
Abstract
Mortalin (mot-2) induces inactivation of the tumor suppressor p53’s transcriptional and apoptotic functions by cytoplasmic sequestration of p53 in select cancers. The mot-2-dependent cytoprotective function enables cancer cells to support malignant transformation. Abrogating the p53-mot-2 interaction can control or slow down the growth of cancer cells. In this study, we report the discovery of a ubiquitin-like (UBX)-domain-containing protein, UBXN2A, which binds to mot-2 and consequently inhibits the binding between mot-2 and p53. Genetic analysis showed that UBXN2A binds to mot-2’s substrate binding domain, and it partly overlaps p53’s binding site indicating UBXN2A and p53 likely bind to mot-2 competitively. By binding to mot-2, UBXN2A releases p53 from cytosolic sequestration, rescuing the tumor suppressor functions of p53. Biochemical analysis and functional assays showed that the overexpression of UBXN2A and the functional consequences of unsequestered p53 trigger p53-dependent apoptosis. Cells expressing shRNA against UBXN2A showed the opposite effect of that seen with UBXN2A overexpression. The expression of UBXN2A and its apoptotic effects were not observed in normal colonic epithelial cells and p53−/− colon cancer cells. Finally, significant reduction in tumor volume in a xenograft mouse model in response to UBXN2A expression was verified in vivo. Our results introduce UBXN2A as a home defense response protein, which can reconstitute inactive p53-dependent apoptotic pathways. Inhibition of mot-2-p53 interaction by UBXN2A is an attractive therapeutic strategy in mot-2-elevated tumors.
Similar content being viewed by others


Introduction
Mot-2 is a member of the heat-shock protein 70 family,1 which increases in tumor cells. Simultaneously, mot-2 changes its subcellular location from mitochondria in normal cells to the cytosol in cancerous cells, where it interacts with p532, 3, 4 in a reversible manner.5 Mot-2 prevents wild-type (WT) p53 from translocating to the nucleus, leading to reduced p53-mediated activation of downstream targets.4, 6, 7, 8 More recently, it has been shown that inhibition of mot-2 induces apoptosis in selective cancer cells with mutant p53.9 The mot-2-based cytoplasmic sequestration of the p53 tumor suppressor is one of the main carcinogenic mechanisms in colorectal cancers.2 In addition, because more than 50% of patients with colon cancer particularly in the early stage have WT-p53,10 targeting mot-2 as a p53 inhibitor in colon cancer cells is considered an efficient treatment strategy along conventional chemotherapy.
Here, we show that the UBXN2A protein is a positive regulator of p53 through its interaction with the C-terminus of mot-2 where the p53-binding site is located. UBXN2A facilitates the translocation of WT-p53 to the nucleus where p53 regulates its target genes, particularly those involved in apoptosis. These results introduce UBXN2A as an important anticancer factor that can contribute to p53 localization and activation as a host defense mechanism against cancerous growth. Because fully functional p53 can reverse tumor formation and progression as well as postpone tumor relapse,11 characterization of mot-2’s endogenous regulators may lead to a new class of therapeutic interventions in tumors with high levels of mot-2.
Results
UBXN2A induces cell death
A number of studies have reported the contribution of the UBXD family of proteins to different cancers.12, 13 Expression of these UBX domain-containing proteins correlate positively or negatively with tumor progression in a tissue-specific manner.14 Our previous study showed that UBXN2A, as a p97-associated protein, is involved in protein quality control.15 We decided to conduct a set of experiments to determine whether UBXN2A might have a role in apoptosis in cancer cells as reported for some other UBXD family members.14 As cell detachment, cell shrinking, and alteration of nuclear morphology are ubiquitous aspects of apoptosis, we examined these apoptotic features in the presence of UBXN2A. We used HCT-116 colon cancer cells transfected with GFP-empty, GFP-p53, or GFP-UBXN2A. As expected for p53 expression,16 GFP-p53 significantly increased the number of cells detaching from the plates at 48 and 72 h post-transfection as compared with the vehicle, which is indicative of typical late stage apoptosis (Figure 1a, Supplementary Figure 1A). Similarly, it turned out that UBXN2A expression induced at least 50% cell detachment as early as 48 h post-transfection (Figure 1a, Supplementary Figure 1A). We next asked whether UBXN2A expression leads to apoptotic characteristics (condensed chromatin and apoptotic bodies), which are ubiquitous aspect of apoptosis. We examined these in the presence of UBXN2A and p53 in fixed cells stained with DAPI. Expression of GFP-UBXN2A and GFP-p53 significantly increased the number of nuclei with condensed chromatin and apoptotic bodies formed in cells transiently transfected with GFP-p53 and GFP-UBXN2A (Figures 1b and c). On the basis of the cell morphology and reduced cell viability, it appeared that UBXN2A leads the cells to apoptosis, which was further confirmed by the large molecular weight DNA fragments in HCT-116 and LoVo colon cancer cells transiently transfected with GFP-UBXN2A or GFP-empty vector (Supplementary Figure 1B).

Cytotoxicity of UBXN2A in colon cancer cells with WT-p53. (a) HCT-116 cells were seeded at 30 000 cells per plate. Cells were transfected with GFP-empty vector, GFP-p53, and GFP-UBXN2A using the Neon transfection system (see Materials and Methods). Many GFP-UBXN2A cells were detached after the first 24 h. As GFP expression reached a maximum level after 24 h, cell detachment was studied after 24. 24, 48, and 72 h after transient transfection, detached cells were removed. Cell detachment was monitored using a Zeiss motorized inverted microscope and measuring the number of adherent live cells (green) using AxioVision software. The numbers of remaining cells were calculated in same five fields under the microscope for each experiment at 24, 48, and 72 h. The data from cell counting show a significant reduction of adherent cells in the presence of p53 or UBX2A in comparison with the empty vector after 72 h. These results suggest the UBXN2A-induced cell detachment due to apoptosis is comparable to that of p53. (b, c) In another set of experiments, HCT-116 cells transiently transfected with GFP-empty, GFP-p53, and GFP-UBXN2A were fixed and stained with DAPI. After 24 h, fluorescent microscopy observation demonstrates typical apoptotic morphology including condensation of the nuclear material (arrowheads) and formation of apoptotic bodies (arrows) when cells express p53 or UBXN2A (b). The bar chart represents the total number of the apoptotic cells counted in five fields for each groups after 24 h. Significant differences between different groups means and control value are indicated by ***P<0.001 (c). Scale bar=50 μm. (d) A cytotoxicity test with the crystal violet staining method was developed to determine cell viability in cancer cell lines transfected with GFP-UBXN2A. The Neon transfection system provides high efficiency of transfection (80–90%) and viability (90%) when electroporation parameters (voltage, pulse width, and number of pulses) are optimized for each individual cell line. Statistically significant cytotoxicity induced by UBXN2A was confirmed in HCT-116, LoVo, and HT-29 cell lines, while UBXN2A overexpression had no induction of cytotoxicity in SW480 cells (mutant p53) and MCF7 (caspase 3 deficient) breast cancer cells with and without suboptimal stress (Eto: etoposide 5 μM). Each value represents mean±S.E. of at least three independent experiments, and each experiment was performed in triplicate (*P<0.05)
UBXN2A induces cell cytotoxicity in a cell type-dependent manner
In another set of experiments, we used crystal violet-based cytotoxicity assays to compare the cytotoxicity of UBXN2A in four colon cancer cell lines as well as MCF7 breast cancer cells (Supplementary Figures 1C and D). Statistically significant cytotoxicity induced by UBXN2A was confirmed in HCT-116 and LoVo cell lines with WT-p53 (Figure 1d). In addition, we chose SW480 and HT-29 colon cancer with mutant p53 for the cytotoxicity assay. We observed significant cell cytotoxicity in the presence of UBXN2A in HT-29 (∼50%), while UBXN2A expression had no significant induction of cytotoxicity in SW480 colon cancer cells (Figure 1d).
