
Abstract
Liver kinase B1 (LKB1) is a tumor suppressor mutationally inactivated in Peutz–Jeghers syndrome (PJS) and various sporadic cancers. Although LKB1 encodes a kinase that possesses multiple functions, no individual hypothesis posed to date has convincingly explained how loss of LKB1 contributes to carcinogenesis. In this report we demonstrated that LKB1 maintains genomic stability through the regulation of centrosome duplication. We found that LKB1 colocalized with centrosomal proteins and was situated in the mitotic spindle pole. LKB1 deficiency-induced centrosome amplification was independent of AMP-activated protein kinase (AMPK), a well-defined substrate of LKB1. Cells lacking LKB1 exhibited an increase in phosphorylated and total Polo-like kinase 1 (PLK-1), NIMA-related kinase 2 (NEK2), and ninein-like protein (NLP). Overexpression of active PLK1 (T210D) reversed the inhibition of LKB1 on centrosome amplification. In contrast, depletion of PLK1 with siRNA or suppression of PLK1 kinase activity with BTO-1 (5-Cyano-7-nitro-2-benzothiazolecarboxamide-3-oxide) abrogated LKB1 deficiency-induced centrosome amplification. We further characterized that LKB1 phosphorylated and activated AMPK-related kinase 5 (NUAK1 or ARK5) that in turn increased the phosphorylation of MYPT1, enhanced the binding between MYPT1–PP1 and PLK1, and conferred an effective dephosphorylation of PLK1. More importantly, we noted that LKB1-deficient cells exhibited multiple nuclear abnormalities, such as mitotic delay, binuclear, polylobed, grape, large, and micronuclear. Immediate depletion of LKB1 resulted in the accumulation of multiploidy cells. Expression of LKB1 is reversely correlated with the levels of PLK1 in human cancer tissues. Thus, we have uncovered a novel function of LKB1 in the maintenance of genomic stability through the regulation of centrosome mediated by PLK1.
Similar content being viewed by others


Main
Liver kinase B1 (LKB1) is a tumor suppressor that is inactivated in patients with Peutz–Jeghers syndrome (PJS), an inherited disorder characterized by hamartomatous polyps within the gastrointestinal tract and a high risk of developing cancers.1, 2, 3 Inactivation of LKB1 has also been found in patients with sporadic cancers.1, 4, 5, 6 LKB1 is involved in multiple cellular functions including tumor suppression, cell metabolic homeostasis, embryogenesis, and cell cycle regulation.7, 8, 9, 10 LKB1 phosphorylates AMP-activated protein kinase (AMPK) and 12 other members of the AMPK superfamily.1, 6, 10, 11, 12 It was found that inactivation of AMPK induces pleiotropic defects in cell mitosis and an increase in S-phase arrest.13 Phosphorylation of AMPKα at T172 is required for association of AMPK with the centrosome, spindle poles, and mid-body of mitotic cells, but this phosphorylation may be independent of LKB1 and likely induced by Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ).14
It was shown in a genome-wide screen that downregulation of LKB1 causes spindle aberrations.13 Several LKB1 downstream kinases or interacting proteins, including aforementioned AMPK, TSC2, p21, and others, have been detected at the centrosomes and have shown to play important roles in the mitotic processes.14, 15, 16 Wei et al.17 recently reported that LKB1 was imperative for spindle orientation through the activation of AMPK during mitosis. When LKB1 is lost, the spindle orientation forms improper angles for cell division and polarity of the dividing cells.17 In addition, another LKB1 substrate, AMPK-related kinase 5 (ARK5, NUAK1), was shown to phosphorylate myosin phosphatase targeting subunit (MYPT1) at Ser 445, 472, and 910 and to inactivate the phosphatase activity of MYPT1 binding partner PP1 by promoting its complex with 14-3-3 adapter, thereby allowing myosin to remain in the activated state during cell adhesion.18 Taken together, these clues indicate that LKB1 is likely involved in mitotic regulation through various mechanisms.
In mammalian cells, the centrosome is critical for the regulation of cellular polarity and cellular division.19 In order to ensure proper cell separation and genomic stability, the centrosome must be tightly regulated so that it is only replicated once during the cell cycle and splits only at the entry of mitosis.20 Overduplication or premature splitting of centrioles produces multipolar cell division, leading to unequal chromosome segregation between daughter cells and inducing aneuploidy, a hallmark in tumorigenesis.20, 21, 22 Lingle et al.23 postulated that centrosome amplification causes chromosomal instability due to the stepwise changes in karyotypes, hence contributing to the initiation and progression of cancers.23, 24, 25
Polo-like kinases (PLK) 1–4 are evolutionarily conserved kinases that function in cell cycle regulation. PLK3 is active during the G1–S transition, whereas PLK2 functions in the S-phase checkpoint before DNA replication. PLK4 regulates centriole duplication whereas PLK1 is one of the primary proteins that coordinate centrosomal spindle assembly and the progression of mitosis.26, 27 PLK1 is considered an oncogene as it is upregulated in multiple cancers including breast, pancreatic, and many others.28, 29, 30 The activity of PLK1 is dictated by how it localizes with substrates through its polo-box domain as well as by phosphorylation that regulates its kinase activity. Currently, there are at least five known kinases that can phosphorylate PLK1. Aurora A is the best known kinase of PLK1.31 In a recent proteomic screen from mitotic cell extracts, Ji et al.32 found that PLK1 is also phosphorylated by p21-activated kinases 1 and 3 (PAK1/3) and mammalian Ste20-like kinase 1 (MST1). These kinases phosphorylate PLK1 at threonine 210 (T210), activating its kinase activity for substrates such as NIMA-related kinase 2 (NEK2) and ninein-like protein (NLP) that cause the centrioles to split,27, 33 and CDC25 to promote mitosis.34 Even when one of these upstream kinases is blocked, phosphorylation of PLK1 at T210 still occurs, suggesting that PLK1 function is governed by multiple kinases to ensure proper advancement of mitosis.32 Although PLK1 phosphorylation is crucial to promote mitosis, its dephosphorylation, which is carried out by the myosin phosphatase targeting subunit–protein phosphotase1 complex (MYPT1–PP1), is also imperative for mitotic exit.35
With all of the evidence that LKB1 is involved in mitosis and its downstream targets interacting with the centrosome, we hypothesized that LKB1 might also play a role in the regulation of centrosome duplication and maturation. In this report, we demonstrate that LKB1 locates to the centrosomes and the spindle poles during mitosis and that loss of LKB1 causes centrosome amplification and nuclear abnormalities. We conclude that LKB1 displays a novel tumor suppressive function by regulating PLK1 and by maintaining genome stability.
