
Abstract
Traumatic brain injury (TBI) produces excessive glutamate, leading to excitotoxicity via the activation of glutamate receptors. Postsynaptic density scaffold proteins have crucial roles in mediating signal transduction from glutamate receptors to their downstream mediators. Therefore, studies on the mechanisms underlying regulation of excitotoxicity by scaffold proteins can uncover new treatments for TBI. Here, we demonstrated that the postsynaptic scaffold protein Homer 1a was neuroprotective against TBI in vitro and in vivo, and this neuroprotection was associated with its effects on group I metabotropic glutamate receptors (mGluRs). Upon further study, we found that Homer 1a mainly affected neuronal injury induced by mGluR1 activation after TBI and also influenced mGluR5 function when its activity was restored. The ability of Homer 1a to disrupt mGluR-ERK signaling contributed to its ability to regulate the functions of mGluR1 and mGluR5 after traumatic injury. Intracellular Ca2+ and PKC were two important factors involved in the mediation of mGluR-ERK signaling by Homer 1a. These results define Homer 1a as a novel endogenous neuroprotective agent against TBI.
Similar content being viewed by others


Main
Traumatic brain injury (TBI) is the most common neurological disease and leads to acute neuronal cell injury, resulting in brain damage and neurological deficits. Glutamate-induced excitotoxicity excessively stimulates glutamate receptors, including the N-methyl-D-aspartate receptor (NMDAR), the α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor and the metabotropic glutamate receptor (mGluR), as well as their downstream signaling cascades, which have prominent roles in TBI.1 Although basic research studies have shown that various antagonists of glutamate receptors are effective in attenuating neuronal cell death and improving neurological function after TBI, these drugs have not performed satisfactorily in clinical investigations and cause some side effects.2
Recently, promising compounds that uncouple postsynaptic scaffold protein PSD-95 from the NMDAR-related neurotoxic signaling pathway have demonstrated effective neuroprotection in the treatment of stroke, another severe acute brain injury.3, 4, 5 Therefore, the disruption of glutamate receptor signaling by targeting postsynaptic scaffold proteins also provides a novel strategy for inhibiting excitotoxicity caused by TBI. Unlike PSD-95, the role of the scaffold protein Homer, which regulates mGluR signaling, in brain injuries has not been elucidated. Homer proteins are characterized by a conserved enabled/vasodilator-stimulated phosphoprotein (Ena/VASP) homology 1 (EVH1) domain.6, 7, 8, 9 However, the variation at the carboxy terminus determines the distinct function of Homer proteins. In mGluR signaling, long Homer variants (Homer 1b/c, Homer 2, and Homer 3) with a coiled-coil homodimerization domain at the carboxy terminus functionally link mGluR and its downstream mediators via their EVH1 domains.7, 8, 10, 11 In contrast, a short Homer variant (Homer 1a) without the coiled-coil domain cannot establish the linkage between the two target proteins via its EVH1 domain;7, 8, 10, 11 thus, it acts as a dominant-negative protein that disrupts the crosslinking action of mGluR-Homer signaling.12 This regulatory model has been demonstrated in several neurological diseases, including inflammatory pain13, 14 and fragile X syndrome,15 which raises the question of how Homer 1a is involved in regulating mGluR signaling in TBI. Here, we reported the neuroprotective role of Homer 1a in TBI and the involvement of group I mGluR signaling.
Results
Effects of Homer 1a on neuronal injury after traumatic injury
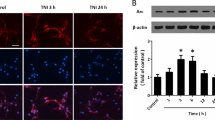
To determine the expression of Homer 1a during traumatic injury in cortical neurons, we used a transection model, which has been reported to result in mechanical neuronal injury and primary neuronal death and considered as an in vitro model of neuronal trauma.16 Homer 1a mRNA expression in mouse cortical neuronal cultures, as assessed by real-time polymerase chain reaction (PCR), increased after traumatic injury (Figure 1a), which was consistent with our previous observation in rat cortical neurons. Homer 1a protein expression also increased after neuronal injury (Figure 1b). Both Homer 1a mRNA and protein followed a similar pattern of expression, peaking at 6 h after traumatic injury. Immunofluorescence staining also showed strong immunoreactivity of Homer 1a in the cytoplasm and dendrites at 6 h after traumatic injury (Figure 1c).

Homer 1a is neuroprotective in vitro. Traumatic injury was induced in mice neuronal cultures for 48 h. The expression of Homer 1a mRNA was measured by real-time PCR (a), and the expression of Homer 1a protein was measured by western blot analysis (b). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus control. The distribution of Homer 1a and MAP2 (neurons) before and after traumatic injury is shown via immunofluorescence staining (c). DAPI was used to stain the nuclei. Scale bar=20 μm. The expression of Homer 1a was assessed by western blot in cortical neurons expressing different lentiviruses (d and e). After transfection and traumatic injury, cell viability was measured by the WST assay (f), cytotoxicity was determined by the lactate dehydrogenase (LDH) assay (g) and the cell death rate was assessed by PI/Hoechst staining (h). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus shRNA H1a
Because elevation of Homer 1a protein expression was observed during traumatic injury, we speculated that this induction of Homer 1a might have some effects on neuronal survival and death. Lentiviral transduction of Flag-tagged Homer 1a (Flag-H1a) increased the expression of Homer 1a protein (Figures 1d and e), which inhibited the reduction of cell viability (Figure 1f), attenuated cytotoxicity (release of LDH) (Figure 1g), and cell death (propidium iodide (PI) positive staining) (Figure 1h) in neurons undergoing traumatic injury. To further clarify the effects of endogenous Homer 1a on traumatic injury, the expression of Homer 1a protein was knocked down by a lentivirally expressed shRNA targeting Homer 1a (shRNA H1a) (Figures 1d and e). The shRNA H1a aggravated the neuronal injury after traumatic injury (Figures 1f–h). Furthermore, the overexpression of human Homer 1a rescued the effects of shRNA H1a, confirming the specificity of the shRNA-mediated knockdown of mouse Homer 1a. Taken together, these results indicate that Homer 1a is an endogenous neuroprotective protein in traumatic injury.
Homer 1a ameliorated brain damage and improved neurological function after TBI
To determine whether Homer 1a has similar effects in vivo, we used a closed head injury model to mimic TBI (Figure 2a). We found that Homer 1a protein expression was also increased in the cortex around the lesion (Figure 2b). Moreover, immunostaining indicated that induction of Homer 1a was also observed in the hippocampus surrounding the lesion area (Figures 2c and d). Next, we infused lentiviruses containing Flag-H1a into the right cortex of mice. After 5 days, western blot and immunofluorescence analyses revealed that Homer 1a was overexpressed in the right cortex of the mice (Figures 2e and f). Then, brain trauma was induced in the right hemisphere, and the water content of the brain was measured at 6 h, 1 day, 3 days, and 7 days after TBI. Neurological severity score (NSS) of the mice was assessed for seven consecutive days after injury. Lentiviral transduction of Flag-H1a in the lesion hemisphere significantly reduced brain edema and neurological deficits (Figures 2g and h). Furthermore, overexpression of Homer 1a decreased the duration of edema and neurological dysfunction. These results indicate the neuroprotective effects of Homer 1a overexpression in brain trauma.

