
Abstract
Lipids are key regulators of cell physiology through the control of many aspects of cellular life and survival. In particular, lipids have been implicated at different levels and through many different mechanisms in the cell death program called apoptosis. Here, we discuss the action of lipids in the regulation of the activation and the integration of Bax into the mitochondrial outer membrane, a key pro-apoptotic member of the BCL-2 family. We describe how, during apoptosis, lipids can act simultaneously or in parallel as receptors or ligands for Bax to stimulate or inhibit its pro-death activity.
Similar content being viewed by others


Facts
-
Lipids and especially Ceramides were among the first known actors of apoptosis.
-
Lipids are involved in the mitochondrial insertion of the pro-apoptotic proteins such as Bax and Bak and in the formation of oligomers.
-
Lipids are constituents of the channels formed by Bax and Bak supramolecular complexes.
-
Recent data have shown that lipids are also instrumental in the control of the activation phase of Bax and Bak.
Open Questions
-
Is the implication of lipids in the activation of Bax and Bak an universal phenomenon?
-
Is this control independent of that of BH3-only proteins (BoPs) controls?
-
When and how are interactions of the lipids/proteins occurring?
Apoptosis (programmed cell death) is an essential physiological process in the development of multicellular organisms and in the maintenance of adult tissue homeostasis. Apoptosis is also involved in many pathologies, such as cardiovascular, neurodegenerative, immunological diseases and cancer.1 There are two distinct cellular pathways leading to apoptosis: the extrinsic pathway (or death receptor pathway) and the intrinsic pathway (also called mitochondrial pathway). The mitochondrial pathway of apoptosis involves proteins, which have a role in other mitochondrial functions such as cytochrome c, but also cytosolic proteins such as the caspases or proteins partly cytosolic and partly mitochondrial such as the members of the BCL-2 family.2 The extrinsic pathway is only partially independent of the mitochondrial pathway, often used as a relay or an amplifier, and is restricted to some tissue, in particular the hematological system. Other actors are implicated in this complex mechanism and pioneer studies on apoptosis have underlined the role of ions (especially calcium) and lipids (especially ceramides) in the initiation and execution phases of apoptosis. Different lipid species like ceramides, free fatty acids, diacylglycerol or cholesterol can lead to pathological cell death through signaling pathways involving proteins intrinsic to cell death programs. It has been established in the 1990s that lipid modifications, trafficking or biosynthesis are closely associated with apoptosis. Several recent reviews have described the importance of mitochondrial lipids in cell survival and death3, 4, 5 and recent results have in particular, shed new light on the regulation of members of the BCL-2 family through direct interactions with lipids in proximity of or in relation with mitochondria. Thus our knowledge on the control by lipids in the early phase of the induction of apoptosis has been challenged by these new findings and this is discussed in this review.
BCL-2 Family of Proteins and Apoptosis
Bcl-2 was the first protein of this family to be identified as a proto-oncogene in human follicular lymphoma.6 The homology between the BCL-2 family of proteins is restricted to certain domains called BH domains (Bcl-2 homology domains), which are implicated in protein–protein interactions as well as post-translational regulation.7, 8, 9
The BCL-2 family of proteins is classically divided into four groups.10
-Anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-W, Mcl-1 and A1): these proteins contain the BH1, BH2, BH3 and BH4 domains. They are located in the cytosol but are also associated with the mitochondrial outer membrane (MOM) and the endoplasmic reticulum (ER) at different proportions. Upon induction of apoptosis, most of these proteins become membrane associated or integrated (mainly with the MOM).
-Pro-apoptotic effector proteins (Bax and Bak): these proteins are activated at the onset of apoptosis. The activation induces protein oligomerization into proteolipidic pores in the MOM, which in turn, directly induce the MOM permeabilization (MOMP).
-Direct activator pro-apoptotic proteins (Bid, Bim and Puma) are able to activate Bax and Bak through a hit and run process.
-Sensitizers or de-repressor pro-apoptotic proteins (Bad, Bik, BMF, HRK, Noxa and Puma) bind to and inhibit the anti-apoptotic proteins, leading to the release of Bax or Bak from anti-apoptotic proteins.
The latter groups (direct activator and sensitizer pro-apoptotic proteins) contain only the BH3 domain and are called BoP.
In healthy cells Bax is maintained in an inactive state under a compact configuration and is mostly located in the cytosol. A fraction of Bax is nonetheless capable of cycling between mitochondria and cytosol. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. However, the precise function of this recycling is not known.11
Bax activation induces an important protein conformational change, which leads to mitochondrial addressing through the interaction of several domains with the MOM components.12, 13 Under pro-apoptotic condition, Bid, Bim and possibly Puma are activated and induce Bax and Bak oligomerization, MOMP and cytochrome c release.10, 14, 15
The mechanism by which MOMP is achieved has been largely discussed and several models of the formation of pores or channels have been proposed: proteolipids, protein only or lipidic channels.14, 15, 16 It has been shown that several members of the BCL-2 family (Bcl-2, Bcl-xL, Bax, Bid...) can form channels or permeabilize artificial liposomes in a manner that recapitulate their function (that is, facilitating or inhibiting the translocation of proteins across lipid bilayers).17, 18, 19
Lipids and Bax: An Apoptotic Mitochondrial Platform?