Results
LKB1 locates to the centrosomes and mitotic spindle poles
To determine whether LKB1 localizes to the centrosome and/or mitotic apparatus, we performed immunofluorescent staining on U2OS cells with either γ-tubulin or α-tubulin along with LKB1. These experiments showed that LKB1 colocalized with γ-tubulin within the centrosomes (Figures 1a and b). It was also noted that during mitosis, LKB1 was recruited to the spindle poles, colocalizing with α-tubulin (Figure 1c). To further confirm the association of LKB1 with the centrosome, we transfected U2OS cells with pEGFP-LKB1 and subsequently fixed and immunostained the cells with pericentrin. As shown in Figure 1d, GFP-LKB1 distributed both in the cytoplasm and the nucleus. Interestingly, punctuated signaling of LKB1 in the cytoplasm colocalized with pericentrin (Figure 1d). Thus, both endogenous and overexpression experiments demonstrated that LKB1 located to the centrosomes.

LKB1 locates to centrosomes and mitotic spindle poles. Proliferating U2OS cells were fixed and immunostained with monoclonal antibody to LKB1 and polyclonal antibody to γ-tubulin (a and b) or α-tubulin (c). The white arrows show the colocalization of the two proteins. (d) U2OS cells were transfected with GFP-LKB1. At 24 h hours after transfection, cells were fixed and stained with anti-pericentrin antibody. The white arrow shows the colocalization of LKB1 and pericentrin. The insets show the enlarged location of GFP-LKB1 in the centrosome
Loss of LKB1 leads to centrosome amplification
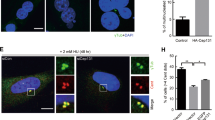
To determine whether LKB1 bears a function coupled to the centrosome localization, we compared primary mouse embryonic fibroblasts (MEFs) that differed only by their LKB1 status. The cells were immunostained with γ-tubulin to view the centrosome (Figure 2a). More than 30% of LKB1-deficient MEFs possessed 3 or more centrosomes, whereas fewer than 5% of LKB1-intact MEFs displayed increased centrosome number (Figure 2b). To determine whether this phenomenon was replicable in cancerous cells, we knocked down LKB1 with short hairpin RNA (shRNA) in U2OS cells and stained the cells with pericentrin. The number of cells with three separated centrioles increased, with more than three times the percentage of cells showing the phenotype as compared with the scrambled shRNA controls (Figure 2c). We also examined the centrosome numbers in LKB1-deficient HeLa cells that ectopically express GFP-LKB1 or GFP. Of the HeLa/pMEP4-GFP cells, 12% possessed increased centrosome numbers (>2). In contrast, when LKB1 was ectopically expressed in HeLa/pMEP4-GFP-LKB1, cells with an increased number of centrosomes were significantly reduced (Figure 2d). Collectively, our results demonstrate that LKB1 regulates the centrosome, and that loss of LKB1 in cells causes increased centrosome number.

Loss of LKB1 causes centrosome amplification. (a) Immunofluorescence staining was performed with primary wild-type and null-LKB1 cells (passage 3) using a monoclonal antibody against γ-tubulin. (b) Percentage of cells with >2 centrosomes in (a). The results shown are the averages of three experiments±S.D. At least 50 cells were counted in each experiment. *P<0.05. (c) U2OS/shRNA-LKB1 and control cells were fixed and immunostained with a polyclonal antibody against pericentrin. Quantification of cells with >2 centrosomes was performed as in (b). *P<0.05. (d) HeLa/pMEP4-GFP-LKB1 or HeLa/pMEP4-GFP cells were induced with 100 μM Zn2SO4 for the induction of LKB1 for 48 h. After the induction, the cells were fixed and immunostained with a polyclonal antibody against pericentrin. Quantification of cells with >2 centrosomes was performed as in (b and c). *P<0.05
LKB1 deficiency-induced centrosome amplification is independent of AMPK
AMPK is a well-characterized downstream target of LKB11, 10, 12, 15 and was reported to locate at centrosomes.14 To determine whether the centrosome regulation by LKB1 is mediated through AMPK, we knocked down LKB1 in wild-type and AMPK-null MEFs with siRNA to determine whether the loss of AMPK had overlapped effects on centrosome numbers as the loss of LKB1 (Supplementary Figure S1). The cells were fixed and stained for pericentrin to examine the number of centrosomes. There were no differences observed in the percentage of cells with increased centrosomes between AMPK-proficient and -deficient cells (Supplementary Figure S1b). However, depletion of LKB1 by RNAi increased the percentage of cells with amplified centrosomes by 2–4-fold in both wild-type and AMPK-null MEFs (Supplementary Figure S1b). A western blot was performed to confirm that LKB1 was knocked down and that the expected effects on the LKB1-AMPK signaling cascade were conveyed (Supplementary Figure S1a). This evidence leads us to believe that AMPK is not involved in LKB1 deficiency-induced centrosome amplification.
