
Abstract
Autophagy has an important role in tumor biology of hepatocellular carcinoma (HCC). Recent studies demonstrated that tissue factor (TF) combined with coagulation factor VII (FVII) has a pathological role by activating a G-protein-coupled receptor called protease-activated receptor 2 (PAR2) for tumor growth. The present study aimed to investigate the interactions of autophagy and the coagulation cascade in HCC. Seventy HCC patients who underwent curative liver resection were recruited. Immunohistochemical staining and western blotting were performed to determine TF, FVII, PAR2 and light chain 3 (LC3A/B) expressions in tumors and their contiguous normal regions. We found that the levels of autophagic marker LC3A/B-II and coagulation proteins (TF, FVII and PAR2) were inversely correlated in human HCC tissues. Treatments with TF, FVII or PAR2 agonist downregulated LC3A/B-II with an increased level of mTOR in Hep3B cells; in contrast, knockdown of TF, FVII or PAR2 increased LC3A/B. Furthermore, mTOR silencing restored the impaired expression of LC3A/B-II in TF-, FVII- or PAR2-treated Hep3B cells and activated autophagy. Last, as an in vivo correlate, we administered TF, FVII or PAR2 agonist in a NOD/severe combined immunodeficiency xenograft model and showed decreased LC3A/B protein levels in HepG2 tumors with treatments. Overall, our present study demonstrated that TF, FVII and PAR2 regulated autophagy mainly via mTOR signaling. The interaction of coagulation and autophagic pathways may provide potential targets for further therapeutic application in HCC.
Similar content being viewed by others

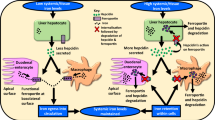

Main
Tissue factor (TF) is a 47-kDa transmembrane glycoprotein that initiates blood coagulation cascade when exposed to activated factor VII (FVIIa).1TF is constitutively expressed in a variety of tumor cells and has been implicated in cellular signaling, angiogenesis and tumor progression. Formation of TF-FVIIa complex and generation of downstream coagulation proteases, including activated factor X and thrombin, initiate signaling by activation of protease-activated receptors.2, 3
Several recent studies indicated that TF has a role in tumor biology4 and increasing evidence suggests that the TF/FVII complex is involved in the development and spread of cancer.5, 6, 7 Especially, a study using specific inhibitory antibodies against TF-FVIIa signaling has demonstrated significant attenuation of tumor growth in a human breast cancer model and was found to be dependent on protease-activated receptor 2 (PAR2) activity.8 Independent evidence also shown that PAR2 deficiency reduced the growth of breast cancer cells in mice and the attenuated tumor development did not further delay in mice with simultaneous deletion of PAR2 and cytoplasmic domain of TF, demonstrating a crosstalk between TF and PAR2 contributes to primary tumor growth in breast cancer.9 The potential of TF-dependent signaling events to be exploited as therapeutic targets and diagnostic biomarkers in cancer was also discussed.10, 11 Furthermore, the plasma TF levels of hepatocellular carcinoma (HCC) patients were closely related to tumor differentiation level, tumor size and hepato-cirrhosis occurrence.12, 13 It also suggested the possible involvement of TF in tumor metastasis and therefore prognosis.14 Accumulating evidences suggest that angiogenesis is a recurring factor and has an important role in the development, progression and metastasis of HCC.15, 16, 17 A prospective study has been conducted to evaluate the expression of TF and its correlation with angiogenesis and tumor invasiveness in HCC, and strong correlation between tumor TF levels and histo-pathological features indicated a significant association between high TF expression and the degree of venous invasion, degree of microsatellite nodules and advanced TNM stage.18 Furthermore, Zhou’s study also demonstrated that TF is overexpressed in both plasma and tissue in HCC patients, and it is closely related with many invasive and metastasis indexes.19
Autophagy is a physiologic process involved in routine turnover of cell constituents and serves as a temporary survival mechanism during starvation where self-digestion becomes an alternative energy source. Autophagy has also been proposed to have another biological function of clearing unfolded protein under certain stress conditions.20, 21 The process of autophagy starts by sequestering a portion of the cytoplasm and intracellular organelles in a double membrane-bound structure known as the autophagosome. The recruitment of both autophagy-related proteins (Atg)-forming Atg12-Atg5 complex and microtubule-associated protein 1 light chain 3 (LC3) are required for the elongation of autophagosomes. These autophagosomes form autolysosomes by fusing with lysosomes and degrade the sequestered contents by lysosomal hydrolases.22 Recent studies showed that autophagy has an active role in cell death.20 Autophagy or autophagic cell death has been known as type II programmed cell death in response to many anti-tumor therapies in various types of cancer.22 Recent data also suggested that autophagy is involved in many aspects of liver diseases,23 and may be important in the regulation of cancer development and progression and determining the response of tumor cells to anticancer therapy.24 However, the role of autophagy in these processes is complicated and may, depending on influence of tissue origins or microenvironments, exhibit straight opposite consequences. The mechanisms responsible for low incidence of autophagy in HCC are not fully elucidated.
