Incubation of anti-WIPI-1 and anti-LC3B antibody for 1 h at room temperature followed by three washing step with PBS-T. Cells were incubated with the second antibody (111-096-045, Jackson Immuno Research, Suffolk, UK) for 1 h at room temperature. Prior mounting in Fluorescence Mounting Medium (S3032; DAKO) cells were washed three times with PBS-T. Images were taken on a confocal microscope at × 60 magnification.
Statistical analysis
Nonparametric Mann–Whitney U-tests were applied to compare the difference between two groups using the program GraphPad Prism 4 (Graph Pad Software, San Diego, CA, USA). P-values <0.05 were considered to be statistically significant.
Abbreviations
- AML:
-
acute myeloid leukemia
- APL:
-
acute promyelocytic leukemia
- ATRA:
-
all-trans retinoic acid
- PI3P:
-
phosphatidylinositol 3-phosphate
- WIPI:
-
WD-repeat protein interacting with phosphoinositides
References
Cecconi F, Levine B . The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 2008; 15: 344–357.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ . Autophagy fights disease through cellular self-digestion. Nature 2008; 451: 1069–1075.
Meijer AJ, Codogno P . Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 2009; 46: 210–240.
Hershko A, Ciechanover A . The ubiquitin system. Annu Rev Biochem 1998; 67: 425–479.
Codogno P, Mehrpour M, Proikas-Cezanne T . Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol 2012; 13: 7–12.
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ . The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 1997; 243: 240–246.
Knævelsrud H, Simonsen A . Lipids in autophagy: constituents, signaling molecules and cargo with relevance to disease. Biochim Biophys Acta 2012; 1821: 1133–1145.
Simonsen A, Tooze SA . Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009; 186: 773–782.
Petiot A, Ogier-Denis E, Blommaart EFC, Meijer AJ, Codogno P . Distinct classes of phosphatidylinositol 3-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275: 992–998.
Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182: 685–701.
Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A et al. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004; 23: 9314–9325.
Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010; 6: 506–522.
Mauthe M, Jacob A, Freiberger S, Hentschel K, Stierhof YD, Codogno P et al. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy 2011; 7: 1448–1461.
Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F et al. The WD40 Repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell 2011; 21: 343–357.
Proikas-Cezanne T, Ruckerbauer S, Stierhof Y-D, Berg C, Nordheim A . Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett 2007; 581: 3396–3404.
Cáceres-Cortés JR . Blastic leukaemias (AML): a biologist’s view. Cell Biochem Biophys 2012; 66: 13–22.
Isakson P, Bjoras M, Boe SO, Simonsen A . Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116: 2324–2331.
Bøe SO, Simonsen A . Autophagic degradation of an oncoprotein. Autophagy 2010; 6: 964–965.
Trocoli A, Mathieu J, Priault M, Reiffers J, Souquère S, Pierron G et al. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy 2011; 7: 1108–1114.
Zhang S-P, Niu YN, Yuan N, Zhang AH, Chao D, Xu QP et al. Role of autophagy in acute myeloid leukemia therapy. Chin J Cancer 2012; 32: 130–135.
Brigger D, Torbett BE, Chen J, Fey MF, Tschan MP . Inhibition of GATE-16 attenuates ATRA-induced neutrophil differentiation of APL cells and interferes with autophagosome formation. Biochem Biophys Res Commun 2013; 438: 283–288.
Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research. Cell 2010; 140: 313–326.
Klionsky DJ . Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 1–100.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–5728.
Bakula D, Takacs Z, Proikas-Cezanne T . WIPI β-propellers in autophagy-related diseases and longevity. Biochem Soc Trans 2013; 41: 962–967.
Kusama Y, Sato K, Kimura N, Mitamura J, Ohdaira H, Yoshida K . Comprehensive analysis of expression pattern and promoter regulation of human autophagy-related genes. Apoptosis 2009; 14: 1165–1175.
He Z, Liu H, Agostini M, Yousefi S, Perren A, Tschan MP et al. p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene. Cell Death Differ 2013; 20: 1415–1424.
Kang Y-A, Sanalkumar R, O’Geen H, Linnemann AK, Chang CJ, Bouhassira EE et al. Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol 2012; 32: 226–239.
Wang K, Wang P, Shi J, Zhu X, He M, Jia X et al. PML/RARα targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell 2010; 17: 186–197.
Zhang P, Zhang X, Iwama A, Yu C, Smith KA, Mueller BU et al. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 2000; 96: 2641–2648.
Rekhtman N, Radparvar F, Evans T, Skoultchi AAI . Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev 2012; 13: 1398–1411.
Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N . Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 2012; 125: 1488–1499.
Itakura E, Mizushima N . Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6: 764–776.
Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M et al. Beclin1: a role in membrane dynamics and beyond. Autophagy 2012; 8: 6–17.
Burman C, Ktistakis NT . Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett 2010; 584: 1302–1312.
Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 2011; 7: 401–411.
Devereaux K, Dall’Armi C, Alcazar-Roman A, Ogasawara Y, Zhou X, Wang F et al. Regulation of mammalian autophagy by Class II and III PI 3-kinases through PI3P Synthesis. PLoS One 2013; 8: 1–17.