Our initial experiments in MCF7 showed no cell cytotoxicity in the presence of UBXN2A overexpression (Figure 1d). Therefore, we examined cell cytotoxicity in MCF7 cells treated with suboptimal toxic dose of 5 μM etoposide for 24 h after transfection with GFP-empty or GFP-UBXN2A. UBXN2A had no effect on cell cytotoxicity in MCF7 cells in the presence of stress (Figure 1d).
UBXN2A binds mot-2 in the cytoplasm
Members of the UBXD family associate with a variety of cargoes that enable them to be involved in many cellular processes.12 We therefore hypothesized that UBXN2A-induced apoptosis in cancer cells is due to UBXN2A’s contribution to specific cancer-related pathways. We sought to identify the binding partners of UBXN2A in the HCT-116 cancer cells in the presence or the absence of the genotoxic agent etoposide (50 μM). HCT-116 cells were collected after a 24-h treatment, followed by IP with an anti-UBXN2A antibody immobilized on protein A magnetic beads (Supplementary Figure 2A). Cytosolic proteins that co-precipitated with UBXN2A, with and without etoposide, were analyzed by mass spectrometry-based proteomic approaches (Supplementary Figure 2B).17 We identified several candidate proteins, including mot-2. More mot-2 was associated with cytoplasmic UBXN2A under conditions of genotoxic stress. Next, we performed iodixanol density gradient centrifugation15, 18 of cytoplasmic extracts from HCT-116 cells treated with etoposide (50 μM). UBXN2A co-sedimented with fractions containing mot-2 (lanes 7–9, Figure 2a). As a control, we probed the collected fractions for another UBXD family member, UBXN2C protein (p47). UBXN2C did not show co-sedimentation with mot-2, indicating the specificity of the co-sedimentation of UBXN2A with mot-2. UBXN2A-containing fractions were additionally enriched for p97 protein, which is a known partner of UBXN2A.12, 15 We performed a set of His-tag pull-down and IP experiments in HEK293T (Figure 2b) and HCT-116 cells (Figure 2c). Cytoplasmic fractions from HEK293T cells transiently transfected with increasing amounts of (His)6-TYG-tagged UBXN2A were incubated with magnetic His-tag beads to isolate the mot-2 protein. Figure 2b shows that upon increased exogenous expression of (His)6-UBXN2A the pulled down mot-2 signal intensities increased proportionally, indicating that (His)6-UBXN2A binds to endogenous mot-2 in a concentration-dependent manner. To determine whether endogenous UBXN2A and mot-2 proteins associate in vivo, we first determined the steady state level of mot-2 and UBXN2A in cells as well as normal and tumor tissues (Supplementary Figure 3). We confirmed that endogenous UBXN2A binds to mot-2 in HCT-116 cells in a p53-independent manner (Figure 2c). Finally, we confirmed UBXN2A and mot-2 interaction in colorectal tumor tissues (Figure 2d). We further confirmed that UBXN2A binds to mot-2 in HT-29 cells carrying mutant p53 (Supplementary Figure 1E). To further verify negative results obtained from MCF7 cells, we conducted an immunoprecipitation (IP) experiment using an anti-UBXN2A antibody. We found UBXN2A does not bind to mot-2 in MCF7 cells with and without stress (Supplementary Figure 1F). The molecular characteristics of mot-2 in MCF7 breast cancer cells versus colon cancer cells and lack of caspase 3 in MCF7 cells, which has specific role in apoptosis and in activation of caspase 6 and 7 are considered as potential reasons for the absence of cell death in MCF7 upon UBXN2A overexpression. Finally, IP experiments with an anti-UBXN2A antibody using normal human umbilical vein endothelial cell (HUVEC) lysates in the absence and the presence of recombinant GST-mot-2 showed UBXN2A only pulls down recombinant mot-2 (sufficient amount of mot-2) and not the endogenous mot-2 expressed in normal HUVEC cells (Supplementary Figure 1G).

UBXN2A interacts with mot-2. (a) HCT-116 cells were treated with etoposide (50 μM) for 24 h and cytoplasmic fractions were prepared. Samples were subjected to the iodixanol gradient centrifugation. The collected fractions (20 total) were analyzed by WB using the indicated antibodies. In HCT-116 cells, co-sedimentation of endogenous UBXN2A and mot-2 was predominantly observed in fractions 7-9 (black box). In contrast, P47 (UBXN2C) did not show co-sedimentation with mot-2, supporting a selective interaction between mot-2 and UBXN2A. In addition, UBXN2A showed co-sedimentation with its known partner, the protein p97 (CDC48). (b) HEK293T cells were transfected with increased amounts of pcDNA-(His)6-TYG-tagged UBXN2A. After 24 h, cell lysates were incubated with 50μl magnetic Dynabeads (His)6-Tag. UBXN2A pulls down endogenous mot-2 in a dose-dependent manner in HEK293T cells. (c, d) Using magnetic Dynabeads protein A, endogenous UBXN2A bound to immobilized anti-UBXN2A antibodies pulls down endogenous mot-2 in HCT-116 (knockout derivative (p53−/−) and wild-type (p53+/+)) colon cancer cells as well as mot-2 in human colon cancer tissues. We particularly observed pulled down mot-2 with tumors in d (Lane 4 versus 3). (e) Fractions shown in a were probed with anti-p53, HSP90α, and HSC70 antibodies. As expected, only some p53 proteins co-sediment with mot-2 (fractions 3–5). Instead, p53 showed strong co-sedimentation with fractions enriched in HSP90α, a known stabilizer of p53, in fractions 3–7. Fractions containing UBXN2A and mot-2 (a, fractions 7–9) have a low level of p53 (black box). As expected, another population of p53 proteins co-sedimented with HSC70, a known p53 regulator, in fractions 12–15. These results suggest that two distinct mot-2-containing complexes exist, one that sediments with p53 (fractions 3–5) and one that sediments with UBXN2A (fractions 7–9)
Switching the protein-binding preference of mot-2 from p53 to UBXN2A
Because mot-2 binds to the cytoplasmic domain of p53 and sequesters WT-p53 in the cytoplasm, we asked whether binding UBXN2A to mot-2 can alter mot-2’s affinity for p53. To test this hypothesis, we probed the fractions collected from the iodixanol gradient (Figure 2a) with an anti-p53 antibody. p53 showed two peaks (Figure 2e) of which the first, at fractions 3–5, dominantly showed co-fractionation with HSP90 protein, as expected, and partially with mot-2.19 The second peak of p53 was at fractions 12 to 15, which may represent p53 association with HSC70/HSP70 complex (Figure 2e). Notably, p53 was not highly abundant in the fractions that contained the majority of the co-sedimented UBXN2A and mot-2 proteins (fractions 7–9 in Figure 2a versus Figure 2e). These results suggest the binding of mot-2 to UBXN2A might inhibit the binding of mot-2 to p53. That is, the two complexes might be mutually exclusive. To further determine whether the binding of mot-2 to UBXN2A decreases the binding of mot-2 to p53, we performed two set of immunoprecipitation experiments with recombinant UBXN2A. Figure 3a describes an in vitro competition immunoprecipitation assay system containing mot-2, p53, and an increasing amount of recombinant UBXN2A. In a competition mechanism, the increasing amounts of recombinant human UBXN2A decreased the intensity of mot-2 bands pulled down by anti-p53 antibodies. The lowest binding between p53-mot-2 was observed when UBXN2A and mot-2 were present in approximately a 1:1 ratio by their molecular