The mammalian target of rapamycin (mTOR) pathway is abnormally activated in a proportion of HCC patients.25 Inhibition of mTOR has been shown to suppress liver tumor growth and metastasis.26, 27 It has also been reported that aberrant activation of the mTOR/ribosomal protein p70 S6 kinase (p70S6K) pathway increased tumor cell proliferation and angiogenesis in an HCC rat model.28, 29 It was recently shown that inhibition of Akt/mTOR significantly induced autophagy and led to cell death in HCC cells by SAHA, a potent inhibitor of histone deacetylases.30
Our goal is to investigate the critical role of autophagy in cancer-associated coagulation cascade of HCC. In the present study, based on the negative correlation between TF/FVII and LC3 expression in 70 HCC patients, we investigated the role of TF/FVII/PAR2-mediated signaling in the regulation of autophagy and the involvement of mTOR activity in human HCC by in vitro and in vivo evidences. Our findings suggest a pivotal role of TF/FVII/PAR2-signaling cascade but not thrombin/PAR1 response in mTOR-dependent blockade of autophagy as further potential therapeutic targets in HCC.
Results
Autophagy is activated in HCC patients with low FVII/PAR2 expression in tumors and vice versa
The formation of lipid-conjugated LC3 (LC3A/B-II) is used as a marker of autophagy. Thus, we performed immunohistochemistry (IHC) staining to evaluate the amount of LC3A/B protein in liver tissue of HCC patients. Tissues were scored for LC3A/B staining from one to four (low to high) for all resected tumor tissues. As shown in Figure 1a, 54 out of 70 normal liver tissues of patients (77.2%) expressed high LC3A/B (scored 3–4) immunogenicity, whereas only 24 out of 70 cancerous liver tissues of patients (35.8%) expressed high LC3A/B. The result demonstrated that LC3A/B is more highly expressed in normal tissues than in cancerous tissues of HCC patients. Furthermore, the expression levels of coagulation factors FVII and PAR2 were analyzed in cancerous tissues by western blot analysis and we found that the expression of both FVII and PAR2 was negatively correlated with that of LC3A/B-II in 70 HCC patients (Figure 1b). We showed that the antibody used to detect endogenous levels of LC3A/B protein has a much stronger reactivity with the type II form of LC3A/B. FVII and PAR2 expression in 45 out of 70 cancerous tissues with low LC3A/B-II (grade 1–2) was significantly higher than in their adjacent normal tissues (P-value=0.006 for FVII, P-value=0.006 for PAR2), whereas in 25 out of 70 high LC3A/B-II cancerous tissues (grade 3–4), the expression of FVII and PAR2 was lower than in the normal counterparts (P-value=0.006 for FVII, P-value=0.006 for PAR2).

(a) IHC staining score for LC3A/B in hepatocellular carcinoma (HCC). The immuno-histochemical staining results for LC3A/B were evaluated with four scores based on the staining intensity: score 4, >76% positive cells; score 3, 51–75% positive cells; score 2, 26–50% positive cells and score 1, ≤25% positive cells. Negative controls that substituted the primary antibodies for the immunoglobulin fraction of non-immune rabbit (for leptin) in HCC are shown in panels. (b) Western blot analysis of FVII, and PAR2 in four representative HCC tissues (T) and their paired non-tumor tissues (N). FVII and PAR2 overexpressed in human HCC tumor compared with adjacent non-tumor tissues, and they are positively correlated. All results are expressed as the mean±S.D. from at least three independent experiments. *P<0.05, **P<0.01 all compared with non-tumor control
Coagulation factors FVII/PAR2 counteract autophagy induction in HCC cells
To investigate the role of coagulation factors FVII and PAR2 in regulating LC3 expression, we treated triplicate Hep3B cells with recombinant (r)TF, rFVIIa, thrombin, PAR1 or a PAR2 peptide agonist for 24 h and observed the LC3A/B-II expression by western blot analysis. As shown in Figure 2a, treatment with rTF, rFVIIa and PAR2 agonist significantly decreased LC3A/B-II expression, whereas no significant change in cells treated with thrombin and PAR1 was observed. Furthermore, knockdown of TF, FVII and PAR2 by means of small interfering RNA (siRNA, 30 nM) leads to a drastic significant increase in protein expression of LC3A/B-II (Figure 2b). Silencing efficiency of siRNAs for TF, FVII and PAR2 genes was determined to be greater than 90% using quantitative RT-PCR. Furthermore, Hep3B cells were analyzed by immunofluorescence (IF) microscopy using anti-LC3 antibody, and the distributed puncta were detected for the autophagy occurrence. The nuclei were stained with 5 μg/ml Hoechst dye and the cytoplasmic localization of the LC3 puncta was visualized. As shown in Figure 3, the LC3-positive IF puncta were suppressed by rTF or rFVIIa treatment, whereas the puncta formation was significantly increased in TF or FVII knockdown cells. These findings indicated that inhibition of coagulation cascade via TF/FVII/PAR2 axis correlated with the induction of autophagic activity.

