
Abstract
24(S)-hydroxycholesterol (24S-OHC), which is enzymatically produced in the brain, has an important role in maintaining brain cholesterol homeostasis. We have previously reported that 24S-OHC induces necroptosis in human neuroblastoma SH-SY5Y cells. In the present study, we investigated the mechanisms by which 24S-OHC-induced cell death occurs. We found that lipid droplets formed at the early stages in the treatment of SH-SY5Y cells with 24S-OHC. These lipid droplets could be almost completely eliminated by treatment with a specific inhibitor or by siRNA knockdown of acyl-CoA:cholesterol acyltransferase 1 (ACAT1). In association with disappearance of lipid droplets, cell viability was recovered by treatment with the inhibitor or siRNA for ACAT1. Using gas chromatography–mass spectrometry, we confirmed that 24S-OHC-treated cells exhibited accumulation of 24S-OHC esters but not of cholesteryl esters and confirmed that accumulation of 24S-OHC esters was reduced when ACAT1 was inhibited. 24S-OHC induced apoptosis in T-lymphoma Jurkat cells, which endogenously expressed caspase-8, but did not induce apoptosis in SH-SY5Y cells, which expressed no caspase-8. In Jurkat cells treated with the pan-caspase inhibitor ZVAD and in caspase-8-deficient Jurkat cells, 24S-OHC was found to induce caspase-independent cell death, and this was partially but significantly inhibited by Necrostatin-1. Similarly, knockdown of receptor-interacting protein kinase 3, which is one of the essential kinases for necroptosis, significantly suppressed 24S-OHC-induced cell death in Jurkat cells treated with ZVAD. These results suggest that 24S-OHC can induce apoptosis or necroptosis, which of the two is induced being determined by caspase activity. Regardless of the presence or absence of ZVAD, 24S-OHC treatment induced the formation of lipid droplets and cell death in Jurkat cells, and this was suppressed by treatment with ACAT1 inhibitor. Collectively, these results suggest that it is ACAT1-catalyzed 24S-OHC esterification and the resulting lipid droplet formation that is the initial key event which is responsible for 24S-OHC-induced cell death.
Similar content being viewed by others


Main
Cell death has traditionally been classified as being either apoptosis or necrosis, description in textbooks implying that these involve opposed mechanisms. Although necrosis has generally been considered to be cell death that is accidental and passive, there is growing evidence that some necrosis is in fact programmed cell death,1, 2 the term ‘necroptosis’.3 Necroptosis is involved in many physiological and pathological processes, such as viral infection,4 neuronal excitotoxicity,5 and ischemic injury.3, 6 Necroptosis is defined as cell death that is necrosis-like, caspase-independent, and mediated through a pathway depending on the receptor-interacting protein kinase 1 (RIPK1)–RIPK3 complex. Necroptosis is further defined as being inhibited by Necrostatin-1 (Nec-1), a specific inhibitor of RIPK1 kinase activity.3 Interestingly, necroptosis can be induced by apoptosis-inducing ligands such as Fas ligand or tumor necrosis factor α (TNFα). These ligands usually bind with so-called death receptors (such as Fas or TNF receptor), resulting in activation of apoptotic machineries. Conversely, it has been found that in some cell lines the presence of caspase inhibitor blocks apoptosis and unveils caspase-independent necroptosis.7, 8 Activation of death receptor can therefore result in either apoptosis or necroptosis, caspase-8 being known to act in a key role for determining which form of cell death will occur.9
We have previously reported that high concentrations of 24(S)-hydroxycholesterol (24S-OHC) induce necroptosis in neuronal cells.10 As the blood–brain barrier prevents cholesterol translocation between the brain and the circulation, brain cholesterol is locally synthesized, and its levels are not affected by dietary cholesterol.11 To maintain a steady-state level of cholesterol in the brain, the neuronal enzyme cholesterol 24-hydroxylase (CYP46A1) converts excess amounts of cholesterol into 24S-OHC, which readily crosses the blood–brain barrier.12 It has been reported that free 24S-OHC is present at concentrations of up to 30 μM in the human brain.13 Several lines of evidence suggest that 24S-OHC is linked to the development of Alzheimer’s disease (AD).14, 15 Higher concentrations of 24S-OHC have been detected in plasma and cerebrospinal fluid of patients with AD or mild cognitive impairment than is present in healthy subjects.16, 17 Selective expression of CYP46A1 around neuritic plaques has also been reported.18 Furthermore, as 24S-OHC has been shown to possess potent neurotoxicity, it is presumed to be involved in the etiology of neurodegenerative disease.19 We have shown that 24S-OHC induces cell death in human neuroblastoma SH-SY5Y cells and rat primary cortical neuronal cells without any of the features typical of apoptosis.10 Instead, we found that Nec-1 or siRNA knockdown of RIPK1 significantly suppressed 24S-OHC-induced cell death, demonstrating that necroptosis may account for 24S-OHC-induced neuronal cell death. We further showed in our previous study that the neuronal cells used in that study did not express caspase-8, suggesting that 24S-OHC induces necroptosis specifically in neuronal cells because of the absence of caspase-8; however, the molecular mechanisms responsible for induction of cell death still remained unclear.
