
Abstract
Cucurbitacin E (CuE) is a natural compound previously shown to have anti-feedant, antioxidant and antitumor activities as well as a potent chemo-preventive action against cancer. The present study investigates its anti-proliferative property using MTT assay; CuE demonstrated cytotoxic activity against malignant glioma GBM 8401 cells and induced cell cycle G2/M arrest in these cells. CuE-treated cells accumulated in metaphase (CuE 2.5–10 μM) as determined using MPM-2 by flow cytometry. We attempted to characterize the molecular pathways responsible for cytotoxic effects of CuE in GBM 8401 cells. We studied the genome-wide gene expression profile on microarrays and molecular networks by using pathway analysis tools of bioinformatics. The CuE reduced the expression of 558 genes and elevated the levels of 1354 genes, suggesting an existence of the common pathways involved in induction of G2/M arrest. We identified the RB (GADD45β and GADD45γ) and the p53 (GADD45α) signaling pathways as the common pathways, serving as key molecules that regulate cell cycle. Results indicate that CuE produced G2/M arrest as well as the upregulation of GADD45 γ and binding with CDC2. Both effects increased proportionally with the dose of CuE, suggesting that the CuE-induced mitosis delay is regulated by GADD45γ overexpression. Our findings suggest that, in addition to the known effects on cancer prevention, CuE may have antitumor activity in glioma therapy.
Similar content being viewed by others


Main
Brain cancer is listed as the 18th most common malignant cancer in Taiwan;1 however, the appropriate means of treatment remains unclear. Many studies have demonstrated that chemotherapeutic drugs synthesized from plants repress tumor growth through the induction of cell cycle arrest or apoptosis.2
Cucurbitacins, derived from the climbing stem of Cucumis melo L., a group of tetracyclic triterpenes that are extensively used in folk and traditional medicine,3 which have selective biological activities and functions against carcinogenesis.4 In recent years, their putative beneficial bromacological/pharmacological effects as anti-inflammatory5 and anticancer agents have been studied.6, 7
Cucurbitacin E (CuE) is an active anti-feedant compound8 with the ability to disrupt cell actin9 and cell adhesion.10 Reports have demonstrated that CuE has an inhibitory effect on cancer cell proliferation, actin polymerization, and permeability.11, 12 However, whether CuE inhibits malignant glioma growth remains unknown. Furthermore, the mechanism underlying the anticancer effect of CuE is yet to be identified.
Human brain malignant gliomas (GBMs) are highly lethal primary brain tumors (grade IV gliomas), which appear to harbor the therapy-resistant cancer stem cells that have been shown to be a major cause of recurrence.13 GBM 8401 cells were isolated and established from a Chinese female patient with brain malignant glioma.2 These cells have been shown to be tumorigenic in athymic nude mice.14 Recent studies have suggested that GBMs contain a subpopulation of tumor cells that display stem cell-like characteristics and could therefore be responsible for tumor growth in vivo.15 For this reason, many studies selected GBM 8401 cells as a model for human brain glioblastoma to analyze the antitumor activity of biologically active compounds.13, 14, 16
Results
CuE inhibits the cell survival/proliferation of GBM 8401 cells
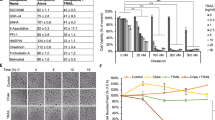
We hypothesized that CuE could mediate the survival of GBM 8401 cells and thus inhibit their proliferation. To explore the antitumor activity of CuE against the GBM 8401 cells, an in vitro study was initiated by treating the GBM 8401 cells to increasing doses of CuE (0, 2.5, 5, and 10 μM) for 24 h. The proliferation of these CuE-treated cancer cells was then measured by the MTT assay. The results summarized in Figure 1a indicate that both the survival and proliferation of GBM 8401 cells, but not that of normal skin fibroblast Hs-68 cells and normal lung fibroblast MRC-5 cells (data not shown), decrease as per increase in the dose of CuE added to the cell culture, in a dose-dependent manner (y=−3.5819x+92.647, R2=0.8374) during the 24-h treatment. Moreover, CuE was noted to induce a morphological change in the GBM 8401 cells. A microscopic examination showed that following the exposure to CuE (5 μM) for 24 h, the GBM 8401 cells have displayed a remarkable change in their morphology and that CuE induced the death of cancer cells, which formed a suspension in the medium (data not shown).