The effects of coagulation factors on the expression of LC3A/B in Hep3B cell lines. (a) Hep3B cells were treatment with TF (200 ng/ml), FVII (200 ng/ml), PAR2 agonist (200 ng/ml), PAR1 agonist (200 ng/ml) or thrombin (200 ng/ml) for 24 h, and total amount of cells and supernatant protein were harvested and detected for LC3A/B and β-actin by western blot assay. (b) Hep3B cells were transfected with TF siRNA (30 nM), FVII siRNA (30 nM) and PAR2 siRNA (30 nM) for 48 h, respectively, and subsequently western blot was performed for LC3A/B and β-actin. All results are expressed as the mean±S.D. from three independent experiments. **P<0.01 compared with control

The effects of coagulation factor on the expression of LC3A/B in Hep3B cell line. Hep3B cells were treated with coagulation factors (TF, 200 ng/ml; FVII, 200 ng/ml) for 24 h or transfected with coagulation factor siRNA (TF siRNA, 30 nM and FVII siRNA, 30 nM) for 48 h detected for LC3A/B by immunofluorescence
Coagulation factors negatively regulate autophagy via mTOR activity
The mTOR protein is the best known regulator of autophagy in eukaryotes.31 It has been found that the activity of mTOR complex 1 is inversely correlated with autophagy induction. To investigate the potential role of mTOR in decreasing constitutive autophagy by activation of the TF/FVII/PAR2 axis, we first examined the expression levels of mTOR in Hep3B cells with exposure to rTF, rFVII or PAR2 agonist. The mTOR protein level was increased in cells treated with rTF, rFVII or PAR2 agonist (Figure 4a), and was almost obliterated in cells with TF, FVII or PAR2 siRNA transfection (Figure 4b). The knockdown efficiency of all siRNA experiments were determined to be greater than 90% using quantitative RT-PCR. We also tested whether mTOR signaling pathway is involved in TF/FVII/PAR2-repressed autophagy. Western blot analysis demonstrated that the level of phosphorylated mTOR (p-mTOR, Ser2448, active form) as well as phosphorylated form of its downstream substrates p-p70S6K and p-4EBP1 (p-p70S6K, Thr389; p-4EBP1, Ser65/Thr70) was increased after rTF, rFVII or PAR2 agonist exposure. However, no significant effect on mTOR expression as well as its phosphorylation in cells treated with thrombin or PAR1 agonist was observed (data nor shown). In the second panel, knockdown of TF/FVII/PAR2 did not alter the expression of phosphorylated mTOR, p70S6K and 4EBP1. These results indicated that induction of TF/FVII/PAR2 leads to activation of mTOR signaling pathway, however, TF/FVII/PAR2 knockdown causes mTOR suppression and blocks the signals toward its downstream substrates. In addition, the drastically suppressed LC3A/B-II levels in Hep3B cells treated with rTF, rFVII or PAR2 agonist were rescued by mTOR siRNA transfection (Figure 4c). The result demonstrated that repression of autophagy by TF/FVII/PAR2 activities relies upon mTOR signaling. Furthermore, the survival of Hep3B cells transfected with PAR2 siRNA or the negative control siRNA was also examined using Cell Counting Kit-8. As shown in Figure 5a, the survival rate decreased significantly in PAR2-knockdown cells compared with those transfected with negative control siRNA (100–59.7%, P<0.05, left panel) in serum-free condition. Correspondingly, the survival was also highly decreased by mTOR knockdown and treatment of PAR2 agonist suppressed cell death and significantly enhanced cell survival (P-value<0.05, right panel). Although 1% serum was added to minimize the apoptotic response induced by starvation, the significant decrease of cell survival in Hep3B cells by PAR2 knockdown (99.7–91.1%, P<0.01, left panel) as well as the significant increase of cell survival by treating PAR2 agonist (91.1–96.7%, P-value<0.05, right panel) in mTOR-silencing Hep3B cells were still observed (Figure 5b). These results indicated that downregulation of mTOR protein expression mediated the induction of autophagic activity by TF/FVII/PAR2 axis of coagulation cascade.

The effects of coagulation factors on the expression and phosphorylation of mTOR and its downstream substrates in Hep3B cell line. (a) Hep3B cells were treated with TF (200 ng/ml), FVII (200 ng/ml), PAR2 agonist (200 ng/ml) for 24 h, and total amount of cells was harvested and detected for mTOR, p-mTOR, p70S6K, p-p70S6K, 4EBP1, p-4EBP1 and β-actin by western blot assay. (b) Hep3B cells were pre-transfected with mTOR siRNA (30 nM) for 24 h, respectively; subsequent treatment of each coagulation factor (TF, 200 ng/ml; FVII, 200 ng/ml; PAR2 agonist, 200 ng/ml) for 24 h and the western blot assay was performed for LC3A/B and β-actin. All results are expressed as the mean±S.D. from three independent experiments

Hep3B cells cultured in (a) serum-free or (b) 1% serum medium were pre-transfected with mTOR siRNA (30 nM) for 24 h, then subsequent treatment of the PAR2 agonist (200 ng/ml) for 24 h was performed and the cell survival rates were measured by cell counting kit (CCK-8). All results are expressed as the mean±S.D. from three independent experiments. *P<0.05, **P<0.01 all compared with control
Coagulation factor-mediated repression of autophagy may occur through an Atg7-dependent pathway
Moreover, the roles of TF/FVII/PAR2 axis on the expression of other Atg involved in the formation and maturation autophagosomes or associated with autolysosomes were examined. We found that protein expression of Atg5 and Atg12, which form the Atg5–Atg12 conjugate as another key regulator of the autophagic process, were not affected by treatment or knockdown of TF/FVII/PAR2. However, the E1 ubiquitin-like modifier-activating enzyme Atg7 was decreased by treatment of rTF, rFVII or PAR2 agonist but not by treatment of thrombin or PAR1 agonist, whereas it was highly induced by knockdown of these genes (Figure 6). We also examined the expression levels of Rab7 and lysosomal-associated membrane protein 1 and found that rTF, rFVII and PAR2 agonist but not thrombin and PAR1 agonist decreased expression of both proteins, whereas knockdown of TF/FVII/PAR2 genes highly induced both proteins.