In the present study, we used not only SH-SY5Y cells but also human T lymphoma Jurkat cells to further investigate the mechanisms responsible for 24S-OHC-induced cell death. We found that cytosolic lipid droplets formed in the early stages in cells treated with 24S-OHC. Lipid droplets are unique intracellular organelles that store neutral lipids for membrane synthesis and energy supply.20 As accumulation of free cholesterol can be toxic to cells, free cholesterol is converted to cholesteryl esters, which mainly exist as lipid droplets.21 Acyl-CoA:cholesterol acyltransferase (ACAT) catalyzes the esterification of free cholesterol to cholesteryl esters in the endoplasmic reticulum.22 Two ACAT isoenzymes, ACAT1 and ACAT2 have been identified. ACAT1 is the main isoenzyme in the brain.22, 23 Here, we demonstrate that the esterified form of 24S-OHC is accumulated in 24S-OHC-treated cells. We also found that 24S-OHC induced either apoptosis or necroptosis, which of the two was induced being determined by caspase activity. We conclude that ACAT1-mediated esterification of 24S-OHC and formation of lipid droplets have important roles in 24S-OHC-induced apoptosis and necroptosis.
Results
Lipid droplet formation mediated by ACAT activity was involved in 24S-OHC-induced cell death in SH-SY5Y cells
Following exposure of SH-SY5Y cells to 24S-OHC for about 6 h, it was noted during monitoring of cell morphology that lipid droplet-like structures had formed. We therefore used the fluorescent probe Nile red24 to examine whether these structures were neutral lipid-enriched droplets. Cells treated with 24S-OHC for 6 h had cytoplasmic structures that stained positive with Nile red, suggesting that 24S-OHC treatment had induced formation of droplets enriched with neutral lipids (Figure 1a upper panel). Pretreatment of cells with F12511, an inhibitor of both ACAT1 and ACAT2,25 suppressed 24S-OHC-induced lipid droplet formation (Figure 1a lower panel). To evaluate whether 24S-OHC-induced lipid droplet formation was correlated with cell death, we examined the effect of F12511 on 24S-OHC-induced cell death by lactate dehydrogenase (LDH) activity assay. The results showed that F12511 almost completely suppressed 24S-OHC-induced cell death (Figure 1b).

Lipid droplet formation by ACAT1 is involved in SH-SY5Y cell death induced by 24S-OHC. (a and b) SH-SY5Y cells were pretreated with or without 5 μM F12511 for 15 min and were then treated with 50 μM 24S-OHC for 6 h (a) or 24 h (b). (a) Cells were subjected to Nile red staining. Representative images are shown. Bar, 20 μm. (b) Cell viability was measured by LDH activity assay. (c and d) SH-SY5Y cells were pretreated with or without 2 μM K-604 for 15 min and were then treated with 50 μM 24S-OHC for 6 h (c) or 24 h (d). (c) Cells were stained with Nile red. Representative images are shown. Bar, 20 μm. (d) Cell viability was measured by LDH activity assay **P<0.01, when compared with cells treated with 24S-OHC alone
Selective inhibitor or siRNA knockdown of ACAT1 suppressed 24S-OHC-induced cell death in SH-SY5Y cells
As it has been reported that ACAT1 is the main isoenzyme in the brain, and real-time PCR analysis revealed that ACAT1 mRNA but not ACAT2 mRNA was expressed in SH-SY5Y cells (Supplementary Figure S1), we tested the effects of the selective ACAT1 inhibitor K-60426 on 24S-OHC-induced lipid droplet formation and cell death. Nile red staining showed that, as was the case with F12511, K-604 suppressed formation of lipid droplets in cells treated with 24S-OHC for 6 h (Figure 1c). K-604 also suppressed 24S-OHC-induced cell death (Figure 1d). Furthermore, we performed siRNA knockdown of ACAT1 in SH-SY5Y cells and confirmed a marked decline in constitutive ACAT1 protein levels (Figures 2a and b). Under these conditions, 24S-OHC-induced lipid droplet formation was suppressed (Figure 2c) and 24S-OHC-induced cell death was significantly inhibited, similar to that which was observed with F12511 treatment (Figure 2d). Taken together, these results suggest that ACAT1 is involved in neuronal cell death induced by 24S-OHC.