Homer 1a is neuroprotective in vivo. TBI was induced in mice. H&E staining was performed on traumatically injured brain tissue (a). Scale bar=200 μM. The expression of Homer 1a was measured by western blot analysis at the indicated times (b). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus control. Immunohistochemistry staining for Homer 1a in the area surrounding the lesion (c). Scale bar=100 μM. The distribution of Homer 1a is revealed by immunofluorescence staining for Homer 1a and NeuN (d). Scale bar=40 μm. Mice were injected with vector or Flag-H1a, and the expression of Homer 1a was measured by western blot analysis (e). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector. Immunofluorescence staining for Flag in an area injected with Flag-H1a (f). Scale bar=40 μm. TBI was induced in mice injected with vector or Flag-H1a. The neurological functions in the vector group (n=10) and the Flag-H1a group (n=12) were assessed by determining the NSS at 6 h and 1, 2, 3, 4, 5, 6, and 7 days after TBI (g). The extent of brain edema in the vector group (n=10) and the Flag-H1a (n=10) group was measured by water content tests at 6 h, 1 d, 3 d, and 7 days (h). The data are represented as the mean±S.E.M. *P<0.05 versus vector
The regulation of neuronal injury by Homer 1a was associated with group I mGluRs
A large number of studies have shown that mGluRs have crucial roles in TBI. After traumatic injury, we found that activation of group I mGluRs contributed to the neuronal injury after traumatic injury, whereas activation of either group II mGluRs or group III mGluRs inhibited the neuronal injury (Supplementary Figure S1). Then, we tested whether Homer 1a regulates the function of mGluRs after traumatic injury. In the presence of an antagonist of group I mGluRs (AIDA), neither overexpression nor knockdown of Homer 1a in neurons had significant effects on neuronal injury after traumatic injury (Figures 3a–c). In contrast, neither an antagonist of group II mGluRs (LY341495) nor an antagonist of group III mGluRs (MSOP) had obvious effects on the changes in neuronal injury induced by Homer 1a overexpression and knockdown (Figures 3d–i). Moreover, overexpression of Homer 1a increased cell viability and decreased cytotoxicity and cell death in neuronal cultures treated with an agonist of group I mGluRs (DHPG), whereas knockdown of Homer 1a enhanced neuronal injury after activation of mGluRs (Figures 3j–l). Taken together, these results indicate that Homer 1a mainly regulates the function of group I mGluRs after traumatic injury.

Homer 1a regulates group I mGluRs in traumatic injury. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, the neuronal cultures were pretreated with (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA) (100 μM) (a–c), (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495) (100 μM) (d–f), or (RS)-α-methylserine-O-phosphate (MSOP) (100 μM) (g–i). Cell viability, cytotoxicity, and cell death rate were measured in neuronal cultures after traumatic injury. The data are represented as the mean±S.E.M. from five experiments. The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus short hairpin RNA (shRNA) Con. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, the neuronal cultures were treated with DHPG (100 μM). Cell viability, cytotoxicity, and cell death rate were measured (j–l). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus shRNA Con
Although the activation of group I mGluRs promoted neuronal injury, two subtypes of group I mGluRs (mGluR1 and mGluR5) are thought to have distinct roles in TBI (Supplementary Figure S2). Here, we hypothesized that Homer 1a might differentially regulate these two subtypes of group I mGluRs. Overexpression of Homer 1a attenuated the neuronal injury in neurons pretreated with mGluR5 antagonists (MPEP) after traumatic injury, whereas knockdown of Homer 1a increased the susceptibility to neuronal injury (Figures 4a–c). Furthermore, mGluR1 activation (Ro 67-7476) induced neuronal injury, which were also influenced by Homer 1a overexpression and knockdown in the same manner (Figures 4d–f). In contrast, in the presence of Bay 36-7620, overexpression of Homer 1a weakened the protective effects of an mGluR5 agonist (CHPG) against traumatic injury, and knockdown of Homer 1a amplified these effects (Figures 4g–i). These results indicate that the inactivation of mGluR1 by Homer 1a overpowered the similar effects of Homer 1a on the activity of mGluR5 after traumatic injury. However, Homer 1a also influenced the function of mGluR5 when its function was restored.

Homer 1a differentially mediates mGluR1 and mGluR5. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, the neuronal cultures were pretreated with 2-methyl-6-(phenylethynyl)pyridine (MPEP) (5 μM). Cell viability, cytotoxicity, and cell death rate were measured in the neuronal cultures after traumatic injury (a–c). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus short hairpin RNA (shRNA) Con. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, cell viability, cytotoxicity, and cell death rate were measured in the neuronal cultures treated with Ro 67-7476 (10 μM) (d–f). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus shRNA Con. Mice cortical neuronal cultures were transfected with different lentiviruses. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, the neuronal cultures were pretreated with a combination of (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) (500 μM) and Bay 36-7620 (10 μM). Cell viability, cytotoxicity, and cell death rate were measured in the neuronal cultures after traumatic injury (g–i). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus shRNA Con
The disruption of mGluR1-ERK signaling by Homer 1a protected against traumatic injury
As an important intracellular signaling pathway, the ERK pathway is a crucial component of downstream mGluR1/5 signal transduction, which has been implicated in the regulation of TBI in vitro and in vivo. Here, we found that an mGluR1 antagonist (Bay 36-7620) inhibited the activation of the ERK pathway in neurons undergoing traumatic injury (Figure 5a), whereas an mGluR1 agonist (Ro 67-7476) increased the phosphorylation of ERK in normal neuronal cultures (Figure 5b). On the other hand, ERK inhibitors (PD98059 and U0126) reduced the neuronal injury in neurons treated with an mGluR1 agonist (Ro 67-7476) (Figures 5c–e). Therefore, activation of mGluR1-ERK signaling might contribute to neuronal injury after traumatic injury.