Several studies have focused recently on bioactive lipids as a new type of key molecular actors accompanying or controlling the functions of members of the BCL-2 family. For example, tBid can permeabilize liposomes by modifying membrane properties (lipid mixing and negative curvature)20 and the efficiency of this permeabilization largely depends on the lipids composition of the membrane (Phosphatidylcholine, Phosphatidyethanolamine, Cardiolipin, Cholesterol).21 In the same way some lipids such as fatty acids (already known for their pro-apoptotic capabilities)22 are able to increase Bax insertion and also the membrane permeabilization analyzed in liposomes.23 However, liposomes with a composition close to ER lipid were not permeabilized by tBid and monomeric Bax, therefore, the mitochondrion-specific lipid composition is essential for membrane permeabilization by Bax or tBid.14
Thus regulation of apoptosis is not only controlled by protein–protein interactions but also by different bioactive lipids. However, two different situations can be envisaged for the Bax/lipids interactions:
Lipids as Receptors for Bax
(a) Cholesterol
Cholesterol is an essential cellular membrane component. It regulates membrane rigidity, membrane protein activity and as such, transmembrane signalization.24 Some cells are able to take up circulating low density lipids (LDL) and high density lipids (HDL) by endocytosis. Cholesterol is also synthesized de novo in the ER.25 Cholesterol is translocated to the MOM by vesicular trafficking or by cholesterol binding and transport proteins. Compared with the plasma membrane, cholesterol is present at low levels in the ER and mitochondria,26 suggesting a tight control of its intracellular transport. Transport of cholesterol from the ER to mitochondria occurs through unique ER in close contact with mitochondria and thus termed the mitochondria-associated ER membrane (MAM).27, 28 It has been postulated that this structure could play a major role during cell death.29 Cholesterol trafficking from the MOM to the mitochondria inner membrane (MIM) requires a protein complex, which includes the translocator protein (TSPO protein).30 Little is known about alteration of cholesterol transport during apoptosis but several studies suggest that inhibitors of cholesterol synthesis have pro-apoptotic properties through activation of the mitochondrial pathway.31, 32, 33, 34
The HMG-CoA reductase inhibitor lovastatin blocks mevalonate and also cholesterol synthesis. A study has shown that lovastatin treatment sensitized colorectal cancer cells to pro-apoptotic stimuli, causing an increase in Bax expression and a decrease in Bcl-2 expression.35 In another study, lovastatin treatment reduced myeloma cell survival by modulation of apoptosis without modifying Bax and Bcl-2 expression.36 These data were confirmed in HeLa cells treated with U18666A; a drug that induces an increase in intracellular and mitochondrial cholesterol without modifying mitochondrial morphology or apoptosis-related protein expression (Smac/Diablo, cytochrome c, Bcl-xL, Bax, Bak).37
Proteins that interact with cholesterol have a conserved sequence called Cholesterol Recognition Amino acid Consensus and recently, this sequence has been identified in the alpha 5 helix of Bax, one of its transmembrane helices.38 The role of cholesterol in Bax activity is still controversial. It has been shown by Christenson et al.39 that the addition of cholesterol to mitochondrial membranes increases Bax binding but markedly reduces its integration into the MOM. Several other studies have suggested that the different roles of cholesterol depend on its concentration. At low concentrations cholesterol acts as a receptor for Bax facilitating its insertion (into liposomes or MOM) but not its activation or oligomerization.37, 38, 40 However, Martinou and colleagues37 have shown that Bax activation is inhibited by cholesterol accumulation, essentially by decreasing its membrane binding property. A direct link between the membrane cholesterol level and Bax activation has been proposed as in liposomes, cholesterol decreases membrane fluidity and inhibits Bax oligomerization induced by tBid without blocking its insertion.37 These data show that Bax activation is not necessarily followed by its oligomerization and MOMP, suggesting that these events depend on other conditions or actors (other mitochondrial lipids or proteins).37 This contention was confirmed by other studies, which showed that a high cholesterol content was an inhibitor of the transition from membrane bound to the membrane embedded status of Bax.39 Thus, mitochondrial cholesterol accumulation inhibits Bax oligomerization and induces partial resistance to pro-apoptotic agents.41 The high mitochondrial cholesterol content found in many tumors may thus account, at least partially, for an inefficient Bax oligomerization, which might contribute to cancer resistance to apoptosis-inducing therapies, providing a new basis for the use of statins as anticancer agents.42
(b) Cardiolipin
Cardiolipin (or diphosphatidylglycerol) is an anionic phospholipid. At first considered as a MIM component, cardiolipin is in fact also present in the MOM even if its percentage remains controversial (23% of total mitochondrial cardiolipin are present at the MOM,43 which could correspond to 0.3-3% of mitochondrial lipids44). However, cardiolipin has an essential role in apoptosis mitochondrial pathway.
It has been shown that the oxidation of cardiolipin present in the MIM is necessary for cytochrome c release.45 Cytochrome c is linked to cardiolipin in the MIM. To facilitate cytochrome c release, cardiolipin is oxidized leading to the separation of the complex cardiolipin–cytochrome c. This cytochrome c pool formed will then be released (after Bax induced-MOMP for example).46 Bax insertion into the MOM is sufficient to induce superoxide production and cardiolipin oxidation.47 Cardiolipin also interacts with tBid and allows its translocation to mitochondria. It has been shown that tBid translocation to mitochondria is strongly reduced in cardiolipin-defective cells.48
In vitro studies in liposomes have shown that cardiolipin is essential for alpha 1 helix of Bax binding to the MOM and then to Bax insertion and oligomerization.49 Interestingly in the absence of cardiolipin some MOM proteins (not yet identified) can help Bax insertion and proteolipidic pores formation induced by Bax or tBid.50
Cholesterol and cardiolipin are two important regulators of mitochondrial pathway of apoptosis, by interacting directly with Bid and Bax to allow or inhibit their insertion into the MOM or by modifying the properties of the mitochondrial membranes.