Knockdown of ACAT1 suppressed 24S-OHC-induced cell death in SH-SY5Y cells. (a and b) SH-SY5Y cells were transfected with ACAT1 (siACAT1) or negative control (NC) siRNA oligo for 48 h. (a) Whole cell lysates were immunoblotted with appropriate antibodies as indicated. (b) Relative expression levels of ACAT1 are shown. *P<0.05, **P<0.01 versus NC siRNA. (c and d) The cells were challenged with 50 μM 24S-OHC for 6 h (c) or 24 h (d). (c) Cells were stained with Nile red. Representative images are shown. Bar, 20 μm. (d) Cell viability was measured by LDH activity assay. **P<0.01, when compared with NC siRNA+24S-OHC
The esterified form of 24S-OHC accumulated in 24S-OHC-treated SH-SY5Y cells
As lipid droplet formation was involved in 24S-OHC-induced cell death, we analyzed lipid components by using gas chromatography–mass spectrometry (GC–MS) in SH-SY5Y cells treated with 24S-OHC for 6 h. A saponification step was used to remove fatty acids from sterol esters and derivatize sterol molecules for detection by GC–MS. Free cholesterol (before saponification) and total cholesterol (after saponification) were both measured, the difference between these values being taken to be the amount of cholesterol esters. The same measurement was performed for 24S-OHC and for 24S-OHC esters. The results showed that neither cholesterol content nor cholesteryl ester content was affected by 24S-OHC treatment (Figure 3a). 24S-OHC was not detected in control cells, and accumulation of 24S-OHC and 24S-OHC esters was observed in 24S-OHC-treated cells (Figure 3b), it being possible to prevent this accumulation of 24S-OHC esters by treatment with F12511 (Figure 3b). In SH-SY5Y cells transfected with ACAT1 siRNA, cholesterol levels remained unaffected (Supplementary Figure S2A), but the production of 24S-OHC esters was inhibited (Supplementary Figure S2B). Together, these data indicate that ACAT1 esterified 24S-OHC but not cholesterol and that this caused formation of lipid droplets in 24S-OHC-treated SH-SY5Y cells.

24S-OHC treatment induced formation of esterified 24S-OHC but not of cholesterol. (a and b) SH-SY5Y cells were pretreated with and without 5 μM F12511 for 15 min and were then treated with 50 μM 24S-OHC for 6 h. The cells were subjected to lipid extraction and measurement of cholesterol (a) and 24S-OHC (b) by using GC–MS. N.D., not detected. *P<0.05
To further confirm the production of the esterified form of 24S-OHC, cell samples without saponification were analyzed by reverse-phase high-performance liquid chromatography (HPLC). As compared with the vehicle control condition (Figure 4a), significant increase was observed at four peaks for 24S-OHC-treated cells (Figure 4b). These four peaks disappeared when cells were co-treated with 24S-OHC and F12511 (Figure 4c). During this HPLC analysis, free 24S-OHC and free cholesterol were detected at 2.5 and 4 min, respectively, and cholesteryl esters were detected at retention times >45 min (data not shown). We then collected these four unidentified peaks as four fractions and analyzed by GC–MS with or without saponification. The results show that all four factions contained the esterified form of 24S-OHC (Figure 4d) but not cholesterol (Figure 4e). Collectively, these results suggest that 24S-OHC treatment induced accumulation of various kinds of 24S-OHC esters but not of cholesteryl esters.