mass (lane 1 versus lane 2). In Figure 3b, cytosolic fractions enriched with mot-2 and p53 proteins (fractions 3-5, Figure 2e) were incubated with recombinant GST-tag human UBXN2A protein. After the initial 2 h of incubation, samples were subjected to immunoprecipitation with anti-p53 antibodies. GST-UBXN2A and endogenous mot-2 ratio was 2.5:1 in the reaction. The presence of UBXN2A decreased the amount of mot-2 protein-bound p53 (Figure 3b). Next, we decided to verify whether endogenous UBXN2A can interfere with mot-2-p53 binding using an ex vivo model. The HCT-116 cell line was identified as one of the best candidates for in vivo experiments, as HCT-116 has minimum expression of UBXN2A (Supplementary Figure 3B) while it has an abundant amount of mot-2-p53 complexes in the absence of stress.6 Figures 3c–f showed that the amounts of UBXN2A mRNA and protein increased in HCT-116 cells treated with etoposide for 24 h, indicating that etoposide can induce upregulation of UBXN2A at RNA and protein levels. Moreover, immunofluorescence staining showed that UBXN2A located at the juxtanuclear region in unstressed HCT-116 cells forms a punctate distribution scattered throughout the cytoplasm in many cells upon etoposide treatment (Figure 3g). This distinct punctate structure of UBXN2A was consistent with punctate p53 and mot-2 formation in colon cancer cell lines.6 As a result, we decided to verify whether UBXN2A decreases p53’s binding to mot-2 in the presence of etoposide (20 and 50 μM). A set of co-immunoprecipitations of mot-2 with UBXN2A as well as mot-2 with p53 showed that an increasing association of UBXN2A and mot-2 correlates with an increased dissociation of p53 and mot-2 in an etoposide dose-dependent manner (Figures 3h and i).

UBXN2A and p53 compete for binding to mot-2. (a) We investigated whether UBXN2A can decrease the association of p53 with mot-2 using an in vitro binding competition assay. First, recombinant human GST-p53 proteins bound to anti-p53 antibodies-IgG magnetic beads were incubated with human GST-mot-2 protein and increasing concentrations of human GST-UBXN2A recombinant proteins. Mot-2 proteins were eluted from the beads and analyzed by western blotting using an anti-mot-2 antibody. The same membrane was re-probed for p53 (lower panel) to show equivalent p53 in each IP. (b) The in vitro competition assay was further confirmed when the human GST-UBXN2A fusion proteins were incubated with cytosolic fractions enriched with mot-2 and p53 proteins (fractions 3-5, Figure 2e) of HCT-116 cells. The level of recombinant protein provided an ∼2.5:1 ratio of UBXN2A to endogenous mot-2. Cell lysates with and without UBXN2A were incubated with anti-p53 antibodies immobilized on magnetic Dynabeads protein G (Lanes 3 and 4). Beads with mouse IgG or beads alone were control groups in this experiment (lane 1 and 2). Western blotting showed that UBXN2A caused displacement of p53 binding from mot-2. These data support that UBXN2A and p53 compete for the mot-2. (c, d) A semiquantitative RT-PCR protocol (SuperScript III One-Step RT-PCR system) and quantitative real-time RT-PCR (d) showed etoposide enhances transcription of UBXN2A in HCT-116 cells. (e, f) At 24 h post-etoposide treatment (20 and 50 μM), total cell lysates were obtained, 50μg of protein was loaded in each lane, and the resulting blots were probed with an anti-UBXN2A antibody. UBXN2A’s signal was normalized with the HSC70 signal (loading control). Together, these data confirmed UBXN2A expression is upregulated at the mRNA and protein levels upon genotoxic stress (P<0.05, n=3). DMSO was used as vehicle control. (g) Immunofluorescent staining reveals that UBXN2A shows a juxtanuclear staining characteristic of the ER/Golgi apparatus in the absence of etoposide. However, UBXN2A showed striking punctate cytoplasmic staining when HCT-116 cells incubated with etoposide (50 μM) for 24 h, suggesting a dynamic change in UBXN2A functional linkage networks in response to genotoxic stress. (h, i) The results presented in f indicate significant upregulation of UBXN2A in the cytoplasm with 20 and 50 μM etoposide after 24 h incubation. To verify whether this upregulated endogenous UBXN2A causes displacement of p53, we conducted a series of IP experiments. (h) Cytoplasmic lysates from e treated with no etoposide or 20 and 50 μM were subjected to IP using anti-UBXN2A (h) or anti-p53 (i) antibodies immobilized on magnetic IgG beads. Samples were resolved by 4–20% gradient SDS-PAGE and detected by WB using the indicated antibodies. The HC indicate the heavy immunoglobulin band. WB results indicate that UBXN2A binds to mot-2 in a etoposide dose-dependent manner, while the amount of mot-2 associated with p53 decreases simultaneously
UBXN2A induces p53 nuclear accumulation
Small molecules, p53 c-terminus peptides, and silenced mot-27, 20, 21, 22 abrogate mot-2-p53 complexes, resulting in p53 nuclear localization. Because UBXN2A is capable of releasing p53 from mot-2, we decided to determine whether UBXN2A can lead to p53 nuclear accumulation in a similar mechanism. HCT-116 cells were transiently transfected with different amounts of UBXN2A plasmid. Exogenous UBXN2A was detected dominantly in the cytoplasm fraction (Figure 4a), and, therefore, it is an ideal model to identify the cellular consequences of UBXN2A gain-of-function. After 48 h, nuclear and cytoplasmic fractions were collected, followed by WB analysis (Figures 4a–d). Panel d in Figure 4 shows an increased level of UBXN2A leads to a significant increase in the amount of p53 in the nucleus. We did not observe any changes in p53 abundance in cytoplasmic fractions after an overexpression of UBXN2A, suggesting that nuclear accumulation of p53 is predominantly due to translocation from the cytoplasm into the nucleus (Figures 4a and b), as previously reported in the absence of active mot-2.7, 22 On the basis of the above data, we hypothesized that etoposide-dependent upregulation of UBXN2A should be linked with an increased level of p53 in the nucleus as well. Hence, we examined the stress-induced p53 nuclear localization in HCT-116. WB analysis of cytoplasm (Figure 4e) and nuclear (Figure 4f) fractions revealed that upregulation and nuclear localization of p53 becomes significant at 20 and 50 μM etoposide, which matches exactly with elevated UBXN2A at the same dosages. Similar to several previously reported scenarios,23 co-upregulation of UBXN2A with p53 may be essential to inhibiting mot-2 and delivering maximum p53 into nuclei during the initiation of genotoxic stress. Similar results were obtained in response to single DNA-strand damage produced by UVB irradiation (data not shown). In the next step, we decided to determine whether UBXN2A increases the transcriptional activities of p53. We found overexpression of UBXN2A led to upregulation of BAX protein24 (Figure 4g). In addition, the level of cleaved PARP increased in the presence of overexpressed UBXN2A (Figure 4g). As expected, silencing UBXN2A (Figure 4h) had the opposite effect, strongly reducing the p53 in the nucleus as well as decreasing the level of p53 in cytoplasm fractions (Figures 4h and i). Collectively, these two gain and loss functions of UBXN2A suggested that UBXN2A can increase the level of functional p53.