LKB1 regulates PLK1 and its downstream targets
To determine the mechanism by which LKB1 regulates the centrosome, we examined the expression and activation of several proteins that are involved in centrosomal regulation using western blot. Phosphorylated PLK1 as well as its downstream targets NEK2 and NLP were markedly increased in U2OS/shRNA-LKB1 cells (Figure 3a). In contrast, inducible expression of LKB1 in HeLa/pMEP4-LKB1 cells reduced the phosphorylation of PLK1, and total levels of NEK2 and NLP (Figure 3b). Consistent with our previous observation that LKB1-mediated centrosome regulation was independent of AMPK (Supplementary Figure S1), phosphorylation of AMPK was slightly present even in LKB1-deficient cells (Figures 3a and b).

LKB1 regulates PLK1 and its downstream targets. (a) U2OS/shRNA-LKB1 and U2OS/Ctrl-shRNA cells were lysed and whole cell extracts (WCEs) were collected for immunoblot analysis. (b) HeLa/pMEP4-GFP or HeLa/pMEP4-GFP-LKB1 cells were treated with 100 μM zinc to induce protein expression for 24 h before WCEs were collected for immunoblot analysis. (c) LKB1 colocalizes with PLK1. Proliferating U2OS cells were fixed and co-immunostained with LKB1 polyclonal antibody and PLK1 monoclonal antibody. Fluorescent images were captured with an Olympus IX53 microscope
PLK1 is a cell cycle-related protein with its highest expression found in the G2 phase.27 To rule out the potential influence of cell cycle distribution resulting from LKB1 alteration, we detected cell cycle distribution in asynchronized U2OS/shRNA-Ctrl and U2OS/shRNA-LKB1 cells. Depletion of LKB1 led to only a marginal reduction in G2/M- and G1-phase cells and a minor increase of S-phase cells (Supplementary Figure S2a), indicating that elevation of PLK1-T210 may not be because of cell cycle differences after LKB1 knockdown. To further validate the observation, we detected the phosphorylation of PLK1 in U2OS/shRNA-LKB1 and control cells after they were serum-starved for 24 h and then released with the addition of FBS for different time points. Phospho-PLK1 was consistently higher in LKB1 knockdown cells as compared with control cells, irrespective of cell cycle distribution (Supplementary Figure S2b). Together, these results support that phospho-PLK1 is negatively regulated by the expression of LKB1.
PLK1 locates at different apparatuses, such as centromere, centrosome, and midbody, through which it regulates mitosis during cell division. Thus, we questioned whether LKB1 could colocalize with PLK1 in mitotic apparatuses and affect its distribution. LKB1 colocalized with PLK1 on the centrosomes, but not on other apparatuses (Figure 3c and data not shown). Furthermore, LKB1 did not affect the localization of PLK1 in cells (Supplementary Figure S3). Together, our results suggest that LKB1 most likely regulates the activity of PLK1, but not its localization.
Overexpression of active PLK1 (T210D) reverses the action of LKB1 on centrosome duplication
To characterize the role of PLK1 in LKB1-induced centrosome regulation, we overexpressed a constitutively active form of PLK1 (T210D) in U2OS/shRNA-LKB1 and control cells. Immunofluorescence of the cells showed that overexpressed PLK1-T210D shared a similar cellular distribution pattern as endogenous PLK1 (Figure 4a). Overexpression of PLK1-T210D increased the number of cells with abnormal centrosomes in LKB1-proficient cells (Figures 4a and b). Overexpression of PLK1-T210D could not further elevate centrosome abnormalities in LKB1-deficient cells (Figure 4b). Immunoblotting confirmed the increased expression of PLK1 as well as the activation of its downstream signaling (Figure 4c). Together, our results suggest that the effect of PLK1 overexpression and that of LKB1 deficiency on centrosome amplification overlap.

Overexpression of constitutively active PLK1 (T210D) reverses the action of LKB1 on centrosome duplication. U2OS/shRNA-LKB1 and U2OS/shRNA-Ctrl cells were transfected with constitutively active form of PLK1 (T210D) or control vector for 48 h. The cells were then fixed and probed with a monoclonal anti-PLK1 antibody and a polyclonal anti-pericentrin antibody (a and b) or lysed for immunoblot analysis with WCEs (c). (b) Percentage of cells with >2 centrosomes after PLK1-T210 overexpression. At least 50 cells were counted for each experimental group. The graph represents the mean and S.D. of three separate experiments. *P<0.05
Inhibition of PLK1 abrogates LKB1 deficiency-induced centrosome amplification
To further validate the role of PLK1 in LKB1-facilitated centrosome regulation, we knocked down PLK1 with siRNA in U2OS/shRNA-LKB1 and control cells and detected centrosome status with immunofluorescent staining. As shown in Figure 5a, ∼10% of control siRNA cells possessed 3 or more centrosomes. In contrast, siRNA of PLK1 dramatically decreased the percentage of cells with amplified centrosomes (Figure 5b). In addition, siRNA of PLK1 did not affect the centrosome number statistically in LKB1-intact cells (Figure 5b).