(a) CuE mediates the survival of GBM8401 cells (n=6 per group) and thus inhibits their proliferation. In vitro study was initiated by treating each of the cell lines to the increasing doses of CuE (0, 2.5, 5 and 10 μM) for 24 h. Reversibility of the growth-inhibitory effect of CuE in Figure 1A. The GBM8401 cells were each first treated with CuE for 24 h. After the treatment was terminated by washing off CuE, the cultures were reincubated for 24–48 h to check the extent of recovery of cancer cells. The survival of these CuE-treated GBM8401 cells was then measured by MTT assay. (b) Influence of CuE on apoptosis in GBM8401 cells. (c) Total apoptosis in GBM8401 cells after 4 h of incubation with CuE. Results were expressed as a percentage of total apoptotic cells (early and late apoptosis). Results were expressed as a percentage of control, which was considered as 100%. All data were reported as the means (±S.E.M.) of at least three separate experiments. Statistical analysis used the t-test, with the significant differences determined at the level of *P<0.05 versus control group, #P<0.05 versus 24 h-treated group
Growth-inhibitory effect of CuE is partially irreversible
To study whether the growth-inhibitory effect of CuE is reversible, the GBM 8401 cells were recultivated in a fresh culture medium, after their exposure to CuE for 24 h, and the recovery of cell proliferation was then assessed for an additional 24–48 h(Figure 1a) and analyzed using the MTT assay. The results in Figure 1a suggest that the cancer cells have substantially lost their ability to proliferate (y=−5.7062x+85.751, R2=0.7744) following their treatment with CuE for 48 and 72 h (y=−6.4054x+85.702, R2=0.8107). These observations could imply that the GBM 8401 cells have undergone an irreversible change, such as apoptosis or cell cycle arrest, at least to a partial extent.
Apoptosis of GBM 8401 cells is not induced by CuE
To identify the role of CuE in the apoptosis of GBM 8401 cells, we used Annexin V–FITC and propidium iodide staining to reveal the formation of apoptotic cells following 4 h of exposure to CuE. The percentage of apoptotic cells was assessed by flow cytometric analysis (Figure 1b). A dot-plot of Annexin V–FITC fluorescence versus propidium iodide (PI) fluorescence indicates a nonsignificant increase in the percentage of apoptotic cells treated with CuE, compared with untreated cells. No significant increase was observed in the percentage of cells undergoing necrosis, apoptosis (Figure 1c) or caspase 3 activation at CuE concentrations of 2.5–10 μM (data not shown). The loss of mitochondrial membrane potential (MMP) is a hallmark for apoptosis. It is an early event coinciding with caspase activation. In non-apoptotic cells, JC-1 exists as a monomer in the cytosol (green) and accumulates as aggregates in the mitochondria, which appear red. In apoptotic and necrotic cells, JC-1 exists in monomeric form and stains the cytosol green. CuE-free GBM 8401 cells are without apoptosis, which have red fluorescing J-aggregates. The percentages of apoptotic GBM 8401 cells were analyzed by flow cytometer in different CuE-treated groups. Taken together, the observations imply that CuE has not significantly reduced the MMP of GBM 8401 cells (data not shown). However, the results summarized in Figure 1 indicate that CuE may mediate the survival of GBM 8401 cells. Thus, we hypothesize that the proliferation of these cells was inhibited by pathways other than apoptosis and necrosis.
CuE treatment induces the accumulation of G2/M phase in GBM 8401 cells
The cell cycle distribution of CuE-treated cells was analyzed by flow cytometry. Cells were exposed to CuE for 24 h before processing and analysis. As shown in Figure 2a, exposure to CuE resulted in an increase in the number of G2/M phase cells, which may imply that the GBM 8401 cells underwent cell cycle arrest. Our results indicate that treatment with CuE increased the cell populations in G2/M phase while simultaneously reducing the number of cells in the S and G1 phases (*P<0.05 versus CuE 0 μM), which show a dose-dependent induction (y=5.8061x+30.567, R2=0.9325) (Figure 2b).

(a) Arrest of cell cycle progression at G2/M in response to CuE treatment. The distribution of the cell cycle of GBM8401 cells was assessed by flow cytometry after staining with PI. (b) Results were expressed as a percentage of G2/M. (c) MPM-2 (anti-phospho-Ser/Thr-Pro) expression in untreated and treated cancer cells. MPM-2 is an antibody that recognizes proteins that are only phosphorylated in mitosis. Cells were dually stained using propidium iodide to analyze DNA content and protein expression was quantified by flow cytometry. As a positive control, separate groups of cells were treated for 24 h with nocodazole (15 μg/ml), an antifungal agent known to induce metaphase arrest. Cell cycle analysis and quantification of MPM-2 expression were performed by flow cytometry following treatment with CuE for 24 h. (d) CuE enhanced the level of MPM-2 in CRC cells. Symbol (*) in each group of bars indicates that the difference resulting from treatment with CuE 0 μM is statistically significant at P<0.05
Effect of CuE on the mitotic index
To distinguish G2 arrest from mitotic arrest, we used an additional marker, MPM-2. This antibody is capable of recognizing proteins whose epitopes are exclusively phosphorylated during mitosis, specifically from early prophase to metaphase.17 MPM-2 is commonly used as an indicator of mitotic disturbance. To provide a positive control, we treated separate groups of GBM 8401 cells with nocodazole (15 μg/ml), an inducer of metaphase arrest.18
Treating the GBM 8401 cells with nocodazole for 24 h resulted in synchronization of entire cell populations in the G2/M phase as well as an increase in MPM-2 labeling (Figure 2c). Among all cells treated with CuE, MPM-2 staining had elevated above negative control levels (Figure 2d) (y=2.6207x+57.239, R2=0.9984).