Lipids as Ligands for Bax
(a) Prostaglandins
The cyclooxygenases (Cox-1 and Cox-2) catalyze arachidonic acid transformation to prostaglandin H2 (PGH2). Then PGH2 can be transformed into several others prostaglandins, predominantly PGE2 and PGD2 by the prostaglandins synthases (PGES and PGDS, respectively).51
Prostaglandins are secreted lipids having a key role in the modulation of the inflammatory response.52 It has been known for many years that chronic inflammation significantly increases the risk of cancer.53 In addition, PGE2 can increase tumor growth by binding to G protein-coupled membrane receptors (EP receptors) leading to the activation of several cellular pathways associated with proliferation, migration, survival and angiogenesis.54
Different Cox-2 inhibitors have also been developed for their anti-tumor activity.53 Recent studies have challenged this view highlighting the important role of intracellular prostaglandins on Bax-dependent apoptosis regulation (independently of their secretion and also of EP receptor activity). An initial study identified two groups of glioblastoma multiforme patients with different expression levels of the microsomal PGES (mPGES) and showed a correlation between high expression of mPGES and increased patient survival.55 In fact, the high expression of mPGES leads to an increase spontaneous as well as staurosporine-induced apoptosis. Furthermore inhibition of the expression of mPGES by siRNA provokes a strong resistance to apoptosis.55 In addition, microinjection of intracellular PGE2 induces an increase in Bax-dependent apoptosis.55 This effect is not dependent on the cell type as the increase in intracellular PGE2 by microinjection or pharmacological inhibition of its export (and degradation) increases Bax-dependent apoptosis in several cancer and non-cancer cells.56 Finally, PGE2 directly induces a conformational change of Bax and its insertion in the MOM (on isolated mitochondria).55 Another study shows that intracellular microinjection of prostaglandin A2 (PGA2), derived from the non-enzymatic dehydration of PGE2, induces apoptosis.57 Like PGE2, PGA2 is also able to induce Bax activation and mitochondrial translocation but not its oligomerization in a cell-free system.57 Linear peptide scan assay and site-directed mutagenesis experiments showed that PGE2 binds to 126cys present between the two transmembrane alpha helix of Bax (alpha 5 and 6 helices) and that this cysteine is necessary for PGE2-induced Bax activation.57 PGD2 can block this interaction between Bax and PGE2 and consequently inhibit PGE2-induced apoptosis. Interestingly, PGE2 and Bid do not activate Bax by the same mechanism, since they do not interact in the same region of Bax, but they can rather cooperate to induce apoptosis.57 As far as PGE2 and PGD2 are concerned, even if their interaction site with Bax is the same,58 they do not trigger the same conformational changes, as visualized by fluorescence spectroscopy experiments (Figure 1). The fluorescence emission of the tryptophan is known to be very sensitive to the polarity of its environment and indicates the degree of exposure of the residue to solvent.59, 60 The tryptophan fluorescence can be used to monitor conformational changes in native protein and protein–protein interactions. There are six tryptophan residues in Bax. Two tryptophan are localized at position 47 and 51, and the remaining four residues are localized in 100–130 region at position 100, 117, 121 and 127, that is, in the close vicinity of the interaction site between Bax and prostaglandins. Bax alone emitted fluorescence at around 339.5 nm at neutral pH. The fluorescence of pH-activated Bax (pH 4) was decreased about 50% in intensity and its emission maximum was slightly shifted to 338.5 nm. Adding PGE2 induced a decrease of about 50% in fluorescence intensity and slightly shifted the emission peak in the same way, even if the shift was not significant. In contrast, PGD2 induced a shift from 339.5–343.5 nm in Bax fluorescence.

The interaction between prostaglandins and Bax in solution can be observed by the change in the fluorescence emission of tryptophans in Bax. Experiments were conducted with purified Bax incubated in the presence of PGE2 or PGD2 at pH 7. Bax protein alone emitted fluorescence around 339 nm at neutral pH, which is consistent with the fact that many tryptophan residues are exposed. The fluorescence of pH-activated Bax (pH 4) decreased about 50% in intensity and its maximum emission is slightly shifted to 338.5 nm. Incubation of Bax with PGE2 induced a decrease of about 50% in fluorescence intensity and slightly shifted the emission peak in the same way, even if the shift was not significant. Incubation of Bax with PGD2 decreased the fluorescence intensity about 50% and shifted the maximum emission peak from 339–343 nm. This suggest that one or several tryptophan residues are more exposed (in a polar environment) upon the interaction. Another explanation for this shift could be a decrease in the fluorescence of buried tryptophan residues induced by their interaction with PGD2. However, it is rather improbable, especially since PGD2 would increase the hydrophobicity around those buried residues, thus shifting the peak to a lower wavelength. These spectra suggest that PGE2 affects the conformation of Bax in a different (or opposite) manner as compared with PGD2, which is consistent with the opposite effect of these ligands on Bax activation53
These data are confirmed by other studies showing that PGA2 inhibits cellular growth, induces cytochrome c release and activates the mitochondrial pathway of apoptosis in HL-60 cells (Human Promyelocytic leukemia cell line) but without establishing a direct link with BCL-2 family of proteins.58, 61
In short, extracellular PGE2 induces an increase in cell proliferation via its binding to its membrane receptors,54 but an increase in intracellular PGE2 induces the activation of the intrinsic pathway of apoptosis by activating Bax.55, 56, 57
The following model was proposed: the induction of apoptosis in cells leads to an increase in the concentration of intracellular PGE2, which actively participates in pro-apoptotic mechanisms. PGE2 is then secreted into the extracellular region where it has the role of a danger signal and induces cell proliferation by activating the EP receptors. This model, applicable both during tissue regeneration after injury62 and in tumor repopulation after treatment,63 has been called the ‘Phoenix rising pathway’. These data also suggest another interesting concept; during tissue regeneration this mechanism causes the proliferation of stem and progenitor cells and involves caspase-3 and -7 and iPLA2.62 It seems conceivable that the role of PGE2 in tumor repopulation after treatment could involve a direct effect on cancer stem cells (Figure 2).