Homer 1a disrupts mGluR1-extracellular signal-regulated kinase (ERK) signaling. Mice cortical neuronal cultures were pretreated with Bay 36-7620 (10 μM). After traumatic injury, phosphorylation of ERK was measured by western blot analysis (a). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Neuronal cultures were treated with Ro 67-7476 (10 μM), and ERK phosphorylation was assessed by western blot analysis (b). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Neuronal cultures were pretreated with PD98059 (40 μM) and U0126 (20 μM). Cell viability (c), cytotoxicity (d), and cell death rate (e) were measured in neuronal cultures treated with Ro 67-7476 (10 μM). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Mice cortical neuronal cultures were transfected with different lentiviruses, and phosphorylation of ERK was measured by western blot analysis after traumatic injury (f) or treatment with Ro 67-7476 (10 μM) (g). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus short hairpin RNA (shRNA) Con
According to previous results, the neuroprotective effect of Homer 1a is mainly dependent on its inhibition of mGluR1 function in traumatic injury. The overexpression of Homer 1a reduced the activation of the ERK pathway after traumatic injury, whereas phosphorylation of ERK was increased by knockdown of Homer 1a under the same conditions (Figure 5f). Similarly, ERK phosphorylation induced by an mGluR1 agonist (Ro 67-7476) was suppressed by the overexpression of Homer 1a and enhanced by Homer 1a knockdown (Figure 5g). Hence, disruption of mGluR1-ERK by Homer 1a could be a potential mechanism underlying the neuroprotective effect of Homer 1a in traumatic neuronal injury.
Homer 1a interfered mGluR5 agonist-induced ERK activation after traumatic injury
Unlike the inactivation of mGluR1, the mGluR5 antagonist MPEP did not affect the ERK pathway in neuronal cultures subjected to traumatic injury (Figure 6a). In contrast, an mGluR5 agonist (CHPG) not only increased the level of ERK phosphorylation in normal neuronal cultures but also enhanced the activation of the ERK pathway after traumatic injury (Figures 6b–c). Using the ERK inhibitors PD98059 and U0126, neuronal injury was increased in neuronal cultures treated with mGluR5 agonist after traumatic injury (Figures 6d–f). These results suggest that mGluR5-ERK signaling has a role in mGluR5 agonist-induced neuroprotection against traumatic injury.

Homer 1a influences mGluR5-extracellular signal-regulated kinase (ERK) signaling. Mice cortical neuronal cultures were pretreated with 2-methyl-6-(phenylethynyl)pyridine (MPEP) (5 μM). After traumatic injury, phosphorylation of ERK was measured by western blot analysis (a). The data are represented as the mean±S.E.M. from five experiments. (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) (500 μM) was administered in normal neuronal cultures or neuronal cultures subjected to traumatic injury. ERK phosphorylation was assessed by western blot analysis (b and c). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Neuronal cultures were pretreated with CHPG (500 μM), PD98059 (40 μM), or U0126 (20 μM). Cell viability (d), cytotoxicity (e), and cell death rate (f) were measured in neuronal cultures after traumatic injury. The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Mice cortical neuronal cultures were transfected with different lentiviruses. Phosphorylation of ERK was measured by western blot analysis after CHPG (500 μM) treatment (g). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus shRNA Con. After transfection with different lentiviruses, neuronal cultures were pretreated with CHPG (500 μM) and Ro 67-7476 (10 μM). Phosphorylation of ERK was measured by western blot analysis after traumatic injury (h)
Owing to the ability of Homer 1a to regulate the effects of the mGluR5 agonist, we assumed that mGluR5-ERK signaling in neuroprotection would also be disrupted by Homer 1a. In normal neuronal cultures, mGluR5 agonist-induced elevation of ERK phosphorylation was inhibited by the overexpression of Homer 1a and augmented by knockdown of Homer 1a (Figure 6g). After pretreatment of neuronal cultures undergoing traumatic injury with an mGluR5 agonist (CHPG) and an mGluR1 antagonist (Bay 36-7620), Homer 1a overexpression and knockdown exerted similar effects on the ERK pathway (Figure 6h). Taken together, these results demonstrate that Homer 1a is also involved in the regulation of mGluR5-ERK signaling-associated neuroprotection.
Involvement of Ca2+ in the regulation of mGluR1-ERK signaling by Homer 1a
Intracellular Ca2+ is an important mediator of mGluR-ERK signaling. Using an intracellular Ca2+ chelator (BAPTA-AM), we found that neuronal injury was attenuated in neuronal cultures undergoing traumatic injury (Figure 7a). Hence, we speculated that Ca2+ was involved in the mediation of mGluR1-ERK signaling owing to its consistent effects on traumatic injury. Next, we observed that an mGluR1 antagonist (Bay 36-7620) inhibited the elevation of intracellular Ca2+ in neuronal cultures undergoing traumatic injury (Figure 7b). Moreover, mGluR1 agonist-induced phosphorylation of ERK was reduced using an intracellular Ca2+ chelator (BAPTA-AM, 10 μM) (Figure 7c). These results suggest that the elevation of intracellular Ca2+ induced by the activation of mGluR1 contributes to mGluR1 signaling to the ERK pathway during traumatic injury.

Homer 1a affects mGluR1-extracellular signal-regulated kinase (ERK) signaling by regulating intracellular Ca2+. Mice cortical neuronal cultures were pretreated with 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) (10 μM). Cell viability, cytotoxicity, and cell death rate were measured in neuronal cultures after traumatic injury (a). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Neuronal cultures were pre-treated with Bay 36-7620 (10 μM). Intracellular Ca2+ concentrations were measured until the indicated times (b). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Neuronal cultures were pretreated with BAPTA-AM. After treatment with Ro 67-7476 (10 μM), phosphorylation of ERK was measured by western blot analysis (c). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, the intracellular Ca2+ concentrations were measured until the indicated times after traumatic injury (d and e). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus short hairpin RNA (shRNA) Con. Mice cortical neuronal cultures were transfected with different lentiviruses. After pretreatment with Bay 36-7620 (10 μM), the intracellular Ca2+ concentrations were measured until the indicated times in neuronal cultures subjected to traumatic injury (f and g). The data are represented as the mean±S.E.M. from five experiments. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, the intracellular Ca2+ concentrations were measured until the indicated times in neuronal cultures treated with Ro 67-7476 (10 μM) (h and i). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus shRNA Con
Next, we further investigated whether Homer 1a disrupted mGluR1-ERK by regulating the levels of intracellular Ca2+. After traumatic injury, overexpression of Homer 1a attenuated Ca2+ overload (Figure 7d), whereas knockdown of Homer 1a enhanced the Ca2+ response in neuronal cultures (Figure 7e). However, in the presence of an mGluR1 antagonist (Bay 36-7620), these effects were no longer significant (Figures 7f and g). In addition, the mGluR1 agonist-induced elevation of Ca2+ was also reduced by Homer 1a overexpression and enhanced by Homer 1a knockdown in normal neuronal cultures (Figures 7h and i). Therefore, we concluded that Homer 1a regulates mGluR1-Ca2+-ERK signaling, providing a mechanism for the role of Homer 1a in traumatic neuronal injury.