UBXN2A induces p53 nuclear translocation in HCT-116 colon cancer cells. HCT-116 cells were transfected with the indicated amount of (His)6-TYG-tagged UBXN2A plasmid. Cytoplasmic (a) and nuclear (c) fractions were subjected to WB. The cell expression pattern of tagged UBXN2A proteins mimics the function of endogenous UBXN2A in the cytoplasm upon etoposide exposure illustrated in Figure 3. (b, d) UBXN2A is capable of significantly increasing the nuclear level of p53 in a dose-dependent manner (fold increase, n=11, *P<0.05). (e, f) HCT-116 cells were incubated with different concentrations of etoposide for 24 h. Cytoplasmic and nuclear protein lysates were prepared and subjected to western blot analysis to monitor p53 and UBXN2A protein levels. GAPDH and Orc-2 antibodies were used as cytoplasmic and nuclear markers, respectively. (g) HCT-116 cells were transfected with empty vector or (His)6-TYG-tagged UBXN2A plasmid. After 48 h, total cell lysates were prepared followed by WB using indicated antibodies. GAPDH was used as loading control. (h, i) Two lentiviral-based shRNAs were able to efficiently decrease the level of endogenous UBXN2A in HCT-116 cells treated for 24 h with 50 μM etoposide. WB of the nuclear cell lysates showed a significant decrease in nuclear p53 (i, n=8, *P<0.05). Taken together, these gain- and loss-of-function approaches in HCT-116 cells indicate that UBXN2A facilitates nuclear localization of transcriptionally active p53
UBXN2A preferentially induces apoptosis in colon cancer cell lines
Figures 5a and b show that the expression of GFP-UBXN2A significantly induces apoptosis in HCT-116 and SW48 cells as assessed by Annexin-V. Unlike HCT-116 and SW48 colon cancer cells, GFP-UBXN2A did not induce a significant level of apoptosis in normal colon fibroblast CCD-18Co cells. Staurosporine-treated uninfected cells were used as apoptosis-positive controls (Supplementary Figures 4A and B). It has already been shown when cancer cells are cultured under high-density condition, cell death is often induced as a consequence of nutritional deficiency.25, 26 With this in mind, HCT-116 cells at ∼80% confluence were transfected with GFP shRNA-expressing lentiviral-based vectors (Clone I and II, Figure 4h), followed by a 48-h incubation. As expected, cells transfected with scrambled shRNA show evidence of apoptosis, while expression of shRNAs (Clone I and II) against UBXN2A resulted in a significant reduction in apoptosis (Figure 5C).

UBXN2A overexpression induces apoptosis in colon cancer cell lines but not in normal, non-transformed cells. CCD-18Co, HCT-116, and SW48 were transfected with GFP-empty or GFP-UBXN2A. 48 h after transfection, cells were stained with Annexin V and early apoptosis was determined using flow cytometry as described in Materials and Methods. (a) Representative flow-cytometry analysis data from an Annexin V assay. The histograms show a comparison of the distribution of Annexin V positive cells (M1) after transient transfection of cells with GFP-empty vector or GFP-UBXN2A. The data were gated on GFP-UBXN2A positive cells prior to Annexin V analysis. (b) UBXN2A expression for 48 h significantly increased the number of apoptotic cells in HCT-116 and SW48 cell lines, while CCD-18Co normal colon cells were unaffected. (c) HCT-116 were transfected with scrambled shRNA or shRNA against UBXN2A (clone I and II). 48 h after silencing, superconfluent cultures of HCT-116 were analyzed by Annexin V assay using flow cytometry. Expression of GFP containing shRNA against UBXN2A resulted in 50% less apoptosis in comparison with cells transfected with scrambled shRNA. Values are expressed as mean (±S.E.M.) from three independent experiments (**P<0.01, ***P<0.001)
UBXN2A induces apoptosis and suppresses cell growth through a p53-dependent pathway
To determine whether the UBXN2A-induced apoptosis observed in colon cancer cells is p53-dependent, we next compared the incidence of early apoptosis as measured by Annexin V and cell viability in HCT-116 p53+/+ and p53−/− in the presence and the absence of UBXN2A expression. HCT-116 cells (p53+/+ and p53−/−) were transfected with GFP-UBXN2A, followed by flow-cytometry analysis. The results showed that UBXN2A induces apoptosis in a p53-dependent manner (Figure 6a), as there was not a significant change between GFP-empty and GFP-UBXN2A in p53−/− using the Annexin V apoptosis assay. The cell viability assay showed that GFP-UBXN2A is effective in HCT-116 p53+/+, indicating a need for the presence of p53 to mediate these effects (Figure 6b).

Induction of apoptosis by UBXN2A is p53 dependent and caspase-mediated in colon cancer cell lines. (a, b) HCT-116 (p53+/+) or HCT-116 (p53−/−) cells were transiently transfected with GFP-empty or GFP-UBXN2A for 48 h. An Annexin V apoptosis assay (a) and Prestoblue cell viability (b) assay show that overexpression of UBXN2A leads to a significant increase in cell apoptosis (c) and reduction of cell viability (d) in HCT-116 with WT-p53 (p53+/+). There was not a significant change between GFP-empty and GFP-UBXN2A in p53-KO cells (*P<0.05, **P<0.01). (c) HCT-116 cells were transfected with GFP-empty or GFP-UBXN2A. After 48 h, levels of caspase-3/7 activity (an indicator of apoptosis) in cells expressing GFP-empty or GFP-UBXN2A were measured using the Apo-ONE homogeneous caspase-3/7 assay kit. Results show 2.5-fold increase in caspase-3/7 activity in cells expressing GFP-UBXN2A relative to GFP-empty cells (*P<0.05). (d) Noncancerous HEK293T cells and HCT-116 colon cancer cell lines were transiently transfected with (His)6-TYG-empty or (His)6-TYG-UBXN2A vectors. A caspase-3 colorimetric assay revealed that UBXN2A exclusively activates caspase-3 only in cancer cells (*P<0.05). (e) Cell migration assay. HCT-116 empty-vector and HCT-116 UBXN2A-expressing cells (300 000 cells/well) were suspended in serum-free medium and seeded on cell culture inserts. After 24 h, cells that migrated through the membrane were stained and photographed at × 20 magnification. Cells were counted in five different fields and the average was plotted. (f) Invasion assay. HCT-116 empty vector and HCT-116 UBXN2A-expressing cells (300 000 cells/well) were suspended in serum-free medium and seeded on Matrigel coated inserts. After 24 h, cells that invaded through the Matrigel insert were stained and photographed at × 20 magnification. Cells were counted in five different fields, and the average was plotted. A representative micrograph (e, f) and quantification (g, h) of invaded cells are shown. The data show a significant decrease in migration/invasion of UBXN2A-expressing cells. For all the assays, data represent the mean of three experiments (Mean±S.E.M.) *P<0.05
UBXN2A induces caspase pathways through p53
Because p53-dependent apoptosis is primarily mediated through the activation of the caspase pathway,27 we next monitored the activity of the caspase pathway in the presence of UBXN2A. Measurement of the caspase 3/7 activity showed that overexpression of GFP-tagged UBXN2A significantly increases caspase activity in HCT-116 cells (Figure 6c). Because cleaved caspase-3 mediates apoptosis and contributes to the chemopreventive functions of several agents in colorectal cancer,28 a caspase colorimetric assay was used to detect the activity of caspase-3 in noncancerous HEK-293T and HCT-116 colon cancer cells transiently transfected with (His)6-TYG-tagged UBXN2A or empty vector. HEK293T cells endogenously express mot-2.29 Only a significant twofold increase in caspase-3 activity was observed in HCT-116 cells expressing UBXN2A versus the empty vector (Figure 6d).