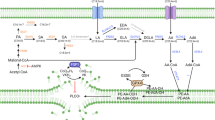
Activation of Bax during apoptosis is controlled by lipids at multiple levels. Inactive Bax (iBax) is predominantly cytosolic in resting cells as a monomer or associated with Bcl-xL in the cytosol or at the MOM (1). At the onset of apoptosis, derepressing BH3-only proteins (dBoP) contribute to Bax activation by releasing either Bax or activator BH3-only proteins (aBoP) from anti-apoptotic proteins (2). PGE2 participates to this activation, while the closely related molecule PGD2 inhibits this action (3). The S1P degradation product, Hexadecenal (Hex), can activate Bax at this step (4). Activated Bax (aBax) binds to mitochondria through its interaction with cholesterol (Chol), which when in excess inhibits Bax oligomerization (5). Oligomerization of Bax (oBax) occurs through its interaction with proteins but also with lipids including cardiolipin (CL) (6), which also facilitate tBid (an activator BH3-only protein) insertion into the MOM (7). Ceramide (Cer) can also facilitate Bax insertion into the MOM through the formation of microdomains and/or channels (8). The oligomerization of Bax and/or the formation of ceramide channels induce MOMP, the release of cytochrome c (c), activation of the caspases and subsequently apoptosis (9). S1P is a bioactive lipid molecule that can act as an intracellular messenger or is secreted where, via its G protein-coupled receptors (S1PRs) it mediates pro-survival signals and cell proliferation (10). The synthesis of PGE2 is enhanced by caspase activation of phospholipase A2 (PLA2) (11). Similarly to S1P, secreted PGE2 promotes resistance to apoptosis via G protein-coupled membrane receptors (EPs) or transactivation of the EGF Receptor (EGFR) (12). Activation of S1PR, EPs and tyrosine kinase receptors (TKR) mediates resistance to apoptosis via over-expression of Bcl-2 or Bcl-xL (13)
(b) Sphingolipids
Sphingolipids are a class of lipids containing a backbone of sphingoid bases and a set of aliphatic amino alcohols that include sphingosine. They are essential structural components of the outer leaflet of the plasma and organelle membranes of eukaryotic cells. In the nineties, their functions were revaluated and were shown to be key actors in signaling pathways leading to inflammatory response, apoptosis, senescence, proliferation and cell migration.64 Sphingolipids may act in signal transduction either as second messengers or by forcing the agglutination of non-soluble raft microdomains enhancing membrane proteins and receptor capping.65 Therefore, sphingolipids contribute to the modulation of the mechanisms of cancer initiation and progression and the sensitivity to anti-tumor treatments.66 The central actor in sphingolipid metabolism is ceramide, composed of a sphingosine and a fatty acid chain of variable length (14–24 carbon atoms), which influences the biological properties of the ceramide.67 The level of intracellular ceramide increases in response to various cellular stresses via de novo synthesis involving one of the six ceramide synthase enzymes or by hydrolysis of sphingomyelin through the activation of neutral or acidic sphingomyelinase enzymes.66 In response to stress, ceramide is considered as a bioactive lipid inducing apoptosis, cell cycle arrest and differentiation.68
Given its pro-apoptotic role, the relationship between ceramide and the proteins of the BCL-2 family have been widely investigated. The over-expression of Bcl-2 or Bcl-xL blocks apoptosis provoked by either exogenous ceramide treatment or TNFα-induced endogenous ceramide, respectively, in ALL-697 leukemia cells69 and MCF-7 breast carcinoma cells.70 On the other hand, exogenous ceramide fails to induce apoptosis in Bax-deficient DU145 cells (prostate carcinoma) and HCT116 (colorectal cancer). Upon Bax transfection DU145 and HCT116 became sensitive to ceramide-induced apoptosis.71 In some cell models, redundancy between Bak and Bax activation was not observed in ceramide-induced apoptosis. For example, Bax- or Bak-deficient cells each individually mimicked ASMase deficiency and exhibited no ceramide-induced endothelial cell death after exposure to ionizing radiation.72 Furthermore, a feed-forward model between BH3-domain proteins and ceramide has been described. Exposure of HeLa cells to UV-radiation leads to ceramide generation and induces Bax-dependent apoptosis.73 In addition, Bak (and to a lesser extent Bax) seems necessary for the long-chain ceramide generation during baby mouse kidney cell apoptosis induced by UV-C radiation or cisplatin treatment.74 Inhibition of the anti-apoptotic Bcl-2 proteins by ABT-263 induces ceramide generation though a Bak-dependent activation of ceramide synthase in human leukemia and myeloma cell lines.75 Ceramide generation and accumulation could be observed at the plasma membrane level as well as in cellular organelles. In fact, TNFα-induced ceramide in MCF-7 cells is concentrated in the mitochondrial membrane and is followed by Bax activation and translocation to the mitochondria. Moreover, the co-incubation of bacterial SMase and isolated mitochondria is sufficient to generate ceramide and increase Bax translocation to mitochondria.76 In the same way, the co-incubation of exogenous C2- and C16-ceramide and isolated rat liver mitochondria potentiates the effect of Bax on the mitochondrial permeability transition.77 In fact, MOMP induced after Bax insertion and oligomerization may be dependent of the formation of ceramide-enriched platforms in the MOM as seen in 10 Gy-irradiated HeLa cells.40 Moreover, activated Bax has been localized preferentially in ceramide-enriched micro-membrane domains.40 Mitochondria pre-incubated with activated Bax (by detergent or tBid) are more sensitive to MOMP induced by ceramide.78