Inhibition of PLK1 rescues LKB1 deficiency-induced centrosome amplification. U2OS/shRNA-LKB1 and U2OS/shRNA-Ctrl cells were transfected with siRNA of PLK1 or scramble control for 48 h. The cells were then fixed for immunofluorescent detection of PLK1 and pericentrin (a and b). (a) Fluorescent images were captured with an Olympus IX53 microscope. (b) Percentage of cells with >2 centrosomes was calculated. The graph represents the mean and S.D. of three independent experiments where at least 50 cells were counted per experiment. *P<0.05. (c) Lysates of siRNA PLK1 or control-transfected cells were harvested and applied for immunoblot. For each lane, 30 μg proteins were loaded. GAPDH served as a loading control
PLK1 is activated by the phosphorylation at T210, and thus we wanted to determine whether inhibiting the activity would be necessary to deplete LKB1 deficiency-induced centrosome amplification. We treated U2OS/shR-LKB1 and control cells with 5-Cyano-7-nitro-2-benzothiazolecarboxamide-3-oxide (BTO-1), an inhibitor that blocks the phosphorylation of PLK1. The quantification of centrosome abnormalities confirmed that ablating the phosphorylation of PLK1 with BTO-1 was sufficient to rescue centrosome amplification resulting from LKB1 deficiency (Supplementary Figure S4). Taken together, our results suggest that inhibition of PLK1 can amend LKB1 depletion-induced centrosome abnormalities.
LKB1 promotes the dephosphorylation of PLK1 through stabilizing MYPT1–PP1–PLK1 complex
To gain an insight into the mechanism by which LKB1 regulates PLK1, we began by screening the kinases and phosphatases that act on PLK1 through immunoblotting in our U2OS and HeLa cell models. In U2OS cells, we noted no differences in the upstream kinases of PLK1 (Supplementary Figure S5a). When LKB1 was overexpressed in HeLa cells, PAK1 and PAK3, two upstream kinases of PLK1, were reduced, but there was no alteration for MST1/2 and Aurora A (Supplementary Figure S5b). To pursue a more generalizable explanation for LKB1-mediated PLK1 regulation, we turned to the dephosphorylation of PLK1. Immunoblot analyses showed that PP1C-γ, the phosphatase responsible for the dephosphorylation of PLK1, was increased in LKB1-intact cells (Supplementary Figures S5a and b). Interestingly, we also observed a similar pattern in both pairs of cells for the expression of NUAK1, a substrate of LKB1 and a partner protein of PP1 (Supplementary Figures S5a and b). Thus, we questioned whether LKB1 might affect PLK1 activity through NUAK1–PP1 complex.
Zagórska et al.18 recently reported that LKB1-activated NUAK1 interacts with and phosphorylates several myosin phosphatases, including MYPT1. MYPT1 possesses a conserved ASSP amino acid motif, which is the preferred recognition site for the polo-box domain of PLK1. Thus, MYPT1 links PLK1 with PP1C and probably enhances the dephosphorylation of PLK1 by PP1C. Therefore, we postulated that LKB1 might inactivate PLK1 through NUAK–MYPT1–PP1-mediated dephosphorylation. We treated U2OS/shRNA-LKB1 or control cells with nocodazole for 12 h to arrest the cells in the G2 phase, when PLK1 is at its peak. Cell lysates were applied for immunoprecipitation with antibodies to PLK1, PP1C-γ, or NUAK1. We then probed the blots with antibodies to PLK1, PP1C-γ, NUAK1, MYPT1, and CDC-2-T161 to determine whether these proteins formed complexes within the cells and whether the phosphatase complexes were affected by LKB1. We found that PLK1 could precipitate PP1C-γ, NUAK1, and MYPT1 in shRNA control cells, although the interaction between PLK1 and MYPT1 was weak. Surprisingly, shRNA of LKB1 enhanced the complex formation between PLK1, MYPT1, and NUAK1, but reduced the binding of PP1C-γ to PLK1. This indicated that reduced kinase activity of NUAK1 in the absence of LKB1 affects the complex formation between MYPT1 and PP1C-γ, thereby disturbing the latter attaching to and dephosphorylating PLK1 (Figure 6a). Thus, we propose that compromised LKB1 reduces the binding of PP1C-γ with the MYPT1–PLK1–NUAK1 complex, enabling the phosphorylation of PLK1 (Figure 6b).

LKB1 regulates PLK1 through stabilizing MYPT1–PP1–PLK1 complex. (a) Nocodazole- or DMSO-treated U2OS/shRNA-LKB1 or control cells were lysed with RIPA buffer and subjected to immunoprecipitation with PLK1, PP1C-γ, or NUAK1. After elution, complexes were subjected to immunoblot detection. (b) PLK1 is regulated by phosphorylation and dephosphorylation. LKB1 may regulate dephosphorylation of PLK1 through the NUAK1–MYPT1–PP1 signaling
Loss of LKB1 results in the accumulation of nuclear abnormalities and multiploidy
Centrosome amplification can cause the daughter cells to gain or lose chromosome content as the result of the misalignment of multiple centrosomes on the polar axis.36, 37, 38, 39 To gain an insight into LKB1-mediated centrosome regulation, we cultured primary LKB1-null MEFs and stained the cells with DAPI at increasing passages. Nuclear abnormalities were observed as early as passage 3 and increased through passage 9 when these experiments were terminated (Figure 7a). To further investigate the outcome of LKB1 deficiency on genome stability, we detected cellular ploidy of MEFs after knockdown of LKB1. Cellular ploidy was determined by propidium iodide staining and flow cytometry (Figure 7b). The results showed that LKB1 depletion led to the accumulation of multiploidy cells with a 34% increase as compared with control cells (Figures 7b and c). Together, our results suggest that LKB1 deficiency exerts a functional impact on the integrity of nucleus and genome stability.