UBXN2A blocks colon cancer migration and invasion in vitro
Recent evidence shows that inactivation of p53 triggers the progression of colorectal tumors from the adenoma to the carcinoma stage and enhances cancer invasiveness and lymph node metastasis.30, 31 Involvement of p53 in cell migration and cell invasion encouraged us to examine a possible role for UBXN2A in these two events. Overexpression of (His)6-UBXN2A decreased migration and invasion by nearly 50% in HCT-116 cells when the results were compared with (His)6-empty transfected cells (Figures 6e–h).
UBXN2A and p53 share a common binding site on the mot-2 protein
Because the p53-binding site of mot-2 located within the substrate-binding domain (SBD domain) in the range of 423 to 450 residues,32, 33 we hypothesized that the p53-mot-2 interaction can be competitively disrupted by the binding of UBXN2A to the SBD domain of mot-2. A yeast two-hybrid (Y2H) strategy15 revealed that UBXN2A uses its SEP domain to interact with mot-2, which is sufficient for this interaction (Figures 7a–f). The binding of UBXN2A to the truncated mot-2 (1-506 aa) and SBD (432-674 aa) and its unsuccessful binding to ATP domain (1-437aa) illustrated that the UBXN2A-binding site is located in the range of 438-506 residues where there is a part of p53’s binding site, according to Utomo et al.33 In addition, a set of IP experiments confirmed UBXN2A only binds to the SBD domain (Supplementary Figure 4C) and not the ATP domain (Supplementary Figure 4).

Interaction of the SEP domain of UBXN2A with the p53-binding site (SBD domain) of mot-2 is sufficient to induce apoptosis. (a, b) Schematic diagram of UBXN2A and mot-2 protein domain structures. (c–f) Comprehensive mapping of protein–protein interaction sites by Y2H method using α-galactosidase activity and nutritional selection verified that (i) WT-UBXN2A interacts with WT-mot-2, (ii) the SEP domain of UBXN2A is sufficient to interact with WT-mot-2, and (iii) a partial section of p53 binding site on the SBD domain of mot-2 (aa:438-506) is sufficient for binding to WT-UBXN2A. (g) An apoptosis assay using Annexin V staining followed by flow cytometry analysis confirmed that only the SEP domain of UBXN2A is required to induce apoptosis in HCT-116 cells, similar to full WT-UBXN2A. No increase in apoptosis was seen with the GFP-UBX domain. (h) HCT-116 cells were transfected with GFP-UBXN2A (WT) or its truncated forms (GFP-SEP or GFP-UBX domains). After 48 h, cells were subjected to a crystal violet cell cytotoxicity assay. Counting the remaining colonies showed both WT-UBXN2A and GFP-SEP domains significantly induce cell cytotoxicity (*P<0.05, n=3)
Kaul et al.34 showed that the Mot-2 amino-acid residues 253-282 Mot-2 are critical for its binding to p53. However, these results are in contrast to results reported by ISosfson et al where their IP experiments showed that the association of p53 occurs via the SBD-binding domain of Mot-2 and not the ATP domain.32 Furthermore, a molecular docking study by Utomo et al. confirmed p53 protein bind to substrate-binding domain of Mot-2 located in the C-terminus.33 We found that some of the binding sites of mot-2 to p53, as predicted by bioinformatics33 and in vitro assays,32 were found to be involved in binding of mot-2 to UBXN2A, suggesting that mot-2-p53 and mot-2-UBXN2A binding may be competitive or even mutually exclusive.
Furthermore, a set of Annexin V apoptosis assays and a crystal violet cell cytotoxicity assay verified that the SEP domain of UBXN2A is sufficient to induce apoptosis in HCT-116 cells, while the UBX domain alone failed to induce apoptosis (Figures 7g and h).
UBXN2A overexpression decreases the growth of HCT-116 human colon carcinoma cells xenografted in mice
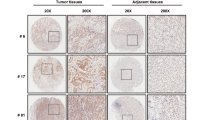
Untransfected HCT-116, as well as UBXN2A, or empty cell suspensions, were injected subcutaneously into the flanks of immunodeficient mice. A portion of the transfected cells was lysed and analyzed by WB to demonstrate that the injected cell lines carried the desired exogenous proteins (Figure 8a). Tumors arose around 16 days after injection and were measured every other day. Our results demonstrate a significant reduction of tumor growth in UBXN2A xenografts compared with empty xenografts and untransfected HCT-116 (Figures 8b–d and Supplementary Figure 5). Immunohistochemical analysis of tumor sections showed that expression of UBXN2A markedly decrease expression of the cell proliferation marker Ki67 (Figure 8e). In addition, a TUNEL assay showed that overexpression of UBXN2A resulted in greater induction of apoptosis compared with tumors expressing empty vector (Figure 8f).

Overexpression of UBXN2A markedly reduces tumor growth in xenograft model. 7 × 106 HCT-116 cells (p53+/+) expressing (His)6-TYG-UBXN2A or an empty vector were subcutaneously injected into nude mice in the lower flank (see Materials and Methods). (a) shows a portion of the transfected HCT-116 cells that was lysed and analyzed by western blotting for expression verification. (b, c) are representative of xenografts experiments with detectable expression of (His)6-tagged UBXN2A proteins. (d) Tumor growth was monitored on the indicated days. Results represent growth rate of tumor volume on the indicated days. Statistical comparisons were done by ANOVA followed by the Bonferroni post hoc test using the GraphPad Prism 6 (**P<0.01, n=3). Each data point is the mean tumor growth on the indicated day, and error bars show the standard error of mean. (e, f) Tumor tissue sections were subjected to immunohistochemical assay for Ki67 expression and TUNEL staining. Alexa-488 labeling of ki67 and 570 red fluorescent labeling for TUNEL assay were captured with the Olympus scanning confocal microscope using the Fluoview 1000 software. Negative controls were processed sections in the absence of primary antibody (e) or before proceeding to the TUNEL staining (f). Scale bar=20 μm. (g, h) The working model of p53 regulation by UBXN2A. Mot-2 protein has been shown to interact with p53 and inhibit its activation in cancerous cells. Upregulation of UBXN2A impedes mot-2-mediated inactivation of p53, which leads to the activation of p53
Discussion
Hsp70 family proteins have been suggested to serve as prognostic and therapeutic markers for cancer cells.2, 14, 35, 36 Mot-2 particularly becomes a tumorigenesis factor in colorectal cancers, and thus is a potential candidate target for cancer therapy.2 We first showed that UBXN2A induces cytotoxicity in a cell type-dependent manner in the presence of wild-type or mutant p53. The level of cytotoxicity (∼50%) induced by UBXN2A in HCT-116, LoVo, and HT-29 was similar to previous reports where cytoplasmic p53 peptides significantly decreased colony formation.21 Interestingly, Gestl and Ann Bottger observed the mutant p53 binds to mot-2 in HT-29 colon cancer cell line.6 Lu et al.7, 9 reported that silencing of mot-2 in hepatocellular carcinoma (HCC) cells with mutant p53 can lead to apoptosis. Therefore, we concluded the induced apoptosis in HT-29 cells in the presence of UBXN2A is mediated through mutant p53 but independent to p53’s transcriptional activation function.