Gene expression profile of GBM 8401 cells following exposure to CuE
By using human genome SurePrint G3 arrays, we studied the genome-wide gene expression profile of GBM 8401 cells exposed for 4 h to the vehicle (DMSO) or CuE (5 μM). Principal component analysis showed that the microarray data derived from the CuE-treated cells and those from DMSO-treated cells constituted two spatially separated planes, suggesting that the treatment with CuE exhibits a much greater impact on gene expression profile than the levels of gene expression changes simply attributable to the technical errors. Therefore, we considered fold changes >2 or <0.5, calculated by dividing the expression levels in the CuE-treated cells by those in the vehicle-treated cells, as substantial upregulation or downregulation, respectively. To identify biologically relevant molecular networks of these genes, the distinct pathway analysis tools of bioinformatics endowed with the comprehensive knowledge base Kyoto Encyclopedia of Genes and Genomes (KEGG) (www.kegg.jp) were used. We identified the KEGG pathway (Table 1) and a battery of downregulated genes (Table 2a) and upregulated genes following the treatment with CuE (Table 2b).
G2/M arrest by CuE in GBM8401 cells via GADD45γ upregulation and dissociation of the cyclin B1/CDC2 complex by GADD45γ binding
CuE elevated the expression of GADD45-α (NM_001924), -β (NM_011575) and -γ (NM_006705), but not in the levels of cyclin B1 (NM_031966) and CDC2 (NM_001786) (Figure 3a). These data suggested the existence of common molecular pathways that were involved in cell cycle G2/M arrest induction. For supporting the microarray analysis data, the RT-PCR (Figure 3b) and qPCR analyses (Figure 3c) validated substantial of cyclin B1 (y=−1.4593x+97.314, R2=0.8402), CDC2 (y=−2.0675x+97.785, R2=0.8694), JunD (y=−1.1851x+98.363, R2=0.9155), GADD45α (y=1.5577x+106.36, R2=0.5179), GADD45β (y=4.1163x+111.09, R2=0.734), and a notable upregulation of GADD45γ (y=191.98x−150.74, R2=0.8786) in GBM8401 cells following exposure to CuE (Figure 3c).

The genes downregulated or upregulated in GBM8401 cells following exposure to CuE. (a) The JunD, cyclinB1, CDC2 and GADD45-α, −β, −γ gene expression profile was studied in GBM8401 cells exposed for 4 h to the vehicle (DMSO) or to the CuE 5 μM. (b) The RT-PCR of JunD, cyclin B1, CDC2 and GADD45-α, −β, −γ mRNAs in GBM8401 cells following exposure to the CuE. The panels (c) indicate quantitative RT-PCR (qPCR) analysis of JunD, cyclin B1, CDC2 and GADD45-α, −β, −γ mRNA expression standardized against the levels of GAPDH in GBM8401 cells exposed for 4 h to DMSO (CuE 0 μM control) or CuE. Statistical analysis used the t-test, with the significant differences determined at the level of *P<0.05 versus the control group
Figure 4 illustrates the immunoblotting of cellular proteins from GBM8401 cells treated with CuE, revealing no effect on CDC2 following incubation with CuE (Figure 4a upper panel). CDC2 protein expression was quantified by measuring relative intensities. We found that CDC2 levels were not significantly changed in cells incubated with CuE. Moreover, the activity of the GADD45γ/CDC2 complex (important for G2–M transition blockade during the cell cycle) was determined by Co-IP (Figure 4a lower panel) and quantified by measuring the relative band intensities. We found that activity of GADD45γ–CDC2 complex was significantly enhanced in cells incubated with CuE (Figure 4c). These results indicate an increase of the cell population in G2/M phase via dissociation of the cyclin B1/CDC2 complex by integrated GADD45γ following incubation with CuE in GBM8401 cells.