Deficiency of LKB1 leads to nuclear abnormalities and multiploidy and correlates with elevated levels of phospho-PLK1 in colon and esophageal cancers. (a) Primary wild-type and null-LKB1 cells were fixed and the DNA was stained with DAPI. Morphology of nucleus was analyzed. Nuclear abnormalities were categorized as: mitotic delay, binuclear, polylobed, grape, large, and micronuclear. White arrows show the micronuclear or lagged nuclear components during the mitosis. (b) MEFs were transfected twice with siRNA LKB1 or control for a total of 96 h. The cells were then fixed and stained with propidium iodide and subjected to flow cytometry analysis for cell cycle distribution. (c) The average fold change of multiploidy cells in siRNA LKB1-treated MEFs as compared with control siRNA-treated MEFs. Error bar represents S.D. of three independent experiments. *P<0.05. (d) Representative IHC staining of LKB1 and PLK1 in serial human esophageal tissues from healthy controls and benign diseases (n=28) or cancers (n=52). Scale bar=80 μm. (e) Quantitation of LKB1 and PLK1 expression in human esophageal disease spectrum tissues. Masked reading was performed by three different investigators with the same criteria to evaluate the staining (low, overall negative, or weak staining; high, overall moderate, or strong staining). Pearson’s χ2-test was used to analyze the distribution difference of PLK1 or LKB1 between benign and cancer tissues (P>0.05). (f) Assessment of phospho-PLK1-T210 in esophageal tissues in (e) by IHC. H-scores of phospho-PLK1 in LKB1 low- and high-expression cells were calculated (see Materials and Methods for an explanation of H-scores). Pearson’s χ2-test was used to analyze the significance of the correlation (*P<0.05)
PLK1 is overexpressed in colon and esophageal tissues with lower expression of LKB1
To investigate the pathological correlation between PLK1 and LKB1 in human cancers, we first analyzed PLK1 and LKB1 expression using IHC in colon and esophageal cancers that commonly lose LKB1.3, 40 We found that healthy tissues displayed low levels of PLK1 in intestinal and esophageal epithelia (Figure 7d and Supplementary Figures S6 and S7). In contrast, the colon and esophageal malignant tissues exhibited relatively higher levels of PLK1 (Figure 7d and Supplementary Figures S6 and S7) and a focal distribution was observed (Supplementary Figures S6 and S7). LKB1 was expressed in epithelia with moderate to strong signals in healthy subjects, whereas expression of LKB1 was markedly weak in cancers (Figure 7d and data not shown). Although there was a negatively correlated trend between PLK1 and LKB1, no statistical difference was observed (Figure 7e). Therefore, we investigated whether there was a relationship between phospho-PLK1 and LKB1 in these tissues as our in vitro data had indicated a reverse association between the two markers (Figures 3, 4, 5). We assessed phospho-PLK1 in colon and esophageal tissues from healthy and cancer subjects with IHC. Although phospho-PLK1 was minimally detectable in most cells, it was markedly higher in some cancerous tissues. More importantly, there was an intense staining of phospho-PLK1 in cancerous tissues with low LKB1 expression. Phospho-PLK1 levels were negatively correlated with LKB1 when an H-score analysis was applied (Figure 7f). Thus, these data support our previous observation that LKB1 suppressed the phosphorylation of PLK1 rather than the total levels of PLK1.
Discussion
Recent studies have shown that several interacting partners of LKB1 and their downstream targets accumulate in and interact with the centrosome.14, 15, 16 In a genome-wide screen of kinases required for cell cycle progress, Bettencourt-Dias et al.13 found that LKB1 might participate in mitotic regulation, and depletion of LKB1 leads to multipolar spindles in the cells. Aberrant centrosomes have been well characterized as a contributing factor to multipolar spindles and genomic instability.38, 41 Consistent with these reports, we found that loss of LKB1 caused an increase in the number of centrosomes. More importantly, we showed that primary LKB1-null MEFs possessed multiple nuclear abnormalities and increased aneuploidy as compared with that in wild-type MEFs. Thus, our results suggest that LKB1 may safeguard genome stability and repress carcinogenesis via the regulation of centrosomes and mitosis.
Whereas AMPK is of central importance as a downstream effector of LKB1, LKB1 is the master upstream kinase of at least 13 AMPK-related kinases, deeming it likely that LKB1 contributes to tumor suppression via various mechanisms.1 AMPK was found to locate to the centrosome and spindle apparatus but this activity of AMPK was independent of LKB1.14 Consistent with these findings, we demonstrated that AMPK was still activated in LKB1-deficient cells (data not shown). This phenomenon can be attributed to the presence of additional kinases of AMPK, such as CaMKK and TAK1.42, 43, 44 Further analysis of AMPK-deficient MEFs showed no increase in centrosome number. However, when siRNA of LKB1 was transduced into the AMPK-proficient or -deficient cells, centrosome amplification was drastically triggered (Supplementary Figure S1), making it unlikely that the centrosome amplification resulting from loss of LKB1 was due to the inactivation of AMPK.