Kaul et al21 reported that overexpressed YFP-tagged p53 carboxyl-terminal peptides bind to mot-2 and lead to translocation of the endogenous p53 to the nucleus in Human osteosarcoma (U2OS) and breast carcinoma (MCF7) cells. However, we obtained no significant cell cytotoxicity in MCF7 cells in the presence of overexpressed UBXN2A. We hypothesized that, like HepG2,7 MCF7 may partially lacked mot-2–p53 interaction in the absence of stress. The cytotoxicity assay in stressed MCF7 cells expressing UBXN2A showed no difference from results obtained without stress in MCF7-expressing UBXN2A. Unsuccessful induction of cell cytotoxicity in certain cell lines (SW480 and MCF7) by UBXN2A suggests that other factors are necessary for the execution of apoptosis induced by UBXN2A.37 To explain the cytotoxic mechanism of UBXN2A, we first investigated UBXN2A partners in HCT-116 colon cancer cells in the presence and the absence of etoposide. This strategy allowed us to find UBXN2A partners within two different protein–protein interaction networks established in cells with and without genotoxic stress.38 We found increased amounts of UBXN2A can bind to mot-2 and subsequently decrease the binding affinity between mot-2 and p53. A similar mechanism has been shown for the ribosomal protein S14 (RPS14) in which RPS14 unties the MDM2-p53 binding, resulting in elevated p53 level and activity.39 These results suggest proteins such as RPS14 and UBXN2A have a protective role for p53 during cancer progression.
It has already been shown that small molecules can bind to the p53-binding site on the substrate binding site of mot-2 and release cytoplasmic p53 for nuclear localization, resulting in a p53-dependent apoptosis.20, 22, 33 The gain and loss of functions indicated that the UBXN2A level in the cytoplasm determines nuclear translocation of p53 in colon cancer cells.
Numerous studies have documented that the activation of caspase-3 is the main executer of apoptotic cell death in several human cancer cell lines, including colon cancers.40, 41 Our data are in agreement with these observations and indicate that, in colon cancer cells, UBXN2A induces the activation of executioner caspases 3/7. Indeed, overexpression of UBXN2A in cells with dysfunctional p53 (HEK293T) and KO p53 (HCT-116 p53−/−) cells had no effect on caspase-3 activity and apoptosis, confirming that UBXN2A induces apoptosis in a p53- and caspase-3 dependent manner. In addition, there was no significant UBXN2A-dependent apoptosis in normal human colon CCD-18Co fibroblasts. Normal levels of mot-2 in noncancerous cells42, 43 and its dominant mitochondrial localization in normal cells,44, 45 as well as the lack of mot-2–p53 interaction in normal cells,7 are likely reasons for ineffective apoptotic function of UBXN2A in normal colon and HEK293T cells. Finally, as previously discussed,46, 47 it is possible that WT-p53 proteins in colon cancer cells are more sensitive to UBXN2A upregulation than normal cells.
Our protein–protein interaction experiments found that the SEP domain of UBXN2A binds to a section of p53’s binding site on mot-2, which contains three binding amino acids used by p53: PRO442, LYS555, and ILE558.32, 33 This finding explains the possible competitive binding mechanism of UBXN2A over p53 when UBXN2A binds to mot-2. A similar competitive binding mechanism has been described for Nutlin-2 where it mimics the three key hydrophobic residues in p53 and releases p53 protein from the MDM2 E3 ubiquitin ligase.48
Besides induction of apoptosis, the UBXN2A-dependent p53 activation led to a significant reduction in cell migration and cell invasion in colon cancer cells. These results suggest that UBXN2A upregulation triggers a complex process involving multiple p53-dependent (both transcriptional and non-transcriptional p53 activities) and possibly p53-independent pathways acting in sequence. UBXN2A binding to p97 complex12, 15 and other UBXN2A partners found in our proteomic work strongly suggests that UBXN2A can initiate multiple biological functions in response to stress.
There is strong evidence that p53 protein levels are regulated by p53-positive regulators that inhibit p53’s negative regulators during the tumor progression.39, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58 While upregulation of p53’s negative regulators, such as mot-2, results in poor prognosis, inhibition of these negative regulators can be a potential home defense mechanism during the initiation and progression of cancer. Collectively, these data suggest that the interaction of UBXN2A with mot-2 enables UBXN2A to enhance p53 activities and suppress tumorigenesis. Because a very high percentage of colorectal cancer expresses high levels of mot-2,2 increased levels of UBXN2A in the cytoplasm could be a unique positive compensatory and/or adaptive mechanism to restore the function of p53 in this cancer. Our results suggest that, by following cell stress, such as that caused by genetic instability in cancer, UBXN2A can further boost p53 nuclear translocation as a home defense mechanism, resulting in an inhibition or slowing down of cancer cell proliferation (Figures 8g and h).
In summary, we showed for the first time that UBXN2A binds to mot-2 and releases sequestrated p53, which leads to p53-dependent apoptosis in cancer cells with high mot-2. Therefore, UBXN2A is a druggable target protein that warrants further investigation in preclinical and clinical trial studies.
Materials and Methods
Antibodies, magnetic beads pull down, and immunoprecipitation
Supplementary Table 1 in the Supplementary Material lists the primary antibodies and the titers used for western blotting, pull down, and immunoprecipitation techniques.
Cell culture, generation of cell lines, chemicals, and drug treatments
The cell lines HEK293, HeLa, HCT-116, MCF7, and SW48, HT-29, SW620, T84, and HUVEC cells were obtained from the ATCC (American Type Culture Collection). Normal colon fibroblasts (CCD-18Co) and SW480 cancer cells were a generous gift of Dr Susanne Talcott (Texas A&M University). The HCT-116 p53+/+ and HCT-116 p53−/− were purchased from GRCF Cell Center, Johns Hopkins University. All cells were grown in the recommended media, supplemented with 10% fetal bovine serum and penicillin/streptomycin (see details in the Supplementary Materials).
Iodixanol gradient analysis, Yeast-two hybrid, and Immunoblot analysis
These techniques were carried out as previously described.15 More details can be found in the Supplementary Materials.
Assessment of apoptosis
Apoptosis in cells was assessed using an Annexin V Apoptosis Detection Kit (BD Pharmingen, San Jose, CA, USA) analyzed by using a BD Accuri C6 flow cytometer according to the manufacturer’s instructions.
Cell viability, caspase assays, and crystal violet cell cytotoxicity assay
Cell viability in cells was measured in cells cultured in 96-well plates using Prestoblue Cell Viability reagent (Life technologies, Grand Island, NY, USA) according to the manufacturer’s protocol. Caspase activity was measured at 24 h using the Apo-ONE Homogeneous Caspase-3/7 Assay (Promega, Madison, WI, USA) following the manufacturer’s instructions. Caspase-3 activity was assessed using the caspase-3 colorimetric assay (BioVision, Milpitas, CA, USA) following the manufacturer’s instructions. For the cytotoxicity assay, we used a technique previously described by Gillies et al and Castro-Garza et al (see Supplementary Materials).