Cell cycle arrest by CuE in GBM8401 cells via GADD45γ/CDC2 complex formation. Cells were treated with CuE followed by (a) western blot analysis (upper) and Co-IP (lower) (b) quantification of intensities by Li-COR near infrared imaging system. (c) Mitosis delay by CuE in GBM8401 cells via inhibition of Cdc2-cyclin B1 complex dissociation by GADD45γ binding with CDC2. Significant differences were determined at a level of *P<0.05 versus the 0 μM control group
Discussion
Cucurbitacins, a family of naturally occurring tetracyclic triterpenes, have been shown to be potent cytotoxic agents in cancer cells.19 In our previous study, CuE-treated cells showed growth arrest and apoptosis in human oral squamous cell carcinoma cell line SAS cells.20 The most common cell death mode in cucurbitacins treatment seems to be apoptosis21 and cell cycle arrest.22 In a previous study, the CuE activated the caspase-dependent pathway, coinciding with the activation of the mitochondrial pathway in bladder cancer cells.23 Throughout Asia, CuE has been used in traditional medicine for cancer therapy and many studies have attempted to elucidate the mechanism underlying its antitumor activity.8 Moreover, triterpepenoids (steroidal compounds) are able to cross blood brain barrier due to their lipophilic nature. So, it can be assumed that such compounds might also be responsible for eliciting antitumor activity in brain.24 Liposomal CuE may conduct the pharmacokinetics of CuE.8, 25
However, the results summarized in Figures 1 and 2 indicate that CuE may mediate the survival of GBM8401 cells. Thus, the proliferation of these cells was inhibited by pathways other than apoptosis. Recent studies have shown that CuE inhibited the growth of tumor precursors26 and enhanced VEGFR2-mediated Jak2–STAT3 pathways27 leading to apoptosis28 and anti-angiogenesis.29
In this study, the CuE demonstrated antitumor activity as well as the ability to induce mitosis delay. The results collected in this series of studies provide experimental evidence supporting the contention that CuE may irreversibly arrest the growth of GBM8401 cells. The results of mechanistic analysis led to the conclusion that both inhibition of proliferation and the induction of mitosis delay are highly dependent on CuE accumulated in the GBM8401 cells. The role of CuE in the inhibition of tumor cell growth was highlighted by the delay of mitosis through the upregulation of GADD45γ, and in the dissociation of the cyclin B1/CDC2 complex in GBM8401 cells.
The Gadd45 gene family encodes three highly conserved nuclear proteins that contribute to cellular homeostasis in response to stress.30 Evidences suggest that GADD45 family fulfills similar functions in survival, cell cycle control, apoptosis, and repair of DNA damage.31, 32 Multiple tumor suppressive genes linked to sporadic tumors have been identified. They can be placed into two groups: the RB group includes GADD45B and GADD45G33 and the p53 group includes GADD45A.34
Among the p53 pathway genes, apoptosis-related genes had markedly increased when compared with controls. Moreover, cell cycle progression genes and DNA-repair genes (that is, GADD45G) had also increased.35
Gadd45γ has also been shown to interact with several key cellular regulators, including cyclin B1, p21, proliferating cell nuclear antigen, and mitogen-activated protein kinase.36 The cellular function of Gadd45 γ is dependent on its interacting partner. Notably, Gadd45 γ is able to suppress G2–M progression in response to stress through its ability to interact with, and suppress the kinase activities, of the cyclin B1/CDC complex.37 Accordingly, RNA silencing of Gadd45 expression impairs G2–M checkpoint activity. It remains to be determined whether interactions between Gadd45 and p21 have a role in G1 arrest.36 Moreover, downregulation of Gadd45 is closely associated with the degree of malignancy in cancers. Therefore, Gadd45 gene family may have an important role in carcinogenesis.
The effect of CuE in GBM8401 cells seemed to be independent of a DNA-damage Chk1-cdc2-mediated pathway, unlike the G2 arrest mediated by radiation, and seemed to be predominantly a metaphase arrest.38 Of interest, our findings suggest that cell cycle G2/M arrests in GBM8401 cell lines at higher CuE doses (7.5 and 10 μM), whereas apoptosis and caspase activation dominate at low doses (5 μM) in SAS cells18 and other cancer cells.27 The results of mechanical analysis have led us to conclude that both inhibition of proliferation and the induction of apoptosis are dependent on the cancer cell types. However, on further investigation, our data suggest a more complex mechanism involving cell cycle deregulation and apoptosis that seem to reflect differences in degree of CuE-induced toxicity between the cancer cell lines.
In conclusion, we have demonstrated for the first time that CuE inhibits tumor growth by arresting the cell cycle at G2/M phase via GADD45γ gene expression and blockade of cyclin B1/CDC2 complex in GBM8401 cells. The role of CuE in the inhibition of tumor growth was highlighted by the delay of mitosis through the upregulation of GADD45 gene family. These findings suggest the applicability of CuE as an anticancer agent.