The effect of coagulation factors on the expression of autophagy-related genes. Hep3B cells were treated with TF (200 ng/ml), FVII (200 ng/ml), PAR2 agonist (200 ng/ml), thrombin (200 ng/ml) or PAR1 agonist (200 ng/ml) for 24 h, and the total amount of cells were harvested and detected for Atg 5 (C1), Atg7, Atg12, Rab7, LAMP-1 and β-actin by western blot assay. All results are expressed as the mean±S.D. from three independent experiments
This result indicated that the Atg5–Atg12 conjugation system may not be involved in the autophagy induction, whereas the LC3-mediated autophagy is regulated by Atg7 through TF/FVII/PAR2 axis in Hep3B cells.
Administration of PAR2 agonist, TF or FVII reduces autophagy induction in xenografts of NOD/severe combined immunodeficiency (SCID) mouse model
To determine whether we were able to recapitulate the molecular events we observed of the TF/FVII/PAR2 axis to autophagy, an HCC xenograft NOD/SCID mouse model was applied. HepG2-inoculated NOD/SCID mice were injected subcutaneously with rTF, rFVIIa or the PAR2 agonist at the site of the tumor. IHC staining of the sections from paraffin-embedded xenograft tumors showed that a significant decrease in LC3A/B expression in tumors treated with rTF, rFVIIa or the PAR2 agonist (Figure 7a). The western blot analysis using lysates from these tumors also demonstrated decreased LC3A/B-II expression by treatment of rTF or rFVIIa as well as the PAR2 agonist (Figure 7b).

The effect of coagulation factors on the expression of LC3A/B protein in mouse xenograft model of tumor grown. TF, FVII or PAR2 agonist was directly injected into NOD/SCID mice bearing HepG2 xenograft tumors and the LC3A/B-II expression was measured by (a) immunohistochemistry or (b) western blot analysis. All results are expressed as the mean±S.D. from three independent experiments
Discussion
The association between cancer and thrombosis has been recognized for over 100 years and the description was refined and extended over the years, culminating in a classic review by Sack et al.,32 and is further supported by the observation that presentation with venous thromboembolism (VTE) may precede the development or diagnosis of cancer.33 The incidence of thrombosis is significantly increased in patients with cancer compared with the general population. Clinical detectable VTE is found in about 15% of all cancer patients and the proportion is even higher when subclinical cases are counted.34, 35 In addition, cancer is associated with a fourfold increase in the risk for VTE and chemotherapy further increases this risk between six and sevenfold compared with that in the general individuals.36 Furthermore, recurrent thromboembolism is twice as likely to occur in patients with cancer as in healthy population, even when established on oral anticoagulant therapy.37, 38 Overall, the occurrence of VTE in patients with cancer is associated with increased morbidity and mortality.39
TF overexpression has been reported in a number of cancers, such as glioma, pancreatic cancer, non-small-cell lung cancer, colorectal cancer, renal cell cancer, ovarian cancer, prostate cancer, hepatocellular cancer and breast cancer.40 Tumor cells themselves are likely to be an important source of the elevated levels of circulating TF. A recent finding showed that cancer cells shed membrane microparticles (MPs) with procoagulant activity.41 Many other studies have now also shown that a variety of tumor cells release TF-positive MPs. It has been widely suggested that these MPs may account for the activation of the coagulation system associated with malignant tumors. Furthermore, it has been found that TF-dependent signaling in promoting tumor progression was mediated by PAR28, 11 and in part was through the activation of thrombin-PAR1 signaling coagulation pathway.42, 43 Interestingly, remarkable studies using specific antibodies or agents against TF have shown that blockade of TF-FVII-mediated signaling but not the TF coagulation response decreases primary tumor growth.8, 44 However, the level of TF expression varies among different types of cancers.45 Some cancers are more likely to be prothrombotic than others,46 but this feature is also suggested to be influenced by disease staging, immobility, as well as by interventions such as surgery or chemotherapy.
In our clinical observations in HCC, the expressions of FVII and PAR2 were inversely correlated with the amount of autophagic effector proteins LC3A/B-II. Our recent data also showed that FVIIa increased invasion and migration properties of HCC cells via PAR2 activation. Consistently, the expression levels of FVII mRNA and protein were significantly correlated with PAR2 gene expression (P=0.011) in tumor tissue of HCC patients, and were associated with increased invasiveness, migration and decreased survival (data not shown). Furthermore, exposure to rTF, rFVII or PAR2 agonist decreased expression of LC3A/B-II protein in vitro indicating that the effect of TF/FVII/PAR2 coagulation cascade may be a pivotal factor for the malignancy around tumor under certain circumstances. The inhibition of mTOR activity has been widely investigated and associated with TF thrombotic complications in patients receiving organ transplantation,47, 48, 49, 50, 51, 52 and has also been implicated in risk of thrombosis after the coronary stents.53 A recent study also elucidated that the TF-FVIIa-activated factor X complex regulates migration of human breast cancer cells.54 However, the mechanism underlying in interplay of TF coagulation cascade and mTOR activity of cancer has rarely been studied. Activation of the mTOR pathway by TF/FVIIa complex was first observed in breast cancer cells, which was associated with enhanced cell migration in vitro.54 Here we found that treatment with rTF, rFVII as well as the PAR2 peptide agonist, but not thrombin or PAR1, increased mTOR expression, whereas knockdown of TF, FVII or PAR2 repressed mTOR expression. The LC3A/B-II expression levels, which drastically inhibited by rTF, rFVII or PAR2 activation, were fully rescued by mTOR knockdown in vitro, suggesting mTOR signaling pathway could have a key role in tumor malignancy through TF/FVII/PAR2-mediated suppression of autophagy. Our study is consistent with the non-overlapping properties of TF-mediated coagulation and signaling studied using specific blocking agents as described above, which might also imply the differential involvement of mTOR-dependent blockade of autophagy via TF/FVII/PAR2 but not thrombin/PAR1 pathway. Autophagy pathway and relationship to mTOR signaling has been well investigated in tumor suppression. However, the contradictory effects of metformin, an mTOR inhibitor by activating adenosine monophosphate kinase, suggests inhibition of mTOR does not always lead to the autophagy induction in cancer55 and could be interrupted by other mechanisms. We further found that the LC3A/B-II expression was induced by sirolimus, a main selective inhibitor of mTOR, and the sirolimus-induced LC3A/B-II was partially reduced by rTF exposure, but was fully suppressed in the presence of rFVII or PAR2 agonist (see Supplementary Figure 1). The results demonstrated that the effects of sirolimus were not consistent with those of mTOR silencing in TF/FVII/PAR2-mediated suppression of autophagy, and suggested that other regulatory pathways may be elicited by sirolimus, which could turn the coagulation cascade-mediated suppression to be mTOR independent. The less reduction of sirolimus-induced LC3A/B-II in cells may also reflect that the TF/FVII/PAR2 signaling mainly operate through endogenous expression of TF. However, the possible mechanism is under our further investigation.