Homer 1a interfered with PKC-dependent ERK activation by an mGluR5 agonist
In addition to Ca2+, PKC is also reportedly involved in mGluR-ERK signaling. After traumatic injury, we observed that the inhibition of PKC activity by chelerythrine chloride (CTC) had no significant effect on neuronal injury (Figure 8a). In contrast, a PKC activator (Bryostatin 1) protected neurons from traumatic injury (Figure 8a). Furthermore, we found that PKC inhibition reduced the mGluR5 agonist-induced elevation of ERK phosphorylation (Figure 8b) and mGluR5 agonist increased the activity of PKC (Figure 8c). These results support the hypothesis that PKC regulates mGluR5-ERK signaling.

Homer 1a influences mGluR5-ERK signaling by mediating protein kinase C (PKC) activity. Mice cortical neuronal cultures were pretreated with chelerythrine chloride (CTC) (10 μM) or Bryostatin 1 (5 μM). Cell viability, cytotoxicity, and cell death rate were measured in neuronal cultures after traumatic injury (a). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Neuronal cultures were pretreated with (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) (500 μM) and CTC (10 μM), and phosphorylation of ERK was measured by western blot analysis (b). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. PKC activity was assessed in neuronal cultures treated with CHPG (500 μM) (c). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vehicle. Mice cortical neuronal cultures were transfected with different lentiviruses. After transfection, the neuronal cultures were treated with CHPG (500 μM). The activity of PKC was measured in neuronal cultures subjected to traumatic injury (d) or normal neuronal cultures (e). The data are represented as the mean±S.E.M. from five experiments. *P<0.05 versus vector; #P<0.05 versus shRNA Con. After Bryostatin 1 (5 μM) treatment, PKC activity was assessed in neuronal cultures transfected with different lentiviruses (f). The data are represented as the mean±S.E.M. from five experiments
Next, we further examined whether Homer 1a influences PKC-dependent ERK activation in the presence of an mGluR5 agonist. After pretreatment with an mGluR5 agonist (CHPG), overexpression of Homer 1a inhibited the activity of PKC, whereas PKC activity was elevated by knockdown of Homer 1a after traumatic injury (Figure 8d). Moreover, the mGluR5 agonist-induced elevation of PKC activity was also decreased by Homer 1a overexpression and increased by Homer 1a knockdown in normal neuronal cultures (Figure 8e). In contrast, the regulation of Homer 1a expression did not alter the activity of PKC induced by a PKC activator (Bryostatin 1) (Figure 8f). Taken together, these results indicate that Homer 1a regulates mGluR5-PKC-ERK signaling by interfering with mGluR5 function.
Discussion
The involvement of mGluRs in models of TBI in vitro and in vivo has been well established in several previous studies.17, 18, 19, 20, 21, 22 In the present study, we revealed that Homer 1a selectively interfered with the actions of mGluRs during TBI. First, the upregulation of Homer 1a attenuated the neuronal injury induced by activation of group I mGluRs, suggesting that the protective effect of Homer 1a against traumatic injury was related to its regulation of group I mGluRs. Second, the beneficial effect on neuronal survival induced by agonists of group II mGluRs and group III mGluRs was not affected by Homer 1a upregulation, which may be because neither group II mGluRs nor group III mGluRs lack the target sequence for Homer (PPXXF) at their C-termini. The discovery that altered Homer 1a was responsible for much of the regulation of group I mGluRs in neuronal injury demonstrated that the neuroprotective effects of Homer 1a against traumatic injury were associated with the regulation of group I mGluR signaling.
There are two subtypes of group I mGluRs, including mGluR1 and mGluR5. Previous studies have reported that mGluR1 contributed to neuronal injury after TBI using mGluR1-specific agonists and antagonists.18, 19, 20, 23 In addition, many studies showed that mGluR5-specific agonists had therapeutic effects on TBI in vitro and in vivo.24, 25, 26, 27 In the present study, we observed that mGluR1 and mGluR5 had different functions, suggesting that the activation of mGluR1 enhanced neuronal injury and the activation of mGluR5 protected neurons from trauma. According to this theory, the inhibition of mGluR5 by antagonists would promote neuronal injury after traumatic injury. However, our results showed that inactivation of mGluR5 had no significant effects on neuronal injury. Hence, we speculated that the activity of mGluR5 was blocked after traumatic neuronal injury; thus, mGluR1 was the major group I mGluR involved in neuronal injury.
Because both mGluR1 and mGluR5 contain the Homer-specific target sequence, they can interact with Homer scaffold proteins.8 Homer 1a has been reported to regulate the function of both mGluR1 and mGluR5.13 In the present study, the upregulation of Homer 1a reduced the neuronal injury induced by activation of mGluR1, supporting the notion that Homer 1a exerts protective effects against traumatic injury. In theory, Homer 1a should promote the function of mGluR5 during traumatic injury because they are both neuroprotective proteins. However, in the case of mGluR1 inhibition, Homer 1a inhibited the protective effects of an mGluR5 agonist. This result was consistent with observations from previous studies, which confirmed that Homer 1a negatively regulates the function of mGluR5 in other neurological diseases.15, 28 Considering that the effects of mGluR1 overpowered the action of mGluR5 after traumatic neuronal injury, it is possible that the enhanced activation of mGluR1 limited the regulatory effect of Homer 1a on mGluR5. Therefore, inactivation of mGluR1 by Homer 1a mainly contributed to its neuroprotective effects against traumatic injury.
Group I mGluRs can activate a large number of downstream modulators and signaling pathways.29 Among these modulators and signaling pathways, the ERK pathway is thought to be closely related to the regulation of TBI. In previous studies, the level of ERK phosphorylation was enhanced, and inhibition of the ERK pathway reduced the cortical lesion and the neuronal injury after TBI.30, 31, 32 In the present study, we observed that mGluR1 was mainly responsible for the activation of ERK pathway by group I mGluRs after traumatic injury. However, this result does not indicate whether the mGluR5-activated ERK pathway would participate in the regulation of TBI. In recent studies, we revealed that an mGluR5 agonist could enhance the activation of the ERK pathway after TBI, and an ERK pathway inhibitor attenuated the protective effects of an mGluR5 agonist against neuronal injury.24, 25 These results suggest that the specific function of mGluR-ERK signaling during trauma is dependent on the subtypes of activated mGluRs.