The importance of sphingolipids in the permeabilization of mitochondria and therefore, in the intrinsic pathway of apoptosis have been confirmed in a recent study wherein sphingolipids present in the ER cooperated with tBid-activated Bax or Bak to facilitate MOMP in vitro.79 In this study, sphingosine-1-phosphate (S1P), a ceramide metabolite, interacts with Bak to induce MOMP. Moreover, the hexadecenal, S1P degradation product, interacts with Bax leading to its oligomerization and mitochondrial permeabilization. S1P have been largely described as a ceramide antagonist inhibiting apoptosis.80 This study challenges the conventional roles of ceramide and S1P as pro-apoptotic and pro-survival agents, respectively. Furthermore, S1P has been described to have anti-proliferative effects on certain cell types such as hepatocytes,81 T-lymphocytes,82, 83 keratinocytes84 and gastric cancer cells,85 even though the exact mechanism is yet to be elucidated.
Bioactive S1P may function in both the intra and extracellular compartments. The extracellular S1P could have pro-survival properties through the binding to one of its specific seven transmembrane receptors.86 In the present hypothesis, the intracellular S1P may have pro-apoptotic functions through its activation of Bak and the resulting MOMP. The potential discrepancies between intra versus extracellular S1P bioactive functions have been partially observed for PGE2 as described in the precedent section.
Ceramides may also form channels in mitochondrial membranes. Ceramides of various chain lengths (C2 and C16) are able, at physiological concentration, to structure pores in phospholipid membranes in vitro.87 These ceramide channels are formed by a ring of anti-parallel columns composed of six ceramide molecules linked by hydrogen bonds.87 The formation of these pores in MOM is in direct correlation with the concentration of ceramide added to the medium88 and results in an increased conductance and therefore, in MOMP.89 Interestingly, ceramide metabolites dihydroceramide and sphingosine inhibit the formation of ceramide pores in mitochondria.90, 91 Again, the modulation of the ceramide channels is connected to BCL-2 protein family. If the pro-apoptotic proteins Bax and Bak do not seem essential for the formation of these channels, the anti-apoptotic protein Bcl-xL inhibits the formation of ceramide channels in isolated mitochondria.92 However, the ceramide channels have been observed in planar phospholipid membranes in vitro and in isolated mitochondria but in the absence of other molecular players and are yet to be characterized in cells. This observation will help to better define their roles in the MOMP and in apoptosis.
Concluding Remarks
We have described some of the possible roles of lipids in apoptosis as modulators of the activity of BCL-2 family members. But lipids may have many other roles during and after cell death and many other questions have to be answered. For example, the rapid appearance of phosphoserine in the outer leaflet of the plasma membrane, at the onset of apoptosis, provides an 'eat me' signal and little is known about its influence over the cell’s survival programs.93 The breakage of lipids, which accompanies the formation of reactive oxygen species, can also lead to modifications of membrane permeability and fluidity and, as such, can dramatically alter cell function. It is also important to find how, in dying cells, the oxidation of lipids can act on kinases implicated in survival or lethal pathways.94 Similarly, owing to the connection between ER stress and the mitochondrial regulation of cell death28, 95 it would be interesting to investigate the modification of lipids present in the MAM in the latter process. Several recent results have pointed out these ER-mitochondria contacts as important in several different pathologies.96, 97
Thus, deciphering the mechanisms by which lipid modulates the activation of survival/death signaling pathways may help to develop therapeutic strategies in the prevention of a number of diseases implicating the dysregulation of apoptosis.
Abbreviations
- BH:
-
Bcl-2 homology domain
- BoP:
-
BH3-only protein
- Cox:
-
cyclooxygenase
- ER:
-
endoplasmic reticulum
- HMG-CoA:
-
hydroxymethylglutaryl coenzyme A
- MAM:
-
mitochondria-associated endoplasmic reticulum membrane
- MIM:
-
mitochondrial inner membrane
- MOM:
-
mitochondrial outer membrane
- MOMP:
-
mitochondrial outer membrane permeabilization
- PGA2:
-
prostaglandin A2
- PGE2:
-
prostaglandin E2
- PGES:
-
prostaglandin synthase
- PGH2:
-
prostaglandin H2
- S1P:
-
sphingosine-1-phosphate
References
Antonsson B, Martinou J-C . The Bcl-2 protein family. Exp Cell Res 2000; 256: 50–57.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Horvath SE, Daum G . Lipids of mitochondria. Prog Lipid Res 2013; 52: 590–614.