Moreover, we found that LC3A/B-II formation induced by inhibition of TF/FVII/PAR2 coagulant axis, especially by PAR2 knockdown, was mediated by Atg7 induction but not involved in Atg12-Atg5 conjugation system. This result indicates the important role of PAR2 activity in regulating autophagic flux of HCC in vitro, and also displays a consistent relevance with our clinical observation and the animal model in vivo. Moreover, a recent study demonstrated the differential inhibitory effects of anti-tumor agents on various human prostate cancer cells were due to the genetically impaired Atg5 expression and suggested further clinical investigations of Atg5 defect as well as other members in this conjugate could be necessary in human subjects.56 Our study may provide another alternative strategy to promote autophagy through induction of Atg7 by PAR2 targeting although more detailed investigations are still needed. Our study provides a novel insight into signaling pathways involved in PAR2 activation, which may be involved in the modulatory mechanisms of autophagy in tumor progression of HCC. We also offer novel evidence for the potential role of PAR2 as a therapeutic target for the treatment of human hepatic cancer.
Materials and Methods
Ethics and sample characteristics
The human study was approved by the Institutional Review Board of Kaohsiung Chang Gung Memorial Hospital, Taiwan (KCGMH-IRB No. 99-3806A3). One hundred and forty human samples collected during liver resection from the Kaohsiung Chang Memorial Hospital. Tissues of HCC and adjacent normal liver 3-cm from the tumor were collected. All the animals were maintained in specific pathogen-free animal facilities with water and commercial rat food provided ad libitum. Our experimental design was reviewed and approved by the Institutional Animal Care and Use Committee of the Kaohsiung Chang Gung Memorial Hospital (approval No: 2010121503), and the Committee recognizes that the proposed animal experiment follows the Animal Protection Law by the Council of Agriculture, Executive Yuan, R.O.C. and the guideline as shown in the Guide for the Care and Use of Laboratory Animals as promulgated by the Institute of Laboratory Animal Resources, National Research Council, USA.
Cell culture
Human HCC cell lines Hep3B and HepG2 were cultivated in an incubator under a 5% carbon dioxide atmosphere at 37 °C in a relative humidity of 95% and were maintained in DMEM (Invitrogen, Karlsruhe, Germany) with 10% FBS (Invitrogen), 100 U/ml penicillin, 100 mg/ml streptomycin and 4 mM L-glutamine (Invitrogen). Recombinant human TF (rTF) and FVII were purchased from R&D Systems (Minneapolis, MN, USA). PAR1 agonist peptide TFLLR-NH2 and PAR2 agonist peptide SLIGKV-NH2 were purchased from Peptides International (Louisville, KY, USA).The working concentration of recombinant proteins and agonist peptides was 200 ng/ml.
IHC staining
Five-micrometer-thick tissue paraffin sections from representative tissue blocks were cut, de-paraffinized in xylene, rinsed and rehydrated with distilled water through graded alcohol. Antigen retrieval was enhanced by autoclaving slides in 10 mM sodium citrate buffer (pH 6.0) for 30 min. Endogenous peroxidase activity was quenched by incubation in 0.3% hydrogen peroxide–methanol buffer for 30 min. The slides were then incubated overnight at 4 °C in humidified chambers with primary rabbit polyclonal anti-LC3A/B (Ab58610, Abcam, Cambridge, UK) at a dilution of 1 : 400. The slides were washed three times in a phosphate-buffered solution and further incubated with a biotinylated secondary antibody for 30 min at room temperature. Antigen–antibody complexes were detected by the avidin–biotin–peroxidase method, using diaminobenzidine as a chromogenic substrate (DAKO, Carpinteria, CA, USA). Finally, the slides were counterstained with hematoxylin and then examined by light microscopy.
Each slide was evaluated for LC3A/B immunostaining using a semi-quantitative scoring system for staining intensity in tumor or non-tumor tissue sections. The tissue sections were scored by manually counting ≥1000 cells based on the percentage of immunostained cells as: ≤25%=S1; 26–50%=S2; 51–75%=S3 and≥76%=S4.