To elucidate the mechanism by which LKB1 regulates centrosome, we began with a screening to characterize centrosomal regulatory proteins in the presence or absence of LKB1. We established that dysregulation of PLK1 is a major factor involved in centrosome amplification due to the loss of LKB1, as reducing PLK1 activity rescued the amplification phenotype and constitutively activated PLK1 overcame LKB1-mediated constriction to centrosome. We further found that PLK1, NUAK1, and PP1C-γ form a complex in LKB1-proficient cells and this complex is disrupted as a result of LKB1 depletion. Zagórska et al.18 recently reported that LKB1-activated NUAK1 interacts with several myosin phosphatases, including MYPT1–PP1β complex. It was also well documented that MYPT1–PP1 complex mediates the dephosphorylation of PLK1.35, 45 Thus, it was postulated that LKB1 might affect the dephosphorylation of PLK1 through NUAK1-associated MYPT1–PP1 complex (Figure 6b). Indeed, our immunoprecipitation assays clearly demonstrated that PLK1, NUAK1, MYPT1, and PP1 were in the same complex in LKB1-intact cells. Interaction between PP1 and PLK1 was remarkably reduced when LKB1 was absent (Figure 6a), supporting the postulation that LKB1 suppresses the activation of PLK1 via dephosphorylation. Taken together, our observations sustain the notion that LKB1 functions upstream of PLK1 and that loss of LKB1 leads to persistent activation of PLK1, producing centrosome amplification and genomic instability.
Considering the frequent mutation or deletion of LKB1 in gastrointestinal cancers, we analyzed the expression of LKB1, total PLK1, and phosphorylated PLK1 in a spectrum of colon and esophageal disease tissues. Although there was no appreciable correlation between LKB1 and total levels of PLK1, we revealed that LKB1 demonstrated a reverse correlation with PLK1-T210 in these cancer tissues (Figure 7f), and this is consistent with our in vitro data showing that LKB1 inhibited the activation rather than the expression of PLK1 (Figures 3, 4, 5). It will be of great interest to begin analyzing the status and correlation of LKB1 and PLK1-T210 in an expanded variety of cancers and determining their functions within these tissues as increased activation of PLK1 has been observed in various cancers.28, 29, 30 This research may also provide further stimulus for examining LKB1 as a marker for prognosis or treatment response to drugs selective for centrosomal regulation, as well as for better selection of patients who would be responsive to PLK1 inhibitor treatment.
Materials and Methods
Cell culture and reagents
The human U2OS (osteosarcoma) and HeLa (cervical cancer) cells, as well as wild-type and AMPK-null or LKB1-null MEFs were maintained in DMEM supplemented with 10% fetal bovine serum (FBS, Thermo Scientific, Waltham, MA, USA) at 37 °C in 5% CO2. BTO-1 and nocodazole were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Antibodies
Polyclonal antibodies against phospho-AMPK-T172, phospho-MST1/2-T180/183, MST1, PAK1, PAK3, and ARK5 were purchased from Cell Signaling (Boston, MA, USA). Polyclonal antibody against phospho-ACC-S79 was purchased from Millipore (Charlottesville, VA, USA). Polyclonal antibodies against γ-tubulin, NLP, and survivin as well as monoclonal anti-LKB1 antibody (Ley 37D/G6) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal antibodies against PLK1-T210 and pericentrin, and an LKB1 monoclonal antibody were obtained from Abcam (Cambridge, MA, USA). GAPDH antibody was obtained from Bethyl Laboratories (Montgomery, TX, USA). NEK2 (monoclonal) antibody was purchased from BD Biosciences (San Jose, CA, USA). PP1C-γ, PLK1, and phospho-CDC2-T161 antibodies were purchased from Novus Biologicals (Littleton, CO, USA). Polyclonal antibody for α-tubulin and monoclonal antibodies against γ-tubulin and α-tubulin were purchased from Sigma-Aldrich.
shRNA and siRNA and transfection
We followed shRNA methods described in our previous report.46 For siRNA, a pool of four siRNA duplexes targeting human or mouse LKB1 and a nontargeting siRNA pool were purchased from Dharmacon, Inc. (Lafayette, CO, USA). Cells were grown in a six-well plate to 40% confluence and incubated with a mixture of a 60 nmol siRNA duplex and 5 μl of transfection reagent 1 (Dharmacon, Inc.). After 4 h, FBS was added to a final concentration of 10% (v/v). Additional transfection was performed at 48-h intervals later for some MEFs.
Immunofluorescent staining
We performed immunofluorescent experiments as previously described.47 Briefly, cells were grown in six-well plates on cover slides and fixed in 4% neutral paraformaldehyde for 30 min on ice, washed with PBS, permeabilized in a solution containing 1% Triton X-100/0.5% Nonidet P-40/PBS, and blocked with 1% bovine serum albumin (BSA). Cells were then incubated with a primary antibody for 2 h, followed by a secondary antibody for 1 h at room temperature and staining of the DNA with DAPI for 5–10 min. Fluorescence signaling was observed with a Zeiss laser scan confocal microscope (LSM 5, Jena, Germany) and/or Olympus IX53 microscope (Center Valley, PA, USA). In some experiments, we used a relatively mild permeability buffer (0.1% Triton X-100, 0.05% NP-40, and 0.1% BSA in PBS) to detect the distribution of LKB1 that allowed us to keep the maximum cytoplasmic signaling of LKB1 while showing the nuclear signaling.
Flow cytometry
Cells were trypsinized to create a single-cell suspension, washed twice in PBS, and fixed with cold 70% ethanol for at least 24 h. The cells were then stained with a mixture of propidium iodide (Roche Diagnostics, Indianapolis, IN, USA) and RNase-A for 45 min on ice before running through a BD FACSCalibur.