Various kinds of 24S-OHC esters accumulated in 24S-OHC-treated SH-SY5Y cells. SH-SY5Y cells were treated with (a) vehicle or (b, d and e) 50 μM 24S-OHC or (c) 50 μM 24S-OHC+5 μM F12511 for 6 h as in Figure 1. (a–c) Lipid extracts were subjected to HPLC analysis. (d and e) The fractions collected by HPLC from panel (b) were further subjected to GC–MS analysis with or without saponification to measure 24S-OHC (d) or cholesterol (e) content
24S-OHC induced necroptosis in caspase-inhibited Jurkat cells
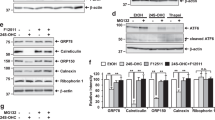
To investigate the effects of caspase-8 expression on induction of cell death by 24S-OHC, we used human T lymphoma Jurkat cells that endogenously expressed caspase-8. We first examined the effect of the pan-caspase inhibitor ZVAD-fmk (ZVAD) on 24S-OHC-induced cell death in Jurkat cells. Jurkat cells were treated with 24S-OHC in the presence or absence of ZVAD for 24 h. Cell viability was measured by WST-8 assay. 24S-OHC treatment significantly decreased cell viability regardless of whether ZVAD was present (Figure 5a). We also used immunoblotting to evaluate the activation of caspase-8 and caspase-3 in cells treated with 24S-OHC for 6 h. The results show that cleaved forms of caspase-8 and caspase-3 were observed in cells treated with 24S-OHC alone (Figure 5b), indicating that 24S-OHC induced apoptosis in Jurkat cells. Interestingly, activation of caspase-8 and caspase-3 was inhibited by ZVAD (Figure 5b), but ZVAD treatment did not exhibit any protective effect on 24S-OHC-induced cell death (Figure 5a). In control experiments, TNFα and cycloheximide (CHX) induced activation of caspase-8 and caspase-3 in Jurkat cells (data not shown), and activation of each was inhibited by ZVAD (Figure 5b). As active caspase-8 has been demonstrated to cleave and inactivate RIPK1, thereby regulating initiation of necroptosis,2 we examined the expression level of RIPK1. As expected, 24S-OHC treatment reduced RIPK1 protein levels, this reduction being suppressed by co-treatment with ZVAD (Figure 5b). These results suggest that in the presence of ZVAD 24S-OHC induces caspase-independent cell death in Jurkat cells. To evaluate whether necroptosis accounts for the caspase-independent cell death induced by 24S-OHC, we examined the effect of the specific necroptosis inhibitor Nec-1 on cell death induced by 24S-OHC in the presence of ZVAD. The results showed that Nec-1 significantly suppressed cell death induced by 24S-OHC in the presence of ZVAD (Figure 5c). We further examined the role of caspase-8 in 24S-OHC-induced cell death using caspase-8-deficient Jurkat cells. 24S-OHC treatment significantly decreased cell viability (Figure 5d), and this decrease in viability was significantly suppressed by Nec-1, suggesting that 24S-OHC induced necroptosis in caspase-8-deficient Jurkat cells. To further confirm the involvement of necroptosis in 24S-OHC-induced cell death, we evaluated the effect of RIPK3 knockdown in Jurkat cells. In the presence of ZVAD, RIPK3 siRNA significantly suppressed cell death induced by 24S-OHC (Figure 5e). Together, these results indicate that 24S-OHC induces either apoptosis or necroptosis in Jurkat cells, which of the two forms of cell death is induced being dependent on caspase activities, especially caspase-8 activity.