I F staining
Hep3B cells were fixed and permeabilized in 4% paraformaldehyde at 4 °C for 30 min. The slides were then incubated overnight at 4 °C in humidified chambers with primary rabbit polyclonal anti-LC3A/B (#4108, Cell Signaling Technology, Danvers, MA, USA) at a dilution of 1 : 400. The slides were washed three times in a phosphate-buffered solution and Texas Red or FITC-conjugated secondary antibody for 2 h at room temperature. Extensive washing with PBS will be performed between each step and before mounting with Fluoromount-G and examined by fluorescence microscope (Zeiss Axioskop2 fluorescent microscope, Oberkochen, Germany).
Western blot analysis
Cells were washed twice with PBS, trypsinized and centrifuged. Cells were washed twice again, transferred to a new tube and lysed with lysis buffer (2% SDS; 68.5 mM Tris-HCl (pH 6.8); 10% glycerin) containing 1 : 100 protease (Sigma-Aldrich, St. Louis, MO, USA) and 1 : 100 phosphatase (Roche Diagnostics, Mannheim, Germany) inhibitors. Lysates were heated at 95 °C for 8 min and centrifuged at 13 000 r.p.m. for 15 min. Total protein concentration was quantified photometrically using the CoomassiePlus Protein Assay reagent (Thermo Fisher Scientific Inc., Rockford, IL, USA) according to the manufacturer’s protocol and compared with a BSA (Sigma-Aldrich) standard curve according to the manufacturer’s instructions. Equal amounts of protein were homogenized with loading buffer, separated by 10% SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). Membranes were blocked for 45 min in 5% nonfat dried milk in TBS and incubated overnight at 4 °C with primary antibodies (TF, 1 : 1000, Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), sc-59714; PAR2, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-13504; FVII, 1 : 1000, Abcam, Ab97614; PAR1, 1 : 1000, Abcam, Ab32611; mTOR, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-1549; p-mTOR, 1 : 1000, Cell Signaling Technology, #5536; p70S6K, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-8418; p-p70S6K, Cell Signaling, 1 : 1000, #9234; 4EBP1, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-6936; p-4EBP1, 1 : 1000, Cell Signaling, #9456; LC3A/B, 1 : 1000, Sigma-Aldrich, L8918; β-actin, 1 : 2000, Millpore (Merck Millipore, Oxford, UK), LV158863; LAMP-1, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-5570; Rab7, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-271608; ATG5, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-133158; ATG7, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-376212; ATG12, 1 : 1000, Santa Cruz Biotechnology, Inc., sc-271688) in 5% milk powder in TBS with 0.05% Tween 20 (TBS/T). After washing in TBS/T three times, the membrane was incubated at room temperature for 2 h with infrared fluorescence-labeled secondary antibody (Cell signaling) in 3% nonfat dried milk 1 : 5000 diluted in TBS/T, then rinsed once in PBS and scanned using G:BOX iChemi XL imaging system (Syngene, Cambridge, UK). The experiments were repeated three times for each experimental condition.
siRNA transfection
siRNAs directed against TF (sc-44984), FVII (sc-40401), PAR-2 (sc-36188) and mTOR (sc-35409) mRNAs were obtained from Santa Cruz Biotechnology Inc. Transient transfection of Hep3B cells was carried out in triplicate experiments in six-well plates using GenMute siRNA transfection reagent (SignaGen Laboratories, Gaithersburg, MD, USA) and standard procedure in accordance with the manufacturer’s recommendations. Briefly, 30 nM of each target siRNA was diluted into free DMEM without serum and antibiotics and mixed in transfection reagent. Cells were then transfected at 37 °C in 5% CO2 for 24 h and changed with DMEM containing 10% FCS without antibiotics.
Cell proliferation assay
To assay for cell proliferation, Hep3B cell suspension of 100 μl were plated at a density of 1 × 104 per well in 96-well plates and serum starved for 24 h. 10 μl Cell Counting Kit-8 solution (Sigma-Aldrich) was added to each well of the plate under various treatment conditions. The absorbance was measured at 450 nm using a microplate spectrophotometer (Dynex Technologies Inc., Chantilly, VA, USA).
Tumor xenograft mouse model
SCID mice (4-week old) were housed under standard conditions and cared for according the institutional guidelines for animal care. For the xenograft tumor growth assay, 5 × 106 HepG2 cells were injected substaneously into the right dorsal flank. Treatment, which was initiated when the tumor reached 5 mm in diameter, was through directly subcutaneous injection of TF, FVIIa or PAR2 agonist every other day. After 30 days, the mice were euthanized and the tumors were excised for study.
Statistical analysis
Each experiment was performed at least three times, and the statistical significance of the contentious data was compared between groups by means of the Mann–Whitney U-test or Student’s t-test. Statistical analysis was performed with the SPSS software package for Windows (SPSS 15.0 for Windows; SPSS Inc., Chicago, IL, USA). A value of P<0.05 was considered to indicate statistical significance.
Abbreviations
- HCC:
-
hepatocelullar carcinoma
- TF:
-
tissue factor
- FVII:
-
factor VII
- PAR2:
-
protease-activated receptor 2
- LC3:
-
light chain 3
- mTOR:
-
mammalian target of rapamycin
- IHC:
-
immunohistochemistry
- Atg:
-
autophagy-related proteins
- rTF:
-
recombinant TF
- rFVII:
-
recombinant FVII
- VTE:
-
venous thromboembolism
- MP:
-
microparticle
- IF:
-
immunofluorescence
- SCID:
-
severe combined immunodeficiency
- siRNA:
-
small interfering RNA
References
Mackman N . Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol 2004; 24: 1015–1022.