Materials and Methods
Materials
CuE, DMSO (dimethyl sulfoxide) and MTT (3-(4,5-dimethylthiazol-2-yl) -2,5-diphenyltetrazolium bromide) were obtained from Sigma (St. Louis, MO, USA). Cell culture medium (DMEM), fetal bovine serum (FBS), antibiotics, sodium pyruvate, trypsin, and phosphate-buffered saline (PBS) were purchased from Gibco, BRL (Grand Island, NY, USA). Polyvinylidene fluoride membrane (PVDF) (Merck Millipore, Darmstadt, Germany), and molecular weight markers were purchased from Bio-Rad (Berkeley, CA, USA). All other reagents and compounds were analytical grades.
Cell culture
GBM 8401 cells were purchased from the Food Industry Research and Development Institute (Hsinchu, Taiwan). The cells were maintained on culture dishes, in RPMI 1640 supplemented with 10% (v/v) FBS and cultured in an incubator at 37 °C in an atmosphere containing 5% CO2.
Cell proliferation assay
Cells were seeded into 96-well culture plates at 10 000 cells/well. Different cell wells were treated with 0, 2.5, 5 and 10 μM of CuE for 1–3 days. MTT dye (1 mg/ml) was added to each well for an additional 4 h following treatment. The reaction was stopped by the addition of DMSO, and optical density was measured at 540 nm using a multi-well plate reader (Powerwave XS, Biotek, Winooski, VT, USA). In the absence of cells, background absorbance of the medium was subtracted. Results were expressed as a percentage of control, which was considered as 100%. Each assay was performed in triplicate and the results were expressed as the mean (±S.E.M.). Changes in cell morphology were determined microscopically with an Olympus CKX41 microscope.
Measurement of apoptosis
GBM 8401 cells were first seeded in six-well plates (Orange Scientific, E.U., Braine-l'Alleud, Belgium). Following the treatment with CuE for 4 h, the cells were harvested and washed in cold PBS. A 1 × annexin-binding buffer (BD Pharmingen, BD, USA) and 100 μg/ml working solution of PI (Sigma) were prepared. The washed cells were recentrifuged (the supernatant was discarded) and resuspended in 1 × annexin-binding buffer. Following this, 5 μl of FITC annexin V (BD Pharmingen) and 1 μl of 100 μg/ml PI working solution were added to each 100 μl of cell suspension, and the cells were incubated at room temperature for 15 min. After the incubation period, the stained cells were analyzed using flow cytometry, with fluorescence emission measurements showing only low levels: apoptotic cells showed green fluorescence, and dead cells showed both red and green fluorescence.
MMP
GBM 8401 cells were first seeded in 24- or 6-well plates (Orange Scientific, E.U.). Following treatment with CuE for 4 h, Rhodamine 123 (10 μg/ml, Sigma-Aldrich, St. Louis, MO, USA) and JC-1 (25 μM) was added to the culture medium (500 μl/well) and incubated (37 °C, 20 min) for mitochondrial staining. After washing twice with warm PBS, the cells were fixed using 2% paraformaldehyde, inspected using a fluorescence microscope (Olympus CKX41 and U-RFLT 50), and the RFU (Relative fluorescence unit) was detected using a BioTek FLx800 TBI. To detect rhodamine 123, the wavelengths were set at 504 and 534 nm. Each assay was carried out in triplicate and the results were expressed as the mean (±S.E.M.) of RFU and reported as the percentage of the RFU for the control group (SFN 0 μM). For JC-1, quantification was performed by flow cytometry (BD FACScalibur, BD) and mitochondria containing red JC-1 aggregates in healthy cells were detectable in the FL2 channel, whereas green JC-1 monomers in apoptotic cells were detectable in the FL1 channel.
Cell cycle analysis
For cell cycle analysis, we used the fluorescent nucleic acid dye PI to identify the proportion of cells in each of the three interphase stages of the cell cycle. Cells were treated with 0, 2.5, 5, and 10 μM CuE for 24 h, and then harvested and fixed in 1 ml of cold 70% ethanol for at least 8 h at −20 °C. DNA was stained in PI/RNaseA solution and the cell cycle (at least 10 000 single cells) was detected using flow cytometry (FACSCalibur, BD). Data were analyzed using WinMDI 2.8 free software (BD).
Mitotic index analysis
The mitotic index was assessed by MPM-2 (anti-phospho-Ser/Thr-Pro) expression. After 24 h of treatment with CuE, cells were harvested and fixed in 70% ethanol overnight. Cells were then washed and suspended in 100 μl of IFA-Tx buffer (4% FCS, 150 nM NaCl, 10 nM HEPES, 0.1% sodium azide, 0.1% Triton X-100) with a primary MPM-2 antibody (1 μg/ml; Upstate Cell Signaling Solutions, Millipore, Watford, UK) at room temperature for 1 h. Cells were washed and resuspended in IFA-TX buffer with a rabbit anti-mouse FITC-conjugated secondary antibody (1 : 50; Serotec, Oxford, UK) for 1 h at room temperature in darkness. Finally, cells were washed and resuspended in 500 μl of PBS with 20 μg/ml of PI (Sigma) for 30 min in the dark. MPM-2 expression was analyzed using flow cytometry (FACSCalibur, BD). Data were analyzed using WinMDI 2.8 free software.