Accumulating evidence has demonstrated that Homer scaffold proteins coordinate mGluR-ERK signaling under various conditions.14, 15, 33 Homer 1a is thought to downregulate the increase in the intracellular Ca2+ response9, 34 and negatively regulate the phosphorylation of ERK,14, 15, 35 both of which are associated with the regulation of mGluR function. Moreover, our previous studies demonstrated that Homer 1a attenuated the overload of Ca2+ induced by injurious stimulations, including excitotoxicity and oxidative stress.36, 37 However, the role of Ca2+ in the regulation of mGluR-ERK by Homer 1a is still not fully understood in the context of neuronal injury. Presumably, Ca2+ mainly engages in mGluR1 signaling after traumatic injury owing to the similar effects of Ca2+ overload and mGluR1 activation in traumatic injury. The present study provides evidence that intracellular Ca2+ overload contributes to the mGluR1-induced activation of ERK after traumatic injury, and upregulation of Homer 1a disrupts mGluR1-ERK signaling by inhibiting the release of intracellular Ca2+ in response to neuronal injury.
Although the mGluR5-induced Ca2+ response could also be negatively regulated by Homer 1a under some conditions,14 this mechanism might be weakened in traumatic injury owing to the opposing effects of Ca2+ response and mGluR5 activation. Therefore, the ability of Homer 1a to modify mGluR1-ERK signaling raises the question as to whether the Homer 1a-mediated phosphorylation of ERK in response to mGluR5 activation occurs in a Ca2+-independent manner. In our previous study, we confirmed the neuroprotective role of mGluR5-PKC-ERK signaling in TBI.24 Accordingly, we further verified the role of Homer 1a in the regulation of mGluR5-PKC-ERK signaling, indicating that Homer 1a also participates in the regulation of ERK phosphorylation by disrupting the signaling from mGluR5 to PKC in traumatic injury. Thus, the effects of Homer 1a on traumatic injury would be reversed once mGluR5 function is restored, indicating that there might be a more complicated mechanism underlying the regulation of group I mGluRs by Homer 1a (Supplementary Figure S4).
Dysregulation of mGluR is implicated in multiple brain disorders.38 Therefore, many selective mGluR antagonists have been developed for clinical therapy. However, these drugs have produced some unacceptable side effects in clinical trials. The present study indicated that Homer 1a may be a potential neuroprotective agent against TBI via its regulation of mGluR1. Because the regulation of mGluR by Homer 1a had more specific and mild effects compared with a direct blockade of mGluR, the side effects caused by inactivation of mGluR might be reduced to some extent. However, the ability of Homer 1a to disrupt mGluR5 signaling indicates that it may have potential side effects in therapy when the function of mGluR5 is restored during recovery phase after TBI. This is because mGluR5 has important roles in regulating normal neurological function. Thus, further investigation is required to elucidate the relationship between Homer 1a and mGluR in neurological diseases.
Materials and Methods
Antibodies and reagents
Primary antibody against Homer 1a was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against phosphor-ERK1/2 (p-ERK) and total ERK1/2 (ERK) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibody against β-actin was obtained from Sigma (St Louis, MO, USA). The secondary antibodies for immunoblotting were horseradish peroxidase-conjugated anti-rabbit and anti-goat IgG (Santa Cruz Biotechnology). The secondary antibodies for immunostaining were Alexa 488 donkey anti-goat IgG and Alex 594 donkey anti-rabbit IgG) (Invitrogen, Carlsbad, CA, USA). Agonists and antagonists were obtained from Tocris Bioscience (Bristol, UK).
Animals
C57BL/6 mice (10–12 weeks, 25–28 g) were obtained from Experimental Center of Fourth Military Medical University, which were kept at a constant temperature (approximately 27 °C) in an air-conditioned room for at least 7 days before the study and were exposed to a 12-h light/dark cycle. All animal studies were performed in adherence with the National Institutes of Health Guidelines on the Use of Laboratory Animals, and were approved by the Fourth Military Medical University Committee on Animal Care.
Primary culture of cortical neurons
Neuronal cortical cultures were prepared as previously described with some modifications. Briefly, cerebral cortices were removed from embryos at 16–18 days. Tissues were dissociated by 0.25% trypsin digestion for 15 min at 37 °C and gentle trituration. Neurons were resuspended in the Neurobasal medium containing 2% B27 supplement and 0.5 mM L-glutamine (Invitrogen) and plated at a density of 3 × 105 cells per cm2. Before seeding, culture vessels, consisting of 96-well plates, 1.5 cm glass slides or 6 cm dishes were coated with poly-L-lysine (50 μg/ml) at room temperature overnight. Neurons were maintained at 37 °C in a humidified 5% CO2 incubator and half of the culture medium was changed every other day. The cultured neurons were used for studies on in vitro days 12–14 (DIV 12–14) and verified to be >95%.
Traumatic neuronal injury model
The in vivo model of TBI used in the present study was previously described by Mukhin et al.16 with some modification. The transection model used a plastic stylet to scrape adherent cells from a culture dish thereby tearing processes and soma while leaving a significant proportion of cells intact. A transection model was used in the present study as described previously.32 Briefly, each dish confluent culture was manually scratched with a sterile plastic pipette tip following a square grid (with 3 mm spacing between the lines). To reduce the inconsistency of damage in different experiments, all traumatic neuronal injury models were established by the same researcher in our group by using a standard square grid module. Cell cultures were placed in an incubator at 37 °C until a designated posttrauma time point was reached. Cultures were incubated without changing the medium. Experiments were performed from 0 h (immediately after mechanical injury) to 48 h after trauma. Uninjured cultures were used as controls. Because scratch injury activates neurons first at the wound edge and later expands to the entire neuron monolayer, the entire culture on each dish was used for all experiments.
Real-time reverse transcription-PCR
RNA was isolated from primary cortical neurons by Trizol (Invitrogen). After equalization of the RNA quantity in each group, the reverse-transcription and real-time PCR were performed using a commercially available kit (TaKaRa, Dalian, China). Following cDNA generation, quantitative PCR was completed with the Bio-Rad iQ5 Gradient Real-Time PCR system (Bio-Rad Laboratories, Hercules, CA, USA). Primers for all PCR experiments was listed as follows: Homer1a primers, 5′-GGCAAACACTGTTTATGGACTGG-3′ (forward), 5′-GTAATTCAGTCAACTTGAGCAACC-3′(reverse); β-actin primers, 5′-CTAAGGCCAACCGTGAAAAGATG-3′ (forward), 5′-ACCGCTCGTTGCCAATAGTGATG-3′ (reverse). Fifty cycles were used for quantitative PCR, using conditions: 94 °C (30 s), 58 °C (30 s) and 72 °C (30 s). The relative intensity against β-actin and the fold change relative to the control were calculated.