Camerer E, Huang W, Coughlin SR . Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci USA 2000; 97: 5255–5260.
Mann KG . Biochemistry and physiology of blood coagulation. Thromb Haemost 1999; 82: 165–174.
Pawlinski R, Mackman N . Use of mouse models to study the role of tissue factor in tumor biology. Semin Thromb Hemost 2008; 34: 182–186.
Abe K, Shoji M, Chen J, Bierhaus A, Danave I, Micko C et al. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc Natl Acad Sci USA 1999; 96: 8663–8668.
Mueller BM, Ruf W . Requirement for binding of catalytically active factor VIIa in tissue factor-dependent experimental metastasis. J Clin Invest 1998; 101: 1372–1378.
Versteeg HH, Spek CA, Richel DJ, Peppelenbosch MP . Coagulation factors VIIa and Xa inhibit apoptosis and anoikis. Oncogene 2004; 23: 410–417.
Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B et al. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008; 111: 190–199.
Rak J, Milsom C, Magnus N, Yu J . Tissue factor in tumour progression. Best Pract Res Clin Haematol 2009; 22: 71–83.
Schaffner F, Ruf W . Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler Thromb Vasc Biol 2009; 29: 1999–2004.
Ruf W, Disse J, Carneiro-Lobo TC, Yokota N, Schaffner F . Tissue factor and cell signalling in cancer progression and thrombosis. J Thromb Haemost 2011; 9 (Suppl 1): 306–315.
Panasiuk A, Zak J, Panasiuk B, Prokopowicz D . Increase in expression of monocytic tissue factor (CD142) with monocytes and blood platelet activation in liver cirrhosis. Blood Coagul Fibrinolysis 2007; 18: 739–744.
Tacke F, Schoffski P, Trautwein C, Manns MP, Ganser A, von Depka M . Tissue factor and thrombomodulin levels are correlated with stage of cirrhosis in patients with liver disease. Blood Coagul Fibrinolysis 2001; 12: 539–545.
Lwaleed BA, Francis JL, Chisholm M . Tissue factor assays as diagnostic tools for cancer? Correlation between urinary and monocyte tissue factor activity. J Hematother Stem Cell Res 1999; 8: 659–668.
Coulon S, Heindryckx F, Geerts A, Van Steenkiste C, Colle I, Van Vlierberghe H . Angiogenesis in chronic liver disease and its complications. Liver Int 2011; 31: 146–162.
Sugimachi K, Tanaka S, Terashi T, Taguchi K, Rikimaru T . The mechanisms of angiogenesis in hepatocellular carcinoma: angiogenic switch during tumor progression. Surgery 2002; 131 (1 Suppl): S135–S141.
Wu XZ . New strategy of antiangiogenic therapy for hepatocellular carcinoma. Neoplasma 2008; 55: 472–481.
Poon RT, Lau CP, Ho JW, Yu WC, Fan ST, Wong J . Tissue factor expression correlates with tumor angiogenesis and invasiveness in human hepatocellular carcinoma. Clin Cancer Res 2003; 9: 5339–5345.
Zhou Q, Huang T, Wang YF, Zhou XB, Liang LJ, Peng BG . Role of tissue factor in hepatocellular carcinoma genesis, invasion and metastasis. Chin Med J 2011; 124: 3746–3751.
Baehrecke EH . Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 2005; 6: 505–510.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169: 425–434.
Kondo Y, Kanzawa T, Sawaya R, Kondo S . The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5: 726–734.
Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R . Autophagy in liver diseases. J Hepatol 2010; 53: 1123–1134.
Carew JS, Kelly KR, Nawrocki ST . Autophagy as a target for cancer therapy: new developments. Cancer Manag Res 2012; 4: 357–365.
Guertin DA, Sabatini DM . Defining the role of mTOR in cancer. Cancer Cell 2007; 12: 9–22.
Wang Z, Zhou J, Fan J, Tan CJ, Qiu SJ, Yu Y et al. Sirolimus inhibits the growth and metastatic progression of hepatocellular carcinoma. J Cancer Res Clin Oncol 2009; 135: 715–722.
Wang Z, Zhou J, Fan J, Qiu SJ, Yu Y, Huang XW et al. Effect of rapamycin alone and in combination with sorafenib in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res 2008; 14: 5124–5130.
Semela D, Piguet AC, Kolev M, Schmitter K, Hlushchuk R, Djonov V et al. Vascular remodeling and antitumoral effects of mTOR inhibition in a rat model of hepatocellular carcinoma. J Hepatol 2007; 46: 840–848.
Bu X, Jia F, Wang W, Guo X, Wu M, Wei L . Coupled down-regulation of mTOR and telomerase activity during fluorouracil-induced apoptosis of hepatocarcinoma cells. BMC Cancer 2007; 7: 208.
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC . Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010; 6: 1057–1065.
He C, Klionsky DJ . Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43: 67–93.
Sack GH Jr, Levin J, Bell WR . Trousseau's syndrome and other manifestations of chronic disseminated coagulopathy in patients with neoplasms: clinical, pathophysiologic, and therapeutic features. Medicine 1977; 56: 1–37.
Prandoni P, Lensing AW, Buller HR, Cogo A, Prins MH, Cattelan AM et al. Deep-vein thrombosis and the incidence of subsequent symptomatic cancer. N Engl J Med 1992; 327: 1128–1133.