Western blot assay
A total of 30–50 μg of proteins were separated by SDS-PAGE (10–12% SDS-polyacrylamide gel electrophoresis), and transferred to PVDF membranes (Merck Millipore) in a tank blotter (in 25 mM Tris/0.192 M glycine, pH 8.3/20% methanol) at 30 V overnight. The membranes were blocked with blocking buffer (Odyddey, LI-COR, Lincoln, NE, USA) overnight, and incubated with anti-β-actin (Sigma-Aldrich), anti-CDC2 (Santa Cruz BioTechnology, Dallas, TX, USA) antibodies for 1.5–2 h. The blots were washed with Tris-HCl (pH 8.0/150 mM NaCl/0.05% Tween-20) for 3 × 10 min, and incubated with a second antibody (anti-rabbit or anti-mouse immunoglobulins) (IRDye Li-COR, USA) at a 1/20 000 dilution for 30 min. The antigen was then visualized using a near infrared imaging system (Odyssey LI-COR) and data were analyzed using Odyssey 2.1 software.
Co-immunoprecipitation (Co-IP)
Co-IP is an effective means of quantifying protein–protein interactions in cells. Briefly, 500 μg of cellular proteins were labeled using anti-CDC2 (Santa Cruz BioTechnology) and GADD45γ (TA505437 OriGene Technologies, Rockville, MD, USA) following overnight incubation at room temperature. The protein–antibody immunoprecipitates were collected by protein A/G plus-agarose (SC-2003 Santa Cruz BioTechnology). Following the final wash, the samples were boiled and centrifuged to pellet the agarose beads. Western blot analysis of the CDC2 protein in the supernatant was then conducted. Antigens were visualized using a near infrared imaging system (Odyssey LI-COR) and data were analyzed using Odyssey 2.1 software.
Gene expression profiling (GEP)
Briefly, the untreated cells or those treated with CuE for 4 h, were harvested and total RNA was isolated using an RNasey kit (Qiagen) as described by the manufacturer. Total RNA was sent to Welgene Company for whole human genome SurePrint G3 arrays GEP analysis (Agilent Technologies).
RT-PCR
A reverse transcriptase system (Promega, Southampon, UK) was used to synthesize cDNA from 1 μg of total RNA. Between 2 and 4 μl of cDNA were used for PCR analysis. The primers used for amplification of the human genes are listed in Tables 3a and 3b. PCR (50 μl) reactions were performed using 100 ng of each primer and 1 U of Dynazyme II (Flowgen, Lichfield, UK). Thermal cycling was conducted for 35 cycles at the following temperature/durations: 94 C for 40 s, 60 C for 40 s, and 72 C for 40 s using a Progene thermal cycler (Cambridge, UK). A final extension of 72 °C was performed for 10 min at the end of 35 cycles. The primers used for amplification of the target genes were checked against all other gene sequences for specificity. PCR reactions were analyzed on 1.5% agarose/TAE minigels and stained using 0.5 μg/ml ethidium bromide. Gels were visualized using an Apligene UV CCD camera system.
Quantitative real-time-PCR (qRT-PCR)
Real-time-PCR was conducted using SYBR Green PCR MasterMix according to the manufacturer’s instructions. QRT-PCR was performed using ∼200 ng of SYBR Green PCR MasterMix in an ABI 7300 system (Applied Biosystems, Foster City, CA, USA). PCR conditions were 95 °C for 5 s, 60 °C for 30 s, and 72 °C for 30 s for 40 cycles. Sample cells from three plates were run in duplicate, using the threshold suggested by the software for the instrument to calculate Ct. To normalize readings, we used Ct values from 18 s as internal controls for each run, obtaining a ΔCt value for each gene.
Statistical analysis
All data were reported as the mean (±S.E.M.) of at least three separate experiments. A t-test or one-way analysis of variance with post hoc test was employed for statistical analysis, with significant differences determined as P<0.05 or 0.01.
Abbreviations
- GBM:
-
human brain malignant glioma
- CuE:
-
cucurbitacin E
- GADD45G:
-
growth arrest and DNA damage-inducible protein gamma
- cDNA:
-
complementary DNA
- qRT-PCR:
-
quantitative real-time-PCR
- PI:
-
propidium iodide
- DMSO:
-
dimethyl sulfoxide
- SDS:
-
sodium dodecyl sulfate
- PAGE:
-
polyacrylamide gel electrophoresis
- CDC2:
-
cell division cycle gene 2
- Co-IP:
-
co-immunoprecipitation
- MMP:
-
mitochondrial membrane potential
- PBS:
-
phosphate-buffered saline
- PVDF:
-
polyvinylidene fluoride membrane
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- GEP:
-
gene expression profiling
References
Lee CC, Wang WH, Lin CF, Chen HH, Chen SC, Lin SC et al. Malignant transformation of supratentorial ganglioglioma. Clin Neurol Neurosurg 2012; 114: 1338–1342.