Immunoblotting and immunoprecipitation
Cell lysis and immunoblotting were performed as described previously.48 For immunoblotting, 30–50 μg of total proteins were used. GAPDH or α-tubulin was used as loading controls. For immunoprecipitation, U2OS/shRNA-LKB1 or control cells were lysed with RIPA buffer for 30 min on ice. PP1C-γ, PLK1, or NUAK1 were immunoprecipitated at 4 °C. Immunoprecipitated complexes were eluted and subjected to immunoblot analysis.
Immunohistochemistry
Esophageal tissues from 52 malignant tumors and 28 benign or normal subjects were purchased from US Biomax, Inc. (no. ES809; Rockville, MD, USA). Colon tissues from University Hospital and US Biomax, Inc. (no. BC05002) were also used for the analysis. Avidin-biotin immunohistochemical analysis was performed as previously described.46 Briefly, serial sections of human colon disease tissue slides and esophageal tissues were subjected to antigen retrieval and probed with anti-LKB1 (Abcam, 1 : 50), anti-PLK1, or anti-PLK1 (T210) antibody (Abcam, 1 : 100) at 4 °C overnight. Biotin-conjugated anti-mouse or -rabbit IgG (Jackson Laboratories, Bar Harbor, ME, USA) was applied in a 1 : 300 dilution for 1 h at room temperature. Bound secondary antibody was further incubated with avidin-HRP (Vector Laboratories, Burlingame, CA, USA). After 3,3′-diaminobenzidine (DAB) staining, slides were counterstained with hematoxylin. Staining regions were reviewed by three different investigators and categorized as negative, weak, moderate, or strong. For determining the H-score, PLK1 (T210)-stained tissues were scored by calculating the product of the percentage of cells staining at each intensity level and the intensity level (0, negative; 1+, weak; 2+, moderate; and 3+, strong). An H-score was then calculated by summing the individual intensity level scores.49
Statistical analysis
Data were analyzed with two-tailed t-tests in the Minitab software package for Windows (Minitab Inc., State College, PA, USA). A P-value of 0.05 was considered as statistically significant.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- ARK5/NUAK1:
-
AMPK-related kinase 5
- BTO-1:
-
5-Cyano-7-nitro-2-benzothiazolecarboxamide-3-oxide
- CaMKKβ:
-
Ca2+/calmodulin-dependent protein kinase kinase β
- LKB1:
-
liver kinase B1
- MST1:
-
mammalian Ste20-like kinase 1
- MYPT1:
-
myosin phosphatase targeting subunit
- NEK2:
-
NIMA-related kinase 2
- NLP:
-
ninein-like protein
- PAK1/3:
-
p21-activated kinases 1 and 3
- PJS:
-
Peutz–Jeghers syndrome
- PLK-1:
-
Polo-like kinase 1
References
Alessi DR, Sakamoto K, Bayascas JR . LKB1-dependent signaling pathways. Annu Rev Biochem 2006; 75: 137–163.
Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391: 184–187.
Bardeesy N, Sinha M, Hezel AF, Signoretti S, Hathaway NA, Sharpless NE et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature 2002; 419: 162–167.
Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell 2010; 17: 547–559.
McCabe MT, Powell DR, Zhou W, Vertino PM . Homozygous deletion of the STK11/LKB1 locus and the generation of novel fusion transcripts in cervical cancer cells. Cancer Genet Cytogenet 2010; 197: 130–141.
Baas AF, Kuipers J, van der Wel NN, Batlle E, Koerten HK, Peters PJ et al. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 2004; 116: 457–466.
Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 2010; 468: 701–704.
Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007; 9: 218–224.
Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J et al. 5'-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 2006; 26: 5336–5347.
Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004; 6: 91–99.
Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 2004; 23: 833–843.
Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003; 13: 2004–2008.
Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F et al. Genome-wide survey of protein kinases required for cell cycle progression. Nature 2004; 432: 980–987.
Vazquez-Martin A, Lopez-Bonet E, Oliveras-Ferraros C, Perez-Martinez MC, Bernado L, Menendez JA . Mitotic kinase dynamics of the active form of AMPK (phospho-AMPKalphaThr172) in human cancer cells. Cell Cycle 2009; 8: 788–791.
Astrinidis A, Senapedis W, Henske EP . Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum Mol Genet 2006; 15: 287–297.
Bompard G, Rabeharivelo G, Cau J, Abrieu A, Delsert C, Morin N . P21-activated kinase 4 (PAK4) is required for metaphase spindle positioning and anchoring. Oncogene 2013; 32: 910–919.
Wei C, Bhattaram VK, Igwe JC, Fleming E, Tirnauer JS . The LKB1 tumor suppressor control spindle orientation and localization of activated AMPK in mitotic epithelial cells. PLoS One 2012; 7: e41118.
Zagórska A, Deak M, Campbell DG, Banerjee S, Hirano M, Aizawa S et al. New roles for the LKB1-NUAK pathway in controlling myosin phosphatase complexes and cell adhesion. Sci Signal 2010; 3: ra25.
Hinchcliffe EH, Sluder G . ‘It takes two to tango’: understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev 2001; 15: 1167–1181.
Hinchcliffe EH, Cassels GO, Rieder CL, Sluder G . The coordination of centrosome reproduction with nuclear events of the cell cycle in the sea urchin zygote. J Cell Biol 1998; 140: 1417–1426.
Pihan GA, Purohit A, Wallace J, Knecht H, Woda B, Quesenberry P et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res 1998; 58: 3974–3985.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2012; 144: 646–674.
Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA 2002; 99: 1978–1983.
Guo HQ, Gao M, Ma J, Xiao T, Zhao LL, Gao Y et al. Analysis of the cellular centrosome in fine-needle aspirations of the breast. Breast Cancer Res 2007; 9: R48.
Jiang F, Caraway NP, Sabichi AL, Zhang HZ, Ruitrok A, Grossman HB et al. Centrosomal abnormality is common in and a potential biomarker for bladder cancer. Int J Cancer 2003; 106: 661–665.
Archamblault V, Glover DM . Polo-like kinases: conservation and divergence in their functions and regulations. Nat Rev Mol Cell Biol 2009; 10: 265–275.
Golsteyn RM, Mundt KE, Fry AM, Nigg EA . Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol 1995; 129: 1617–1628.
Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ et al. Prognostic significance of polo-like kinase (PLK) expression in nonsmall cell lung cancer. Oncogene 1997; 14: 543–549.
Maire V, Némati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G et al. Polo-like kinase 1: a potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple negative breast cancer. Cancer Res 2013; 73: 813–823.
Zhang C, Sun X, Ren Y, Lou Y, Zhou J, Liu M et al. Validation of Polo-like kinase 1 as a therapeutic target in pancreatic cancer cells. Cancer Biol Ther 2012; 13: 1214–1220.
Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 445: 119–123.
Ji JH, Hwang HI, Lee HJ, Hyun SY, Kang HJ, Jang YL . Purification and proteomic identification of putative upstream regulators of polo-like kinase-1 from mitotic cell extracts. FEBS Lett 2010; 584: 4299–4305.
Rapley J, Baxter JE, Blot J, Wattam SL, Casenghi M, Meraldi P et al. Coordinate regulation of the mother centriole component nlp by nek2 and plk1 protein kinases. Mol Cell Biol 2005; 25: 1309–1324.
Toyoshima-Morimoto F, Taniguchi E, Nishida E . Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep 2002; 3: 341–348.
Matsumura F, Yamakita Y, Yamashiro S . Myosin light chain kinases and phosphatase in mitosis and cytokinesis. Arch Biochem Biophys 2011; 510: 76–82.
Gordon DJ, Resio B, Pellman D . Causes and consequences of aneuploidy in cancer. Nat Rev Genet 2012; 13: 189–203.
Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH . Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011; 333: 1895–1898.
Ganem NJ, Godinho SA, Pellman D . A mechanism linking extra centrosomes to chromosomal instability. Nature 2009; 460: 278–282.
Thompson SL, Bakhoum SF, Compton DA . Mechanisms of chromosomal instability. Curr Biol 2010; 20: R285–R295.
Gu Y, Lin S, Li JL, Nakagawa H, Chen Z, Jin B et al. Altered LKB1/CREB-regulated transcription co-activator (CRTC) signaling axis promotes esophageal cancer cell migration and invasion. Oncogene 2012; 31: 469–479.
Vitale I, Galluzzi L, Castedo M, Kroemer G . Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12: 385–392.
Chen WL, Kang CH, Wang SG, Lee HM . alpha-Lipoic acid regulates lipid metabolism through induction of sirtuin 1 (SIRT1) and activation of AMP-activated protein kinase. Diabetologia 2012; 55: 1824–1835.
Chen D, Pamu S, Cui Q, Chan TH, Dou QP . Novel epigallocatechin gallate (EGCG) analogs activate AMP-activated protein kinase pathway and target cancer stem cells. Bioorg Med Chem 2012; 20: 3031–3037.
Yee SW, Chen L, Giacomini KM . The role of ATM in response to metformin treatment and activation of AMPK. Nat Genet 2012; 44: 359–360.
Chiyoda T, Sugiyama N, Shimizu T, Naoe H, Kobayashi Y, Ishizawa J et al. LATS1/WARTS phosphorylates MYPT1 to counteract PLK1 and regulate mammalian mitotic progression. J Cell Biol 2012; 197: 625–641.
Lu C, Chen J, Xu HG, Zhou X, He Q, Li YL et al. MIR106B and MIR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology 2014; 146: 188–199.
Xu ZX, Zou WX, Lin P, Chang KS . A role for PML3 in centrosome duplication and genome stability. Mol Cell 2005; 17: 721–732.
Wang Y, Wang JW, Xiao X, Shan Y, Xue B, Jiang G et al. Piperlongumine induces autophagy by targeting p38 signaling. Cell Death Dis 2013; 4: e824.
Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med 2012; 18: 1052–1059.
Acknowledgements
We thank Dr. Frazier at MD Anderson Cancer Center and Dr. Mao in the Baylor College of Medicine for generously providing the primary mouse cells, Dr. KR Laderoute from SRI International for providing wild-type and AMPK-null MEFs, Dr. Lizhong Wang from University of Alabama at Birmingham for providing wild-type and LKB1fl/fl MEFs, and Dr. RJ Muschel at the Children’s Hospital of Philadelphia for providing the PLK1-T210D plasmid via Dr. E Nigg. We thank Dr. Boris Pasche for helpful discussion in the project. This work was supported by grants from National Cancer Institute R01CA133053, the Cervical Cancer SPORE Career Development Award and Pilot Award from NCI P50CA098252, and the Biomedical Research Foundation (to Z-XX).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U Simon
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Werle, K., Chen, J., Xu, HG. et al. Liver kinase B1 regulates the centrosome via PLK1. Cell Death Dis 5, e1157 (2014). https://doi.org/10.1038/cddis.2014.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.135