Nishioka C, Ikezoe T, Yang J, Gery S, Koeffler HP, Yokoyama A et al. Inhibition of mammalian target of rapamycin signaling potentiates the effects of all- transretinoic acid to induce growth arrest and differentiation of human acute myelogenous leukemia cells. Int J Cancer 2009; 125: 1710–1720.
Breems DA, Boogaerts MA, Dekker AW, Van Putten WL, Sonneveld P, Huijgens PC et al. Autologous bone marrow transplantation as consolidation therapy in the treatment of adult patients under 60 years with acute myeloid leukaemia in first complete remission: a prospective randomized Dutch-Belgian Haemato-Oncology Co-operative Group (HOVON) and Swiss Group for Clinical Cancer Research (SAKK) trial. Br J Haematol 2005; 128: 59–65.
Federzoni EA, Valk PJ, Torbett BE, Haferlach T, Löwenberg B, Fey MF et al. PU.1 is linking the glycolytic enzyme HK3 in neutrophil differentiation and survival of APL cells. Blood 2012; 119: 4963–4970.
Lowenberg B, Boogaerts MA, Daenen SM, Verhoef GE, Hagenbeek A, Vellenga E et al. Value of different modalities of granulocyte- macrophage colony-stimulating factor applied during or after induction therapy of acute myeloid leukemia. J Clin Oncol 1997; 15: 3496–3506.
Lowenberg B, van Putten W, Theobald M, Gmür J, Verdonck L, Sonneveld P et al. Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N Engl J Med 2003; 349: 743–752.
Ossenkoppele GJ . The value of fludarabine in addition to ARA-C and G-CSF in the treatment of patients with high-risk myelodysplastic syndromes and AML in elderly patients. Blood 2004; 103: 2908–2913.
Rockova V, Abbas S, Wouters BJ, Erpelinck CA, Beverloo HB, Delwel R et al. Risk stratification of intermediate-risk acute myeloid leukemia: integrative analysis of a multitude of gene mutation and gene expression markers. Blood 2011; 118: 1069–1076.
Duprez E, Benoit G, Flexor M, Lillehaug JR, Lanotte M . A mutated PML/RARA found in the retinoid maturation resistant NB4 subclone, NB4-R2, blocks RARA and wild-type PML/RARA transcriptional activities. Leukemia 2000; 14: 255–261.
Ochs HD, Igo RP . The NBT slide test: a simple method for detecting chronic granulomatous disease and female carriers. J Pediatr 1973; 83: 77–82.
Tschan MP, Shan D, Laedrach J, Eyholzer M, Leibundgut EO, Baerlocher GM et al. NDRG1/2 expression is inhibited in primary acute myeloid leukemia. Leuk Res 2010; 34: 393–398.
Britschgi C, Rizzi M, Grob TJ, Tschan MP, Hügli B, Reddy VA et al. Identification of the p53 family-responsive element in the promoter region of the tumor suppressor gene hypermethylated in cancer 1. Oncogene 2005; 25: 2030–2039.
Ogier-Denis E, Houri J-J, Bauvy C, Codogno P . Guanine Nucleotide Exchange on Heterotrimeric Gi3 Protein Controls Autophagic Sequestration in HT-29 Cells*. J Biol Chem 1996; 271: 28593–28600.
Kaeser MD, Iggo RD . Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Cell 2002; 99: 95–100.
Tschan MP . Alternative splicing of the human cyclin d-binding myb-like protein (hdmp1) yields a truncated protein isoform that alters macrophage differentiation patterns. J Biol Chem 2003; 278: 42750–42760.
Acknowledgements
We gratefully acknowledge Dr. PJM Valk and Dr. B Löwenberg and the HOVON (Dutch-Belgian Hematology-Oncology) cooperative group for providing primary AML patient samples. Deborah Shan is gratefully acknowledged for excellent technical support. We thank Drs BE Torbett and MS Soengas for providing ATRA-resistant NB4-R2 APL cell lines and an EGFP-Cherry-LC3 lentiviral vector, respectively. This study was supported by grants from the Foundation Cancer Research Switzerland (KFS-02486-08-2009 to MPT), the Swiss National Science Foundation (31003A_143739) (to MPT), the German Research Society DFG (SFB 773, TP A03 to TP-C), the Hedy Berger-Janser Foundation of Cancer Research (to MPT) and the Bern University Research Foundation (to MPT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Brigger, D., Proikas-Cezanne, T. & Tschan, M. WIPI-dependent autophagy during neutrophil differentiation of NB4 acute promyelocytic leukemia cells. Cell Death Dis 5, e1315 (2014). https://doi.org/10.1038/cddis.2014.261
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.261
This article is cited by
-
The dual role of autophagy in acute myeloid leukemia
Journal of Hematology & Oncology (2022)
-
Role of cardiolipins, mitochondria, and autophagy in the differentiation process activated by all-trans retinoic acid in acute promyelocytic leukemia
Cell Death & Disease (2022)
-
Reducing FASN expression sensitizes acute myeloid leukemia cells to differentiation therapy
Cell Death & Differentiation (2021)
-
Intracellular alpha-fetoprotein interferes with all-trans retinoic acid induced ATG7 expression and autophagy in hepatocellular carcinoma cells
Scientific Reports (2021)
-
RACK1 deficiency synergizes with all-trans retinoic acid to induce apoptosis in human acute promyelocytic leukemia cells
Molecular and Cellular Biochemistry (2019)