Huang TY, Chang WC, Wang MY, Yang YR, Hsu YC . Effect of sulforaphane on growth inhibition in human brain malignant glioma GBM 8401 cells by means of mitochondrial- and MEK/ERK-mediated apoptosis pathway. Cell Biochem Biophys 2012; 63: 247–259.
Chen X, Bao J, Guo J, Ding Q, Lu J, Huang M et al. Biological activities and potential molecular targets of cucurbitacins: a focus on cancer. Anticancer Drugs 2012; 23: 777–787.
Ríos JL, Andújar I, Escandell JM, Giner RM, Recio MC . Cucurbitacins as inducers of cell death and a rich source of potential anticancer compounds. Curr Pharm Des 2012; 18: 1663–1676.
Recio MC, Andujar I, Rios JL . Anti-inflammatory agents from plants: progress and potential. Curr Med Chem 2012; 19: 2088–2103.
Lee DH, Iwanski GB, Thoennissen NH . Cucurbitacin: ancient compound shedding new light on cancer treatment. ScientificWorldJournal 2010; 10: 413–418.
Chen JC, Chiu MH, Nie RL, Cordell GA, Qiu SX . Cucurbitacins and cucurbitane glycosides: structures and biological activities. Nat Prod Rep 2005; 22: 386–399.
Habib L, Khreich N, Jraij A, Abbas S, Magdalou J, Charcosset C et al. Preparation and characterization of liposomes incorporating cucurbitacin E, a natural cytotoxic triterpene. Int J Pharm 2013; 448: 313–319.
Abbas S, Vincourt JB, Habib L, Netter P, Greige-Gerges H, Magdalou J . The cucurbitacins E, D and I: investigation of their cytotoxicity toward human chondrosarcoma SW 1353 cell line and their biotransformation in man liver. Toxicol Lett 2013; 216: 189–199.
Zhang T, Li J, Dong Y, Zhai D, Lai L, Dai F et al. Cucurbitacin E inhibits breast tumor metastasis by suppressing cell migration and invasion. Breast Cancer Res Treat 2012; 135: 445–458.
Sörensen PM, Iacob RE, Fritzsche M, Engen JR, Brieher WM, Charras G, Eggert US . The natural product cucurbitacin E inhibits depolymerization of actin filaments. ACS Chem Biol 2012; 7: 1502–1508.
Huang WW, Yang JS, Lin MW, Chen PY, Chiou SM, Chueh FS et al. Cucurbitacin E Induces G(2)/M phase arrest through STAT3/p53/p21 signaling and provokes apoptosis via Fas/CD95 and mitochondria-dependent pathways in human bladder cancer T24 cells. Evid Based Complement Alternat Med 2012; 2012: 952762.
Tang JY, Chang HW, Chang JG . Modulating roles of amiloride in irradiation-induced antiproliferative effects in glioblastoma multiforme cells involving Akt phosphorylation and the alternative splicing of apoptotic genes. DNA Cell Biol 2013; 32: 504–510.
Lee WH, Yeh MY, Tu YC, Han SH, Wang YC . Establishment and characterization of a malignant glioma cell line, GBM8401/TSGH,NDMC. J Surg Oncol 1988; 38: 173–181.
Aldaz B, Sagardoy A, Nogueira L, Guruceaga E, Grande L, Huse JT et al. Involvement of miRNAs in the Differentiation of Human Glioblastoma Multiforme Stem-Like Cells. PLoS One 2013; 8: e77098.
Weng HY, Hsu MJ, Wang CC, Chen BC, Hong CY, Chen MC, Chiu WT et al. Zerumbone suppresses IKKα, Akt, and FOXO1 activation, resulting in apoptosis of GBM 8401 cells. J Biomed Sci 2012; 19: 86.
Glatz K, Hartmann C, Antic M, Kutzner H . Frequent mitotic activity in banal melanocytic nevi uncovered by immunohistochemical analysis. Am J Dermatopathol 2010; 32: 643–649.
Seo HR, Lee DH, Lee HJ, Baek M, Bae S, Soh JW et al. Cyclin G1 overcomes radiation-induced G2 arrest and increases cell death through transcriptional activation of cyclin B1. Cell Death Differ 2006; 13: 1475–1484.