Western blot analysis
Western blot analysis was performed as described previously. Briefly, the entire neurons on each well in vitro and the impaired cortexes of the mice in vivo were lysed with a lysis buffer with protease inhibitor (Roche, Penzberg, Germany). Equivalent amounts of protein were loaded and separated by 8–12% SDS-PAGE gels, and transferred to nitrocellulose membranes (Invitrogen). The membranes were incubated overnight at 4 °C with the appropriate primary antibody (Homer 1a, 1:200; β-actin, 1:2500; p-ERK, 1:800; ERK, 1:1000). Membranes were washed in TBST and incubated for 1 h at room temperature with the secondary antibodies. Immunoreactivity was detected by SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA). The membranes were stripped for 15 min at room temperature with Re-blot Plus Strong Solution (Thermo Scientific) and retested with tubulin or β-actin antibodies to normalize for protein loading. The optical densities of the bands were quantified by using an image analysis system with ImageJ (National Institutes of Health, Bethesda, MD, USA).
Immunocytochemistry
After fixing with 4% paraformaldehyde for 15 min at 37 °C, mice neuronal cultures were washed with phosphate-buffered saline (PBS) and permeabilized with 0.2% Triton X-100, followed by incubation of primary antibodies overnight at 4 °C. The primary antibodies were diluted as follows: Homer 1a (1:50) and MAP-2 (1:200). Then, cells were incubated with secondary antibodies (Alexa 488 donkey-anti-goat IgG (Invitrogen), 1:300; Alexa-594-conjugated goat-anti-mouse IgG, 1:300) for 2 h. Cultures were dehydrated with ethanol and mounted with 4,6-diamidino-2-phenylindole (DAPI) for nuclear staining (Sigma). Images were captured using an Olympus FV10i Confocal Microscope (Olympus, Tokyo, Japan). All images from one experiment were acquired using the same exposure time to allow comparisons of relative levels of immunoreactivity between the different treatment conditions. At least six images of each group were taken by an evaluator blinded to the experimental conditions.
Cell viability assay
Cell viability assay was performed by using The Cell Proliferation Reagent WST-1 (Roche) following the manufacturer’s protocol. Cells (at a concentration of 0.5–5 × 105) were cultured in 24-well plates with a final volume of 500 μl per well in the culture medium. After treatment, 50 μl cell proliferation reagent WST-1 was added to each well and incubated for 4 h at 37 °C in 5% CO2, and then shaken thoroughly for 1 min on a shaker. One hundred and ten microliters of supernatant from each well was collected to a 96-well microplate. The absorbance of the samples was measured by using a microplate reader (Bio-Rad Laboratories).
Cytotoxicity assay
LDH release assay was used for the measurement of cytotoxicity. LDH release into the culture medium was detected using a diagnostic kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions. Briefly, 50 μl of supernatant from each well was collected to assay LDH release. The samples were incubated with reduced form of nicotinamide-adenine dinucleotide and pyruvate for 15 min at 37 °C and the reaction was stopped by adding 0.4 mol/l NaOH. The activity of LDH was calculated from the absorbance at 440 nm, and background absorbance from the culture medium that was not used for any cell cultures was subtracted from all absorbance measurements. The results were normalized to the maximal LDH release, which was determined by treating control wells for 60 min with 1% Triton X-100 to lyse all cells. LDH release was defined as the ratio of LDH in the media to total LDH and normalized to the fold of control.
Cell death assessment
Following exposure of neuronal cultures to the various treatments, neuronal survival was quantified and presented as percent of cell death rate. Neuronal survival was determined by staining treated neuronal cultures with 5 μM Hoechst 33342 and 2 μM propidium iodide (PI) (Sigma) for 10 min. Culture plates were placed on a mechanized stage of a Nikon microscope (Nikon Corporation, Tokyo, Japan) and photomicrographs were collected by a blinded observer. The numbers of total and injured (PI positive) cells were counted by automated computer-assisted software (Image-Pro Plus version 6.0; Media Cybernetics Inc., Rockville, MD, USA). The raw counts are presented in an Excel file for the generation of percent cytotoxicity and statistical analysis.
Lentiviral preparation for RNAi and overexpression
For developing shRNA lentiviruses, an siRNA oligo directed to the coding region +1190–1208 was subcloned into a lentiviral expression vector, pFU-GW-RNAi (GeneChem Company, Shanghai, China). Following digestion and ligation, clones were selected and verified for the inserted sequence. The lentiviral construct coexpressed EGFP driven by the ubiquitin C promoter, in addition to U6 promoter driving the shRNA. To control for off-target and nonspecific effects of shRNA, a negative control shRNA (5′-TTCTCCGAACGTGTCACGT-3′) was used. The overexpression of lentiviral system was developed by removing the EGFP open reading frame from the pGC-FU-EGFP-3FLAG construct (GeneChem Company) by an AgeI/NheI digestion and replacing it with the cDNA of Homer 1a. DNA fragments encoding mouse Homer 1a and human Homer 1a were PCR-amplified using mouse and human brain cDNA library as a template (Supplementary Figure S3).
Calcium imaging
Intracellular Ca2+ concentration were measured using the ratiometric calcium indicator Fura-2-AM (Invitrogen). Cultured neurons grown on glass slides were loaded with 5 μM Fura-2-AM for 45 min before glutamate treatment at room temperature. Neurons were then placed in the open-bath imaging chamber containing Dulbecco’s PBS (0.901 mM CaCl2, 0.493 mM MgCl2·6H2O, 2.67 mM KCl, 1.47 mM KH2PO4, 137.93 mM NaCl and 8.06 mM Na2HPO4·7H2O, pH 7.2–7.4) supplemented with 20 mM glucose at ambient temperature. Using the Nikon inverted epifluorescence microscope (Nikon), neurons were excited at 345 and 385 nm and the emission fluorescence at 510 nm was recorded. Images were collected and analyzed with the MetaFluor image-processing software (Molecular Devices, Sunnyvale, CA, USA). The Ca2+ concentration values were then calculated and background fluorescence was subtracted from each wavelength before calculations.
PKC activity assay
PKC activity assay was performed by using PKC kinase activity assay (Enzo Life Sciences, Farmingdale, NY, USA) following the manufacturer’s protocol. Briefly, samples were added to wells of a PKC substrate microtiter plate pretreated with 50 μl of kinase assay dilution buffer for 10 min at room temperature. Ten microliters of diluted ATP was added to each well and incubated for 90 min at 30 °C. Then, supernatants were removed from each well and 40 μl of phosphospecific substrate antibody was added to each well for 60 min at room temperature. Each well was washed with washing buffer and 40 μl of diluted anti-rabbit IgG:HRP conjugate was added to each well for 30 min at room temperature. Later, each well was washed with washing buffer and 60 μl of TMB substrate was added to each well for 45 min at room temperature. Twenty microliters of acid stop solution was added to each well and the absorbance of the samples was measured by using a microplate reader.