Cell migration and invasion assays
These two techniques were conducted as previously described (see Supplementary Materials).
Xenograft models and immunohistochemistry
The animal study was approved by the Animal Use and Care Committee of the University of South Dakota (study protocol 02-01-12-15C). In vivo and immunohistochemistry experiments were carried out as previously described.59 For details on in vivo techniques, see the Supplementary Materials.
Statistical analysis of data
Unless indicated otherwise, at least three biological repeats were performed for all the cell culture experiments. Statistical values were analyzed with either the Student’s t test or by one-way ANOVA and Tukey multiple comparison post hoc tests, when appropriate. The means were compared considering a P-value of ≤0.05 as a significant difference (mean±S.E.). Data presented in Figures 1d and 8d were analyzed by GraphPad Prism VI software (GraphPad Software, La Jolla, CA, USA), and statistical significance was determined with the two-way ANOVA with the Bonferroni post hoc test. Error bars in Figures 1d and 8d represent S.E.M.
Abbreviations
- cDNA:
-
complementary DNA
- DMSO:
-
dimethyl sulfoxide
- Eto:
-
etoposide
- FBS:
-
fetal bovine serum
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- mot-2:
-
heat-shock protein 70 Mortalin2
- Orc-2:
-
origin recognition complex protein-2
- PBS:
-
phosphate-buffered saline
- RT-PCR:
-
reverse transcription-PCR
- SDS-PAGE:
-
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- shRNA:
-
short hairpin RNA
- TUNEL:
-
TdT-mediated dUTP nick end labeling assay
- WB:
-
western blot
- Y2H:
-
yeast two-hybrid
References
Luo WI, Dizin E, Yoon T, Cowan JA . Kinetic and structural characterization of human mortalin. Protein Expr Purif 2010; 72: 75–81.
Dundas SR, Lawrie LC, Rooney PH, Murray GI . Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J Pathol 2005; 205: 74–81.
Takano S, Wadhwa R, Yoshii Y, Nose T, Kaul SC, Mitsui Y . Elevated levels of mortalin expression in human brain tumors. Exp Cell Res 1997; 237: 38–45.
Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR et al. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J Cancer 2006; 118: 2973–2980.
Ostermeyer AG, Runko E, Winkfield B, Ahn B, Moll UM . Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide. Proc Natl Acad Sci USA 1996; 93: 15190–15194.
Gestl EE, Anne Bottger S . Cytoplasmic sequestration of the tumor suppressor p53 by a heat shock protein 70 family member, mortalin, in human colorectal adenocarcinoma cell lines. Biochem Biophys Res Commun 2012; 423: 411–416.
Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R et al. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ 2011; 6: 1046–1056.
Wadhwa R, Takano S, Robert M, Yoshida A, Nomura H, Reddel RR et al. Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J Biol Chem 1998; 273: 29586–29591.
Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R et al. Induction of mutant p53-dependent apoptosis in human hepatocellular carcinoma by targeting stress protein mortalin. Int J Cancer 2011; 129: 1806–1814.
Iacopetta B . TP53 mutation in colorectal cancer. Hum Mutat 2003; 21: 271–276.
Cheok CF, Verma CS, Baselga J, Lane DP . Translating p53 into the clinic. Nat Rev Clin Oncol 2011; 8: 25–37.
Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ . UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell 2008; 134: 804–816.
Haines DS . p97-containing complexes in proliferation control and cancer: emerging culprits or guilt by association? Genes Cancer 2010; 1: 753–763.
Lee JJ, Kim YM, Jeong J, Bae DS, Lee KJ . Ubiquitin-associated (UBA) domain in human Fas associated factor 1 inhibits tumor formation by promoting Hsp70 degradation. PLoS One 2012; 7: e40361.
Rezvani K, Teng Y, Pan Y, Dani JA, Lindstrom J, Garcia Gras EA et al. UBXD4, a UBX-containing protein, regulates the cell surface number and stability of alpha3-containing nicotinic acetylcholine receptors. J Neurosci 2009; 29: 6883–6896.
Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J . Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci USA 1992; 89: 4495–4499.
Fang L, Kaake RM, Patel VR, Yang Y, Baldi P, Huang L . Mapping the protein interaction network of the human COP9 signalosome complex using a label-free QTAX strategy. Mol Cell Proteomics 2012; 11: 138–147.
Rezvani K, Baalman K, Teng Y, Mee MP, Dawson SP, Wang H et al. Proteasomal degradation of the metabotropic glutamate receptor 1alpha is mediated by Homer-3 via the proteasomal S8 ATPase: Signal transduction and synaptic transmission. J Neurochem 2012; 122: 24–37.
Wadhwa R, Yaguchi T, Hasan MK, Mitsui Y, Reddel RR, Kaul SC . Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp Cell Res 2002; 274: 246–253.
Grover A, Priyandoko D, Gao R, Shandilya A, Widodo N, Bisaria VS et al. Withanone binds to mortalin and abrogates mortalin-p53 complex: computational and experimental evidence. Int J Biochem Cell Biol 2012; 44: 496–504.
Kaul SC, Aida S, Yaguchi T, Kaur K, Wadhwa R . Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J Biol Chem 2005; 280: 39373–39379.
Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R et al. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res 2000; 60: 6818–6821.
Deng WG, Kawashima H, Wu G, Jayachandran G, Xu K, Minna JD et al. Synergistic tumor suppression by coexpression of FUS1 and p53 is associated with down-regulation of murine double minute-2 and activation of the apoptotic protease-activating factor 1-dependent apoptotic pathway in human non-small cell lung cancer cells. Cancer Res 2007; 67: 709–717.
el-Deiry WS . Regulation of p53 downstream genes. Semin Cancer Biol 1998; 8: 345–357.
Yang SY, Sales KM, Fuller BJ, Seifalian AM, Winslet MC . Inducing apoptosis of human colon cancer cells by an IGF-I D domain analogue peptide. Mol Cancer 2008; 7: 17.
Ishii T, Fujishiro M, Masuda M, Okudela K, Kitamura H, Teramoto S et al. Nutritional deficiency affects cell cycle status and viability in A549 cells: role of p27Kip1. Cancer Lett 2004; 213: 99–109.
Schuler M, Green DR . Mechanisms of p53-dependent apoptosis. Biochem Soc Trans 2001; 29 (Pt 6): 684–688.
Kim WH, Yeo M, Kim MS, Chun SB, Shin EC, Park JH et al. Role of caspase-3 in apoptosis of colon cancer cells induced by nonsteroidal anti-inflammatory drugs. Int J Colorectal Dis 2000; 15: 105–111.
Burbulla LF, Schelling C, Kato H, Rapaport D, Woitalla D, Schiesling C et al. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease: functional impact of disease-related variants on mitochondrial homeostasis. Hum Mol Genet 2010; 19: 4437–4452.
Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013; 23: 93–106.
Hwang CI, Matoso A, Corney DC, Flesken-Nikitin A, Korner S, Wang W et al. Wild-type p53 controls cell motility and invasion by dual regulation of MET expression. Proc Natl Acad Sci USA 2011; 108: 14240–14245.
Iosefson O, Azem A . Reconstitution of the mitochondrial Hsp70 (mortalin)-p53 interaction using purified proteins-identification of additional interacting regions. FEBS Lett 2010; 584: 1080–1084.
Utomo DH, Widodo N, Rifa'i M . Identifications small molecules inhibitor of p53-mortalin complex for cancer drug using virtual screening. Bioinformation 2012; 8: 426–429.