Kim SR, Seo HS, Choi HS, Cho SG, Kim YK, Hong EH, Shin YC et al. Trichosanthes kirilowii ethanol extract and Cucurbitacin D inhibit cell growth and induce apoptosis through inhibition of STAT3 activity in breast cancer cells. Evid Based Complement Alternat Med 2013; 2013: 975350.
Hung CM, Chang CC, Lin CW, Ko SY, Hsu YC . Cucurbitacin E as inducer of cell death and apoptosis in human oral squamous cell carcinoma cell line SAS. Int J Mol Sci 2013; 14: 17147–17156.
Lang KL, Deagosto E, Zimmermann LA, Machado VR, Campos Bernardes LS, Schenkel EP et al. Chemical modification produces species-specific changes in cucurbitacin antifeedant effect. J Agric Food Chem 2013; 61: 5534–5539.
Alghasham AA . Cucurbitacins—a promising target for cancer therapy. Int J Health Sci (Qassim) 2013; 7: 77–89.
Spear SA, Burns SS, Oblinger JL, Ren Y, Pan L, Kinghorn AD et al. Natural compounds as potential treatments of NF2-deficient schwannoma and meningioma: cucurbitacin D and goyazensolide. Otol Neurotol 2013; 34: 1519–1527.
Librowaski P, Czarapki R, Mendyk A, Jastrzebska M . Influence of new monoterpene homologous of GABA on the CNS activity in mice. Pol J Pharmacol 2000; 52: 317–321.
Habib L, Jraij A, Khreich N, Fessi H, Charcosset C, Greige-Gerges H . Morphological and physicochemical characterization of liposomes loading cucurbitacin E, an anti-proliferative natural tetracyclic triterpene. Chem Phys Lipids 2013; 177C: 64–70.
Dong Y, Lu B, Zhang X, Zhang J, Lai L, Li D et al. Cucurbitacin E, a tetracyclic triterpenes compound from Chinese medicine, inhibits tumor angiogenesis through VEGFR2-mediated Jak2-STAT3 signaling pathway. Carcinogenesis 2010; 31: 2097–2104.
Sun C, Zhang M, Shan X, Zhou X, Yang J, Wang Y et al. Inhibitory effect of cucurbitacin E on pancreatic cancer cells growth via STAT3 signaling. J Cancer Res Clin Oncol 2010; 136: 603–610.
Sadzuka Y, Hatakeyama H, Sonobe T . Enhancement of doxorubicin concentration in the M5076 ovarian sarcoma cells by cucurbitacin E co-treatment. Int J Pharm 2010; 383: 186–191.
Sultan FA, Sweatt JD . The role of the gadd45 family in the nervous system: a focus on neurodevelopment, neuronal injury, and cognitive neuroepigenetics. Adv Exp Med Biol 2013; 793: 81–119.
Niehrs C, Schäfer A . Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol 2012; 22: 220–227.
Tamura RE, de Vasconcellos JF, Sarkar D, Libermann TA, Fisher PB, Zerbini LF . GADD45 proteins: central players in tumorigenesis. Curr Mol Med 2012; 12: 34–51.
Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H et al. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev 2013; 12: 661–684.
Zhou Y, Zhang X, Klibanski A . Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol Cell Endocrinol 2013; S0303: 7207.
Moskalev AA, Smit-McBride Z, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A et al. Gadd45 proteins: relevance to aging, longevity and age-related pathologies. Ageing Res Rev 2012; 11: 51–66.
Calenic B, Yaegaki K, Ishkitiev N, Kumazawa Y, Imai T, Tanaka T . p53-Pathway activity and apoptosis in hydrogen sulfide-exposed stem cells separated from human gingival epithelium. J Periodontal Res 2013; 48: 322–330.
Shih RS, Wong SH, Schoene NW, Zhang JJ, Lei KY . Enhanced Gadd45 expression and delayed G2/M progression are p53-dependent in zinc-supplemented human bronchial epithelial cells. Exp Biol Med (Maywood) 2010; 235: 932–940.
Lee YH, Bedford MT, Stallcup MR . Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev 2011; 25: 176–188.
Cretu A, Sha X, Tront J, Hoffman B, Liebermann DA . Stress sensor Gadd45 genes as therapeutic targets in cancer. Cancer Ther 2009; 7: 268–276.
Acknowledgements
We gratefully acknowledge the financial support of Tainan Sin-Lau Hospital (SLH 101-01 and SLH 102-01).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Stephanou
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hsu, YC., Chen, MJ. & Huang, TY. Inducement of mitosis delay by cucurbitacin E, a novel tetracyclic triterpene from climbing stem of Cucumis melo L., through GADD45γ in human brain malignant glioma (GBM) 8401 cells. Cell Death Dis 5, e1087 (2014). https://doi.org/10.1038/cddis.2014.22
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.22