Model of TBI in vivo
The in vivo model of TBI used in the present study was previously described by Flierl et al.39 with some modifications. After induction of anesthesia with 4% isoflurane, a mouse was placed on the platform directly under the weight of the weight-drop device. A midline longitudinal scalp incision was made and the skull exposed. After identification of the right anterior frontal area (1.5 mm lateral to the midline in the mid-coronal plane) as impact area, the weight was released and dropped onto the skull. Then, the scalp wound was closed by standard suture material and the wound area was treated with lidocain cream. Mice were returned to cages at the end of the surgical procedures, where access to water and food was freely available.
Immunohistochemistry
The animals were transcardially perfused and postfixed with 4% paraformaldehyde in 0.1 M phosphate buffer. Brain tissues were embedded in paraffin. Serial sections were cut at 5 μm on a cryostat and mounted on the slides. For hematoxylin and eosin (H&E) staining, deparaffinized sections were stained with H&E according to the routine protocol. For immunostaining, deparaffinized sections were boiled in unmasking antigen buffer (10 mM sodium citrate buffer, pH 6.0) and incubated in 3% hydrogen peroxide for 10 min. After treating with blocking buffer for 1 h at room temperature, diluted primary antibody was added and incubated overnight at 4 °C. For DAB staining, sections were incubated with secondary antibodies (HRP-conjugated IgG) for 30 min. DAB dilution was added on sections and the reaction was stopped according to appropriate staining intensity. Sections were counterstained in hematoxylin. For double-labeled immunofluorescence staining, sections were incubated with secondary antibodies (Alexa 488 donkey-anti-goat IgG (Invitrogen), 1:300; Alexa-594-conjugated goat-anti-mouse IgG, 1:300) for 2 h. After staining, sections were coversliped with different mounting mediums. All the sections were observed using a fluorescence microscope (Olympus BX51, DP71).
Stereotaxic injection of lentivirus
Delivery of lentiviral vectors in vivo was carried out by using a stereotaxic cortical injection as described previously. Three cortical injections were performed in the right hemisphere (ipsilateral to the lesion) as follows: point 1, 1.0 mm anterior to the bregma, 1.5 mm lateral, 1.5 mm deep; point 2, 0 mm anteroposterior to the bregma, 1.5 mm lateral, 1.5 mm deep; point 3, 1.0 mm posterior to the bregma, 1.5 mm lateral, 1.5 mm deep. All the target points were in the right hemisphere (ipsilateral to the lesion). Each injection contained 1.5 μl of 1 × 109 TU/ml lentivirus suspension at a rate of 0.2 μl/min. The needle was withdrawn over a course of 10 min. Five days after injection of lentivirus, mice were subjected to brain trauma as described above.
Neurological severity score
The NSS highly correlates with the severity of brain damage and was measured as described previously.39 NSS was conducted by an investigator who was blinded to the experimental groups. The score consists of 10 individual neurological parameters, including tasks on motor function, alertness and physiological behavior. One point is awarded for the lack of a tested reflex or for the inability to perform the tasks, and no point for succeeding. A maximal NSS of 10 points thus indicates severe neurological dysfunction, with failure of all tasks.
Water content measurement
The extent of cerebral edema is evaluated by determining the tissue water content in the injured hemisphere as reported previously.39 Briefly, mice were decapitated under deep anesthesia with 100 mg/kg pentobarbital. The brains were quickly removed and the hemispheres were separated in the sagittal plane. Coronal sections of 3 mm thickness were prepared from the area bordering the lesion. These sections were weighed (wet weight, Ww) and dried in a desiccator oven for 24 h at 95 °C. After weighing the dried sections (dry weight, Wd), the water content was calculated as: % H2O=((Ww−Wd) × 100)/Ww.
Statistical analysis
All of the experiments were performed a minimum of three times. Statistical evaluation was carried out with GraphPad Prism software, version 5.0 (GraphPad, San Diego, CA, USA). Significant differences between experiments were assessed by univariate ANOVA (more than two groups), followed by Bonferroni’s multiple comparisons or unpaired t-test (two groups).
Abbreviations
- TBI:
-
traumatic brain injury
- NMDAR:
-
N-methyl-D-aspartate receptor
- AMPAR:
-
α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor
- mGluR:
-
metabotropic glutamate receptor
- EVH1:
-
enabled/vasodilator-stimulated phosphoprotein (Ena/VASP) homology 1
- NSS:
-
neurological severity score
- PI:
-
propidium iodide
- AIDA:
-
(RS)-1-aminoindan-1,5-dicarboxylic acid
- LY341495:
-
(2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid
- MSOP:
-
(RS)-α-methylserine-O-phosphate
- MPEP:
-
2-methyl-6-(phenylethynyl)pyridine
- CHPG:
-
(RS)-2-chloro-5-hydroxyphenylglycine
- BAPTA-AM:
-
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- CTC:
-
chelerythrine chloride
References
Luo P, Fei F, Zhang L, Qu Y, Fei Z . The role of glutamate receptors in traumatic brain injury: implications for postsynaptic density in pathophysiology. Brain Res Bull 2011; 85: 313–320.
Loane DJ, Faden AI . Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci 2010; 31: 596–604.
Zhou L, Li F, Xu HB, Luo CX, Wu HY, Zhu MM et al. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat Med 2010; 16: 1439–1443.
Cook DJ, Teves L, Tymianski M . Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 2012; 483: 213–217.
Bach A, Clausen BH, Moller M, Vestergaard B, Chi CN, Round A et al. A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1-2 and protects against ischemic brain damage. Proc Natl Acad Sci USA 2012; 109: 3317–3322.
Kato A, Ozawa F, Saitoh Y, Hirai K, Inokuchi K . Vesl, a gene encoding VASP/Ena family related protein, is upregulated during seizure, long-term potentiation and synaptogenesis. FEBS Lett 1997; 412: 183–189.
Brakeman PR, Lanahan AA, O'Brien R, Roche K, Barnes CA, Huganir RL et al. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 1997; 386: 284–288.
Xiao B, Tu JC, Petralia RS, Yuan JP, Doan A, Breder CD et al. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron 1998; 21: 707–716.
Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M et al. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 1998; 21: 717–726.
Sun J, Tadokoro S, Imanaka T, Murakami SD, Nakamura M, Kashiwada K et al. Isolation of PSD-Zip45, a novel Homer/vesl family protein containing leucine zipper motifs, from rat brain. FEBS Lett 1998; 437: 304–308.