Rickles FR, Edwards RL . Activation of blood coagulation in cancer: Trousseau's syndrome revisited. Blood 1983; 62: 14–31.
Donati MB . Cancer and thrombosis: from Phlegmasia alba dolens to transgenic mice. Thromb Haemost 1995; 74: 278–281.
Heit JA . Cancer and venous thromboembolism: scope of the problem. Cancer Control 2005; 12 (Suppl 1): 5–10.
Levitan N, Dowlati A, Remick SC, Tahsildar HI, Sivinski LD, Beyth R et al. Rates of initial and recurrent thromboembolic disease among patients with malignancy versus those without malignancy. Risk analysis using Medicare claims data. Medicine 1999; 78: 285–291.
Bona RD, Hickey AD, Wallace DM . Warfarin is safe as secondary prophylaxis in patients with cancer and a previous episode of venous thrombosis. Am J Clin Oncol 2000; 23: 71–73.
Sorensen HT, Mellemkjaer L, Olsen JH, Baron JA . Prognosis of cancers associated with venous thromboembolism. N Engl J Med 2000; 343: 1846–1850.
Rickles FR, Hair GA, Zeff RA, Lee E, Bona RD . Tissue factor expression in human leukocytes and tumor cells. Thromb Haemost 1995; 74: 391–395.
Dvorak HF, Van DeWater L, Bitzer AM, Dvorak AM, Anderson D, Harvey VS et al. Procoagulant activity associated with plasma membrane vesicles shed by cultured tumor cells. Cancer Res 1983; 43: 4434–4442.
Ruf W, Mueller BM . Thrombin generation and the pathogenesis of cancer. Semin Thromb Hemost 2006; 32 (Suppl 1): 61–68.
Nierodzik ML, Karpatkin S . Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 2006; 10: 355–362.
Carneiro-Lobo TC, Schaffner F, Disse J, Ostergaard H, Francischetti IM, Monteiro RQ et al. The tick-derived inhibitor Ixolaris prevents tissue factor signaling on tumor cells. J Thromb Haemost 2012; 10: 1849–1858.
Kakkar AK, Lemoine NR, Scully MF, Tebbutt S, Williamson RC . Tissue factor expression correlates with histological grade in human pancreatic cancer. Br J Surg 1995; 82: 1101–1104.
White RH, Chew H, Wun T . Targeting patients for anticoagulant prophylaxis trials in patients with cancer: who is at highest risk? Thromb Res 2007; 120 (Suppl 2): S29–S40.
Guba M, Yezhelyev M, Eichhorn ME, Schmid G, Ischenko I, Papyan A et al. Rapamycin induces tumor-specific thrombosis via tissue factor in the presence of VEGF. Blood 2005; 105: 4463–4469.
Trotter JF . Sirolimus in liver transplantation. Transplant Proc 2003; 35 (3 Suppl): 193S–200S.
Fortin MC, Raymond MA, Madore F, Fugere JA, Paquet M, St-Louis G et al. Increased risk of thrombotic microangiopathy in patients receiving a cyclosporin-sirolimus combination. Am J Transplant 2004; 4: 946–952.
Paramesh AS, Grosskreutz C, Florman SS, Gondolesi GE, Sharma S, Kaufman SS et al. Thrombotic microangiopathy associated with combined sirolimus and tacrolimus immunosuppression after intestinal transplantation. Transplantation 2004; 77: 129–131.
Barone GW, Gurley BJ, Abul-Ezz SR, Gokden N . Sirolimus-induced thrombotic microangiopathy in a renal transplant recipient. Am J Kidney Dis 2003; 42: 202–206.
Robson M, Cote I, Abbs I, Koffman G, Goldsmith D . Thrombotic micro-angiopathy with sirolimus-based immunosuppression: potentiation of calcineurin-inhibitor-induced endothelial damage? Am J Transplant 2003; 3: 324–327.
Choi SB . CYPHER coronary stents and risk of thrombosis. Can Med Assoc J 2003; 169: 218.
Jiang X, Zhu S, Panetti TS, Bromberg ME . Formation of tissue factor-factor VIIa-factor Xa complex induces activation of the mTOR pathway which regulates migration of human breast cancer cells. Thromb Haemost 2008; 100: 127–133.
Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Research 2010; 70: 2465–2475.
Ouyang DY, Xu LH, He XH, Zhang YT, Zeng LH, Cai JY et al. Autophagy is differentially induced in prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing profiles of ATG5. Autophagy 2013; 9: 20–32.
Acknowledgements
This work was supported in part by grants from the National Science Council (NSC101-2314-B-182A-031-MY3 to CL Chen; NSC101-2320-B-182-037-MY3 to T Nakano) and the Chang Gung Memorial Hospital (CMRPG891062 and CMRPG8A0431 to CL Chen; CMRPG891301 and CMRPG8B0911 to KD Chen; NMRPG8A0081, CMRPG8A1201, CMRPG8A1202 and CMRPG8B0091 to CC Lin) of Taiwan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Chen, KD., Wang, CC., Tsai, MC. et al. Interconnections between autophagy and the coagulation cascade in hepatocellular carcinoma. Cell Death Dis 5, e1244 (2014). https://doi.org/10.1038/cddis.2014.212
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.212
Keywords
This article is cited by
-
Coagulation factor VII and malignant progression of hepatocellular carcinoma
Cell Death & Disease (2016)
-
Factor VII promotes hepatocellular carcinoma progression through ERK-TSC signaling
Cell Death Discovery (2015)

{kind=link}