Crimi M, Esposti MD . Apoptosis-induced changes in mitochondrial lipids. Biochim Biophys Acta 2011; 1813: 551–557.
Schug ZT, Gottlieb E . Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta 2009; 1788: 2022–2031.
Tsujimoto Y, Cossman J, Jaffe E, Croce CM . Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985; 228: 1440–1443.
Borner C, Olivier R, Martinou I, Mattmann C, Tschopp J, Martinou JC . Dissection of functional domains in Bcl-2 alpha by site-directed mutagenesis. Biochem Cell Biol 1994; 72: 463–469.
Yin XM, Oltvai ZN, SJ Korsmeyer . BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 1994; 369: 321–323.
Farrow SN, Brown R . New members of the Bcl-2 family and their protein partners. Curr Opin Genet Dev 1996; 6: 45–49.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR . The BCL-2 Family Reunion. Mol Cell 2010; 37: 299–310.
Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011; 145: 104–116.
Lalier L, Cartron P-F, Juin P, Nedelkina S, Manon S, Bechinger B et al. Bax activation and mitochondrial insertion during apoptosis. Apoptosis 2007; 12: 887–896.
Walensky LD, Gavathiotis E . BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci 2011; 36: 642–652.
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002; 111: 331–342.
Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 2000; 14: 2060–2071.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 2005; 17: 525–535.
Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M, Reed JC . Channel formation by antiapoptotic protein Bcl-2. Proc Natl Acad Sci USA 1997; 94: 5113–5118.
Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I et al. Inhibition of Bax channel-forming activity by Bcl-2. Science 1997; 277: 370–372.
Schendel SL, Azimov R, Pawlowski K, Godzik A, Kagan BL, Reed JC . Ion channel activity of the BH3 only Bcl-2 family member, BID. J Biol Chem 1999; 274: 21932–21936.
Epand RF, Martinou J-C, Fornallaz-Mulhauser M, Hughes DW, Epand RM . The apoptotic protein tBid promotes leakage by altering membrane curvature. J Biol Chem 2002; 277: 32632–32639.
Zhai D, Miao Q, Xin X, Yang F . Leakage and aggregation of phospholipid vesicles induced by the BH3-only Bcl-2 family member, BID. Eur J Biochem 2001; 268: 48–55.
Penzo D, Tagliapietra C, Colonna R, Petronilli V, Bernardi P . Effects of fatty acids on mitochondria: implications for cell death. Biochim Biophys Acta 2002; 1555: 160–165.
Epand RF, Martinou J-C, Montessuit S, Epand RM . Fatty acids enhance membrane permeabilization by pro-apoptotic Bax. Biochem J 2004; 377: 509.
Ikonen E . Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 2008; 9: 125–138.
Miller WL, Bose HS . Early steps in steroidogenesis: intracellular cholesterol trafficking thematic review series: genetics of human lipid diseases. J Lipid Res 2011; 52: 2111–2135.
Mesmin B, Maxfield FR . Intracellular sterol dynamics. Biochim Biophys Acta 2009; 1791: 636–645.
Kornmann B . The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol 2013; 25: 443–448.
Rowland AA, Voeltz GK . Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 2012; 13: 607–625.
Grimm S . The ER-mitochondria interface: the social network of cell death. Biochim Biophys Acta 2012; 1823: 327–334.
Scarf AM, Kassiou M . The Translocator protein. J Nucl Med 2011; 52: 677–680.
Jiang Z, Zheng X, Lytle RA, Higashikubo R, Rich KM . Lovastatin-induced up-regulation of the BH3-only protein, Bim, and cell death in glioblastoma cells. J Neurochem 2004; 89: 168–178.
Marcuzzi A, Tricarico PM, Piscianz E, Kleiner G, Brumatti LV, Crovella S . Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis 2013; 4: e585.
Herrero-Martin G, López-Rivas A . Statins activate a mitochondria-operated pathway of apoptosis in breast tumor cells by a mechanism regulated by ErbB2 and dependent on the prenylation of proteins. FEBS Lett 2008; 582: 2589–2594.
Cafforio P, Dammacco F, Gernone A, Silvestris F . Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 2005; 26: 883–891.
Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR . Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin Cancer Res 1999; 5: 2223–2229.
N W C J van de Donk, Kamphuis MMJ, Lokhorst HM, Bloem AC . The cholesterol lowering drug lovastatin induces cell death in myeloma plasma cells. Leukemia 2002; 16: 1362–1371.
Lucken-Ardjomande S, Montessuit S, Martinou J-C . Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ 2007; 15: 484–493.
Martínez-Abundis E, Correa F, Rodríguez E, Soria-Castro E, Rodríguez-Zavala JS, Pacheco-Alvarez D et al. A CRAC-like motif in BAX sequence: relationship with protein insertion and pore activity in liposomes. Biochim Biophys Acta 2011; 1808: 1888–1895.
Christenson E, Merlin S, Saito M, Schlesinger P . Cholesterol effects on BAX pore activation. J Mol Biol 2008; 381: 1168–1183.