Kaul SC, Reddel RR, Mitsui Y, Wadhwa R . An N-terminal region of mot-2 binds to p53 in vitro. Neoplasia 2001; 3: 110–114.
Ando K, Oki E, Zhao Y, Ikawa-Yoshida A, Kitao H, Saeki H et al. Mortalin is a prognostic factor of gastric cancer with normal p53 function. Gastric Cancer 2013; 3 Print ISSN 1436–3291.
Luk JM, Lam CT, Siu AF, Lam BY, Ng IO, Hu MY et al. Proteomic profiling of hepatocellular carcinoma in Chinese cohort reveals heat-shock proteins (Hsp27, Hsp70, GRP78) up-regulation and their associated prognostic values. Proteomics 2006; 6: 1049–1057.
Lopergolo A, Pennati M, Gandellini P, Orlotti NI, Poma P, Daidone MG et al. Apollon gene silencing induces apoptosis in breast cancer cells through p53 stabilisation and caspase-3 activation. Br J Cancer 2009; 100: 739–746.
Pines A, Kelstrup CD, Vrouwe MG, Puigvert JC, Typas D, Misovic B et al. Global phosphoproteome profiling reveals unanticipated networks responsive to cisplatin treatment of embryonic stem cells. Mol Cell Biol 2011; 31: 4964–4977.
Zhou X, Hao Q, Liao J, Zhang Q, Lu H . Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress. Oncogene 2013; 32: 388–396.
Vakifahmetoglu H, Olsson M, Orrenius S, Zhivotovsky B . Functional connection between p53 and caspase-2 is essential for apoptosis induced by DNA damage. Oncogene 2006; 25: 5683–5692.
Noble P, Vyas M, Al-Attar A, Durrant S, Scholefield J, Durrant L . High levels of cleaved caspase-3 in colorectal tumour stroma predict good survival. Br J Cancer 2013; 108: 2097–2105.
Kaul SC, Duncan EL, Englezou A, Takano S, Reddel RR, Mitsui Y et al. Malignant transformation of NIH3T3 cells by overexpression of mot-2 protein. Oncogene 1998; 17: 907–911.
Kaula SC, Reddelb RR, Sugiharac T, Mitsuia Y, Wadhwac R . Inactivation of p53 and life span extension of human diploid fibroblasts by mot-2. FEBS Lett 2000; 474: 159–164.
Wadhwa R, Takano S, Kaur K, Aida S, Yaguchi T, Kaul Z et al. Identification and characterization of molecular interactions between mortalin/mtHsp70 and HSP60. Biochem J 2005; 391 (Pt 2): 185–190.
Wadhwa R, Taira K, Kaul SC . An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell Stress Chaperones 2002; 7: 309–316.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848.
Villalonga-Planells R, Coll-Mulet L, Martinez-Soler F, Castano E, Acebes JJ, Gimenez-Bonafe P et al. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS One 2011; 6: e18588.
Wang S, Zhao Y, Bernard D, Aguilar A, Kumar S . Targeting the MDM2-p53 protein-protein interaction for new cancer therapeutics In: Wendt MD, (ed) Protein-Protein Interactions. Springer: Berlin Heidelberg, 2012; vol. 8 pp 57–79.
Chan AL, Grossman T, Zuckerman V, Campigli Di Giammartino D, Moshel O, Scheffner M et al. c-Abl phosphorylates E6AP and regulates its E3 ubiquitin ligase activity. Biochemistry 2013; 52: 3119–3129.
Zuckerman V, Lenos K, Popowicz GM, Silberman I, Grossman T, Marine JC et al. c-Abl phosphorylates Hdmx and regulates its interaction with p53. J Biol Chem 2009; 284: 4031–4039.
Su CH, Zhao R, Zhang F, Qu C, Chen B, Feng YH et al. 14-3-3sigma exerts tumor-suppressor activity mediated by regulation of COP1 stability. Cancer Res 2011; 71: 884–894.
Zhang Y, Xiong Y, Yarbrough WG . ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998; 92: 725–734.
Zhang Y, Wang J, Yuan Y, Zhang W, Guan W, Wu Z et al. Negative regulation of HDM2 to attenuate p53 degradation by ribosomal protein L26. Nucleic Acids Res 2010; 38: 6544–6554.
Huang Q, Raya A, DeJesus P, Chao SH, Quon KC, Caldwell JS et al. Identification of p53 regulators by genome-wide functional analysis. Proc Natl Acad Sci USA 2004; 101: 3456–3461.
Halaby MJ, Hakem A, Li L, El Ghamrasni S, Venkatesan S, Hande PM et al. Synergistic interaction of Rnf8 and p53 in the protection against genomic instability and tumorigenesis. PLoS Genet 2013; 9: e1003259.
Toiyama Y, Inoue Y, Yasuda H, Saigusa S, Yokoe T, Okugawa Y et al. DPEP1, expressed in the early stages of colon carcinogenesis, affects cancer cell invasiveness. J Gastroenterol 2011; 46: 153–163.
Kudo T, Ikeda M, Nishikawa M, Yang Z, Ohno K, Nakagawa K et al. The RASSF3 candidate tumor suppressor induces apoptosis and G1-S cell-cycle arrest via p53. Cancer Res 2012; 72: 2901–2911.
Hsu TI, Wang MC, Chen SY, Yeh YM, Su WC, Chang WC et al. Sp1 expression regulates lung tumor progression. Oncogene 2012; 31: 3973–3988.
Goyeneche AA, Caron RW, Telleria CM . Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin Cancer Res 2007; 13: 3370–3379.
Acknowledgements
KR is supported by start-up package (Division of Basic Biomedical Sciences, University of South Dakota), PQCD Research Center (Sanford School of Medicine of the University of South Dakota), TRACK to Success Grant/RFP-FY12 (Office of Research and Sponsored Programs, University of South Dakota) and the National Institute of General Medical Sciences of the National Institutes of Health under award number 5P20GM103548 (Miskimins). We thank Dr. Zhenkun Lou (Division of Oncology Research, Department of Oncology, Mayo Clinic, Rochester) for critical input. We thank Dr. Said Soleman in histopathology core and Dr. Kathleen Eyster in the Genomics Core at USD. We thank Dr. Robin Miskimins (Division of Basic Biomedical Sciences, University of South Dakota) for critical reading and editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by D Aberdam
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Sane, S., Abdullah, A., Boudreau, D. et al. Ubiquitin-like (UBX)-domain-containing protein, UBXN2A, promotes cell death by interfering with the p53-Mortalin interactions in colon cancer cells. Cell Death Dis 5, e1118 (2014). https://doi.org/10.1038/cddis.2014.100
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.100
Keywords
This article is cited by
-
UBXN2A suppresses the Rictor-mTORC2 signaling pathway, an established tumorigenic pathway in human colorectal cancer
Oncogene (2023)
-
Pre-clinical safety and therapeutic efficacy of a plant-based alkaloid in a human colon cancer xenograft model
Cell Death Discovery (2022)
-
Epigenetic silencing of UBXN8 contributes to leukemogenesis in t(8;21) acute myeloid leukemia
Experimental & Molecular Medicine (2021)
-
P53 functional abnormality in mesenchymal stem cells promotes osteosarcoma development
Cell Death & Disease (2016)
-
Structural studies of UBXN2A and mortalin interaction and the putative role of silenced UBXN2A in preventing response to chemotherapy
Cell Stress and Chaperones (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}