24S-OHC induced apoptosis and necroptosis in Jurkat cells. (a) Jurkat cells were pretreated with 20 μM ZVAD for 1 h and were then treated with 50 μM 24S-OHC for 24 h. Cell viability was measured by WST-8 assay. N.S., not significant. **P<0.01, when compared with cells without ZVAD and 24S-OHC; ††P<0.01, when compared with ZVAD-treated cells. (b) Jurkat cells were pretreated with 20 μM ZVAD for 1 h and were then incubated with 50 μM 24S-OHC for 6 h. As a control, cells were treated with 100 ng/ml CHX and 100 μg/ml TNFα. Whole cell lysates were immunoblotted with appropriate antibodies as indicated. (c) Jurkat cells were pretreated with 20 μM ZVAD and 100 μM Nec-1 for 1 h and were then treated with 50 μM 24S-OHC for 16 h. Cell viability was measured by flow cytometry using propidium iodide. *P<0.05. (d) Caspase-8-deficient Jurkat cells were pretreated with 100 μM Nec-1 for 1 h and were then treated with 50 μM 24S-OHC for 16 h. Cell viability was measured by flow cytometry using propidium iodide. **P<0.01. (e) Jurkat cells were transfected with RIPK3 (siRIPK3) or negative control (NC) siRNA oligo for 72 h. The cells were pretreated with 20 μM ZVAD for 1 h and were then treated with 50 μM 24S-OHC for 24 h. Cell viability was measured by WST-8 assay. **P<0.01 when compared with NC siRNA+24S-OHC+ZVAD
Lipid droplet formation mediated by ACAT1 activity was involved in 24S-OHC-induced apoptosis and necroptosis in Jurkat cells
To investigate whether ACAT1 is involved in 24S-OHC-induced apoptosis and necroptosis, we carried out a series of experiments on Jurkat cells in the presence and absence of ZVAD. Real-time PCR analysis revealed that ACAT1 mRNA but not ACAT2 mRNA was expressed in Jurkat cells as well as in SH-SY5Y cells (Supplementary Figure S1), suggesting that ACAT1 is the main isoenzyme in Jurkat cells. Nile red staining showed that in the absence of ZVAD 24S-OHC treatment induced the formation of lipid droplets (Figure 6a). It was further observed that formation of these Nile red-positive lipid droplets could be reduced by treatment with K-604 (Figure 6a) or F12511 (Supplementary Figure S3A). We next evaluated whether 24S-OHC-induced lipid droplet formation was observed under necroptosis-inducing condition (ZVAD+). The results showed that 24S-OHC treatment induced lipid droplet formation and that this could be suppressed by inhibition of ACAT1 activity (Figure 6b and Supplementary Figure S3B). These results revealed that, regardless of the presence or absence of ZVAD, 24S-OHC treatment induces ACAT1-mediated esterification of sterols and lipid droplet formation in Jurkat cells.

24S-OHC induced lipid droplet formation in Jurkat cells treated with or without ZVAD. (a and b) Jurkat cells were pretreated with or without 2 μM K-604 for 15 min and were then treated with 50 μM 24S-OHC for 6 h in the absence (a) or presence (b) of ZVAD. Cells were stained with Nile red. Representative images are shown. Bar, 20 μm
We then evaluated the effect of ACAT inhibitor on 24S-OHC-induced cell death in Jurkat cells. Under both apoptosis-inducing condition (ZVAD−) and necroptosis-inducing condition (ZVAD+), F12511 inhibited 24S-OHC-induced cell death (Figure 7a). The selective ACAT1 inhibitor K-604 also suppressed both types of cell death (Figure 7b). These results suggest that ACAT1-mediated lipid droplet formation is a common early-stage event that occurs in 24S-OHC-induced cell death regardless of whether that cell death is caspase dependent or caspase independent.

ACAT inhibitor suppressed 24S-OHC-induced cell death in Jurkat cells treated with or without ZVAD. (a and b) Jurkat cells were pretreated with or without 5 μM F12511 (a) or 2 μM K-604 (b) for 15 min and were then treated with 50 μM 24S-OHC for 24 h in the absence or presence of ZVAD. Cell viability was measured by WST-8 assay. **P<0.01, when compared with the cells treated with 24S-OHC in the absence of ACAT inhibitor
Discussion
Lipid droplets have significant biological roles in many cell types.20 Their hydrophobic cores store neutral lipids such as triglyceride and cholesteryl esters for supplying metabolic energy and membrane components. Lipid droplets also serve in protein storage and degradation. It had generally been thought that ACAT-mediated esterification of cholesterol with fatty acid results in the formation of cholesteryl ester-enriched lipid droplets, which serve in a protective role by preventing cytotoxic accumulation of free cholesterol.21 It has been reported that an unsaturated fatty acid, oleic acid, promotes the formation of triglyceride-enriched lipid droplets while barely inducing apoptosis; and that a saturated fatty acid, palmitic acid, poorly forms lipid droplets while significantly inducing apoptosis.27 Contrary to such conventional findings, our present data showed that formation of lipid droplets upon 24S-OHC treatment correlates with induction of cell death. Although it was not clear in the present study whether the esterified form of 24S-OHC itself was cytotoxic or whether it was the formation of lipid droplets that triggered cell death signaling, we showed that esterification of 24S-OHC by action of ACAT1 was definitely responsible for initiating cell death signaling. It is possible to hypothesize that certain proteins which suppress cell death, for example, cellular FLICE inhibitory protein28 or cellular inhibitor of apoptosis,29 might collect within lipid droplets. It is also possible that lipid droplets may provide an intracellular platform permitting association of certain proteins that promote cell death signaling.