Martínez-Abundis E, Correa F, Pavón N, Zazueta C . Bax distribution into mitochondrial detergent-resistant microdomains is related to ceramide and cholesterol content in postischemic hearts. FEBS J 2009; 276: 5579–5588.
Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G et al. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res 2008; 68: 5246–5256.
Corcos L, Le Jossic-Corcos C . Statins: perspectives in cancer therapeutics. Dig Liver Dis 2013; 45: 795–802.
Hovius R, Lambrechts H, Nicolay K, de Kruijff B . Improved methods to isolate and subfractionate rat liver mitochondria. Lipid composition of the inner and outer membrane. Biochim Biophys Acta 1990; 1021: 217–226.
AIPM De Kroon, Dolis D, Mayer A, Lill R, de Kruijff B . Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim Biophys Acta 1997; 1325: 108–116.
Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y . Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J 2000; 351: 183–193.
Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S . Cytochrome C Release from mitochondria proceeds by a two-step process. PNAS 2002; 99: 1259–1263.
Jiang J, Huang Z, Zhao Q, Feng W, Belikova NA, Kagan VE . Interplay between bax, reactive oxygen species production, and cardiolipin oxidation during apoptosis. Biochem Biophys Res Commun 2008; 368: 145–150.
Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X . Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol 2000; 2: 754–761.
Sani M-A, Dufourc EJ, Gröbner G . How does the Bax-α1 targeting sequence interact with mitochondrial membranes? the role of cardiolipin. Biochim Biophys Acta 2009; 1788: 623–631.
Schafer B, Quispe J, Choudhary V, Chipuk JE, Ajero TG, Du H et al. Mitochondrial outer membrane proteins assist Bid in Bax-mediated lipidic pore formation. Mol Biol Cell 2009; 20: 2276–2285.
Cha YI, Solnica-Krezel L, DuBois RN . Fishing for prostanoids: deciphering the developmental functions of cyclooxygenase-derived prostaglandins. Dev Biol 2006; 289: 263–272.
Portranova et al. Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J Exp Med 1996; 184: 883–891.
Wang D, DuBois RN . Prostaglandins and cancer. Gut 2006; 55: 115–122.
Rundhaug JE, Simper MS, Surh I, Fischer SM . The role of the EP receptors for prostaglandin E2 in skin and skin cancer. Cancer Metastasis Rev 2011; 30: 465–480.
Lalier L, Cartron P-F, Pedelaborde F, Olivier C, Loussouarn D, Martin SA et al. Increase in PGE2 biosynthesis induces a Bax dependent apoptosis correlated to patients’ survival in glioblastoma multiforme. Oncogene 2007; 26: 4999–5009.
Lalier L, Pedelaborde F, Braud C, Menanteau J, Vallette FM, Olivier C . Increase in intracellular PGE2 induces apoptosis in Bax-expressing colon cancer cell. BMC Cancer 2011; 11: 153.
Lalier L, Cartron P-F, Olivier C, Logé C, Bougras G, Robert J-M et al. Prostaglandins antagonistically control Bax activation during apoptosis. Cell Death Differ 2011; 18: 528–537.
Kim H-S, Rhim H, Jeong S-W, Kim JW, Kim I-K . Induction of apoptosis dependent on caspase activities and growth arrest in HL-60 cells by PGA2. Prostaglandins Other Lipid Mediat 2002; 70: 169–183.
Lakowicz JR . Principles of Fluorescence Spectroscopy. Springer, 2007.
Callis PR, Burgess BK . Tryptophan fluorescence shifts in proteins from hybrid simulations: an electrostatic approach. J Phys Chem B 1997; 101: 9429–9432.
Lee S-Y, Ahn J-H, Ko KW, Kim J, Jeong SW, Kim I-K et al. Prostaglandin A2 activates intrinsic apoptotic pathway by direct interaction with mitochondria in HL-60 cells. Prostaglandins Other Lipid Mediat 2010; 91: 30–37.
Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS et al. Apoptotic Cells activate the ‘phoenix rising’ pathway to promote wound healing and tissue regeneration. Sci Signal 2010; 3: ra13.
Huang Q, Li F, Liu X, Li W, Shi W, Liu F-F et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 2011; 17: 860–866.
Hannun YA, Obeid LM . Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008; 9: 139–150.
Corre I, Niaudet C, Paris F . Plasma membrane signaling induced by ionizing radiation. Mutat Res 2010; 704: 61–67.
Ogretmen B, Hannun YA . Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 2004; 4: 604–616.
Grösch S, Schiffmann S, Geisslinger G . Chain length-specific properties of ceramides. Prog Lipid Res 2012; 51: 50–62.
Taha TA, Mullen TD, Obeid LM . A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim Biophys Acta 2006; 1758: 2027–2036.
Zhang J, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA . Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc Natl Acad Sci USA 1996; 93: 5325–5328.
El-Assaad W, El-Sabban M, Awaraji C, Abboushi N, Dbaibo GS . Distinct sites of action of Bcl-2 and Bcl-xL in the ceramide pathway of apoptosis. Biochem J 1998; 336 (Pt 3): 735–741.
Von Haefen C, Wieder T, Gillissen B, Stärck L, Graupner V, Dörken B et al. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene 2002; 21: 4009–4019.
Rotolo JA, Maj JG, Feldman R, Ren D, Haimovitz-Friedman A, Cordon-Cardo C et al. Bax and Bak do not exhibit functional redundancy in mediating radiation-induced endothelial apoptosis in the intestinal mucosa. Int J Radiat Oncol Biol Phys 2008; 70: 804–815.