Kato A, Ozawa F, Saitoh Y, Fukazawa Y, Sugiyama H, Inokuchi K . Novel members of the Vesl/Homer family of PDZ proteins that bind metabotropic glutamate receptors. J Biol Chem 1998; 273: 23969–23975.
Xiao B, Tu JC, Worley PF . Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol 2000; 10: 370–374.
Hu JH, Park JM, Park S, Xiao B, Dehoff MH, Kim S et al. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron 2010; 68: 1128–1142.
Tappe A, Klugmann M, Luo C, Hirlinger D, Agarwal N, Benrath J et al. Synaptic scaffolding protein Homer1a protects against chronic inflammatory pain. Nat Med 2006; 12: 677–681.
Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci 2012; 15: 431–440 S431.
Mukhin AG, Ivanova SA, Knoblach SM, Faden AI . New in vitro model of traumatic neuronal injury: evaluation of secondary injury and glutamate receptor-mediated neurotoxicity. J Neurotrauma 1997; 14: 651–663.
Gong QZ, Delahunty TM, Hamm RJ, Lyeth BG . Metabotropic glutamate antagonist, MCPG, treatment of traumatic brain injury in rats. Brain Res 1995; 700: 299–302.
Camon L, Vives P, de Vera N, Martinez E . Seizures and neuronal damage induced in the rat by activation of group I metabotropic glutamate receptors with their selective agonist 3,5-dihydroxyphenylglycine. J Neurosci Res 1998; 51: 339–348.
Faden AI, O'Leary DM, Fan L, Bao W, Mullins PG, Movsesyan VA . Selective blockade of the mGluR1 receptor reduces traumatic neuronal injury in vitro and improves outcome after brain trauma. Exp Neurol 2001; 167: 435–444.
Mukhin A, Fan L, Faden AI . Activation of metabotropic glutamate receptor subtype mGluR1 contributes to post-traumatic neuronal injury. J Neurosci 1996; 16: 6012–6020.
Zhou F, Hongmin B, Xiang Z, Enyu L . Changes of mGluR4 and the effects of its specific agonist L-AP4 in a rodent model of diffuse brain injury. J Clin Neurosci 2003; 10: 684–688.
Berent-Spillson A, Robinson AM, Golovoy D, Slusher B, Rojas C, Russell JW . Protection against glucose-induced neuronal death by NAAG and GCP II inhibition is regulated by mGluR3. J Neurochem 2004; 89: 90–99.
Fei Z, Zhang X, Bai HM, Jiang XF, Wang XL . Metabotropic glutamate receptor antagonists and agonists: potential neuroprotectors in diffuse brain injury. J Clin Neurosci 2006; 13: 1023–1027.
Chen T, Cao L, Dong W, Luo P, Liu W, Qu Y et al. Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. Neurochem Res 2012; 37: 983–990.
Chen T, Zhang L, Qu Y, Huo K, Jiang X, Fei Z . The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int J Mol Med 2012; 29: 630–636.
Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI . Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. J Neurotrauma 2013; 30: 403–412.
Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI . Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation 2012; 9: 43.
Hu JH, Yang L, Kammermeier PJ, Moore CG, Brakeman PR, Tu J et al. Preso1 dynamically regulates group I metabotropic glutamate receptors. Nat Neurosci 2012; 15: 836–844.
Niswender CM, Conn PJ . Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 2010; 50: 295–322.
Lu KT, Cheng NC, Wu CY, Yang YL . NKCC1-mediated traumatic brain injury-induced brain edema and neuron death via Raf/MEK/MAPK cascade. Crit Care Med 2008; 36: 917–922.
Mori T, Wang X, Jung JC, Sumii T, Singhal AB, Fini ME et al. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Metab 2002; 22: 444–452.
Zhao Y, Luo P, Guo Q, Li S, Zhang L, Zhao M et al. Interactions between SIRT1 and MAPK/ERK regulate neuronal apoptosis induced by traumatic brain injury in vitro and in vivo. Exp Neurol 2012; 237: 489–498.
Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ . The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci 2005; 25: 2741–2752.
Kammermeier PJ, Worley PF . Homer 1a uncouples metabotropic glutamate receptor 5 from postsynaptic effectors. Proc Natl Acad Sci USA 2007; 104: 6055–6060.
Luo P, Zhao Y, Li D, Chen T, Li S, Chao X et al. Protective effect of Homer 1a on tumor necrosis factor-alpha with cycloheximide-induced apoptosis is mediated by mitogen-activated protein kinase pathways. Apoptosis 2012; 17: 975–988.
Chen T, Fei F, Jiang XF, Zhang L, Qu Y, Huo K et al. Down-regulation of Homer1b/c attenuates glutamate-mediated excitotoxicity through endoplasmic reticulum and mitochondria pathways in rat cortical neurons. Free Radic Biol Med 2012; 52: 208–217.
Luo P, Chen T, Zhao Y, Xu H, Huo K, Zhao M et al. Protective effect of Homer 1a against hydrogen peroxide-induced oxidative stress in PC12 cells. Free Radic Res 2012; 46: 766–776.
Ribeiro FM, Paquet M, Cregan SP, Ferguson SS . Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol Disord Drug Targets 2010; 9: 574–595.
Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E . Mouse closed head injury model induced by a weight-drop device. Nat Protoc 2009; 4: 1328–1337.
Acknowledgements
The work was supported by National Natural Science Foundation of China (Nos. 30930093, 81301037 and 81200949), National Science and Technology Major Project of China (2013ZX 09J13109-02C); National Science and Technology Pillar Program of China (No. 2012BAI11B02), Science and Technology Project of Shaanxi (No. 2012SF2-01-4), and Research Foundation of PLA (Nos. 2010gxjs078 and AWS11J008).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Luo, P., Chen, T., Zhao, Y. et al. Postsynaptic scaffold protein Homer 1a protects against traumatic brain injury via regulating group I metabotropic glutamate receptors. Cell Death Dis 5, e1174 (2014). https://doi.org/10.1038/cddis.2014.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.116
Keywords
This article is cited by
-
Arc silence aggravates traumatic neuronal injury via mGluR1-mediated ER stress and necroptosis
Cell Death & Disease (2020)
-
Preso regulates NMDA receptor-mediated excitotoxicity via modulating nitric oxide and calcium responses after traumatic brain injury
Cell Death & Disease (2019)
-
Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage
Cell Death & Disease (2018)
-
Pu-erh Tea Protects the Nervous System by Inhibiting the Expression of Metabotropic Glutamate Receptor 5
Molecular Neurobiology (2017)
-
Homer1a attenuates glutamate-induced oxidative injury in HT-22 cells through regulation of store-operated calcium entry
Scientific Reports (2016)

{kind=link}
{kind=link}
{kind=link}