Kashkar H, Wiegmann K, Yazdanpanah B, Haubert D, Krönke M . Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J Biol Chem 2005; 280: 20804–20813.
Siskind LJ, Mullen TD, Romero Rosales K, Clarke CJ, Hernandez-Corbacho MJ, Edinger AL et al. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J Biol Che 2010; 285: 11818–11826.
Beverly LJ, Howell LA, Hernandez-Corbacho M, Casson L, Chipuk JE, Siskind LJ . BAK activation is necessary and sufficient to drive ceramide synthase-dependent ceramide accumulation following inhibition of BCL2-like proteins. Biochem J 2013; 452: 111–119.
Birbes H, Luberto C, Hsu Y-T, El Bawab S, Hannun YA, Obeid LM . A mitochondrial pool of sphingomyelin is involved in TNFalpha-induced Bax translocation to mitochondria. Biochem J 2005; 386: 445–451.
Pastorino JG, Tafani M, Rothman RJ, Marcinkeviciute A, Hoek JB, Farber JL et al. Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. J Biol Chem 1999; 274: 31734–31739.
Ganesan V, Perera M, Colombini D, Datskovskiy D, Chadha K, Colombini M . Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010; 15: 553–562.
Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012; 148: 988–1000.
Spiegel S, Milstien S . Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 2003; 4: 397–407.
Ikeda H, Satoh H, Yanase M, Inoue Y, Tomiya T, Arai M et al. Antiproliferative property of sphingosine 1-phosphate in rat hepatocytes involves activation of Rho via Edg-5. Gastroenterology 2003; 124: 459–469.
Dorsam G, Graeler MH, Seroogy C, Kong Y, Voice JK, Goetzl EJ . Transduction of multiple effects of sphingosine 1-phosphate (S1P) on T cell functions by the S1P1 G protein-coupled receptor. J Immunol 2003; 171: 3500–3507.
Jin Y, Knudsen E, Wang L, Bryceson Y, Damaj B, Gessani S et al. Sphingosine 1-phosphate is a novel inhibitor of T-cell proliferation. Blood 2003; 101: 4909–4915.
Kim D-S, Kim S-Y, Kleuser B, Schäfer-Korting M, Kim KH, Park K-C . Sphingosine-1-phosphate inhibits human keratinocyte proliferation via Akt/protein kinase B inactivation. Cell Signal 2004; 16: 89–95.
Yamashita H, Kitayama J, Shida D, Yamaguchi H, Mori K, Osada M et al. Sphingosine 1-phosphate receptor expression profile in human gastric cancer cells: differential regulation on the migration and proliferation. J Surg Res 2006; 130: 80–87.
Young N, Van Brocklyn JR . Signal transduction of sphingosine-1-phosphate g protein—coupled receptors. ScientificWorldJournal 2006; 6: 946–966.
Siskind LJ, Colombini M . The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J Biol Chem 2000; 275: 38640–38644.
Siskind LJ, Kolesnick RN, Colombini M . Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion 2006; 6: 118–125.
Siskind LJ, Kolesnick RN, Colombini M . Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem 2002; 277: 26796–26803.
Stiban J, Fistere D, Colombini M . Dihydroceramide hinders ceramide channel formation: implications on apoptosis. Apoptosis 2006; 11: 773–780.
Elrick MJ, Fluss S, Colombini M . Sphingosine a product of ceramide hydrolysis, influences the formation of ceramide channels. Biophys J 2006; 91: 1749–1756.
Siskind LJ, Feinstein L, Yu T, Davis JS, Jones D, Choi J et al. Anti-apoptotic Bcl-2 family proteins disassemble ceramide channels. J Biol Chem 2008; 283: 6622–6630.
Clark MR . Flippin’ lipids. Nat Immunol 2011; 12: 373–375.
Newton AC . Lipid activation of protein kinases. J Lipid Res 2009; 3: ra13.
Vannuvel K, Renard P, Raes M, Arnould T . Functional and morphological impact of ER stress on mitochondria. J Cell Physiol 2013; 228: 1802–1818.
Paillard M, Tubbs E, Thiebaut P-A, Gomez L, Fauconnier J, Da Silva CC et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013; 128: 1555–1565.
Hedskog L, Pinho CM, Filadi R, Rönnbäck A, Hertwig L, Wiehager B et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci USA 2013; 110: 7916–7921.
Acknowledgements
This work was sponsored by a grant from INCA (PLBIO-2010). We thank Professor F Fleury (FRE CNRS 3478/ Université de Nantes) for his help with the circular dichroism experiments and Dr L Oliver for fruitful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Agostini
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Mignard, V., Lalier, L., Paris, F. et al. Bioactive lipids and the control of Bax pro-apoptotic activity. Cell Death Dis 5, e1266 (2014). https://doi.org/10.1038/cddis.2014.226
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.226
Keywords
This article is cited by
-
RNAi-mediated knockdown of MCM7 gene on CML cells and its therapeutic potential for leukemia
Medical Oncology (2017)
-
Enhanced therapeutic efficacy of LHRHa-targeted brucea javanica oil liposomes for ovarian cancer
BMC Cancer (2016)
-
Prostate tumor attenuation in the nu/nu murine model due to anti-sarcosine antibodies in folate-targeted liposomes
Scientific Reports (2016)