Interestingly, employment of inhibitor or siRNA knockdown of ACAT1 almost completely inhibited the cytotoxic effects of 24S-OHC in both SH-SY5Y and Jurkat cells. ACAT1 esterifies various oxysterols such as 7-ketocholesterol and 25-OHC.30, 31 It is therefore plausible that 24S-OHC might also be available as a substrate for ACAT1. Indeed, GC–MS analysis revealed that there was accumulation of 24S-OHC esters but not cholesteryl esters in 24S-OHC-treated cells. In macrophages, ACAT promotes 7-ketocholesterol-induced caspase-3-dependent apoptosis via accumulation of 7-ketocholesterol esters.32 In mast cells, ACAT is implicated in 24(S),25-epoxycholesterol-induced apoptosis.33 In contrast, a plant sitosterol, which was poorly esterified by ACAT, was found to trigger caspase-independent necroptosis in macrophages.34 We have previously shown that 7-ketocholesterol, 7α-OHC, 7β-OHC, and 22R-OHC induce SH-SY5Y cell death and that this was not prevented by Nec-1.10 In the present study, we further examined whether ACAT1 was involved in cell death induced by these oxysterols, as a result of which we found that Nile red staining showed that these oxysterols did not induce lipid droplet formation (Supplementary Figure S4A), and we found that ACAT inhibitor did not suppress cell death induced by these oxysterols (Supplementary Figure S4B). Together, the current data suggest that differences in the structures of oxysterols and their esterified forms are important factors for determining the pathway by which cell death occurs. As HPLC analysis showed that at least four kinds of 24S-OHC esters accumulated in 24S-OHC-treated cells, studies to identify which 24S-OHC esters are responsible for the induction of cell death may provide further insight. Because standards for each 24S-OHC ester are not commercially available, further experiments will be required to characterize and quantify the individual 24S-OHC esters. It should be noted that the amount of free 24S-OHC in cells co-treated with 24S-OHC and F12511 was lower than that in cells treated with 24S-OHC alone (Figure 3b). There is a possibility that F12511 may affect influx and/or efflux of 24S-OHC. However, ACAT1 inhibition almost completely inhibited esterification of 24S-OHC, and this was accompanied by a corresponding suppression of 24S-OHC-induced cell death. We therefore concluded that it is the esterification of 24S-OHC by ACAT1 and the resulting lipid droplet formation that triggers cell death signaling. It has been reported that ACAT1 gene ablation or ACAT inhibitor ameliorates amyloid pathology in AD mice.23 In the light of our present results, it is thought that ACAT1 inhibitors may hold promise for treating neurodegenerative diseases due also to their presumed role in cell death inhibition.
Our results show that in Jurkat cells ACAT inhibition suppressed both 24S-OHC-induced formation of lipid droplets and cell death regardless of whether or not caspase inhibitor was present (Figure 6 and Figure 7). These results suggest that esterification of 24S-OHC is an early-stage event in the cell death machinery and more particularly that it occurs upstream of the switching point for apoptosis and necroptosis. As ACAT1 inhibition also suppresses 24S-OHC-induced lipid droplet formation and cell death in SH-SY5Y cells, we propose that esterification of 24S-OHC by ACAT1 is the common mechanism responsible for 24S-OHC-induced cell death in different cell types. It has been reported that there are two key pathways for apoptosis signaling, one being the extrinsic pathway which is initiated through cell surface death receptors and which results in caspase-8 activation, and the other being the intrinsic pathway which is initiated through a mitochondrion-centered control mechanism.35 Drug-induced caspase-8 activation in death receptor-independent apoptosis has been reported in several types of cells.36, 37 Our results in Jurkat cells showed that 24S-OHC induced activation of caspase-8, resulting in activation of apoptotic machineries. The possibility that certain death receptor was involved in 24S-OHC-induced cell death should be addressed in the near future. It has been known that autocrine secretion of death ligands such as TNFα upon stimulation with a cytotoxic compound induces apoptosis.38 We therefore evaluated TNFα secretion from 24S-OHC-treated SH-SY5Y cells and found no secretion of TNFα into the culture medium after 6 and 12 h treatment (data not shown). Further experiments will be required to address whether other ligands might be secreted to stimulate cell death receptors. In the cells treated with caspase inhibitor or in caspase-8-deficient cells, 24S-OHC still caused cell death, and this was partially but significantly inhibited by Nec-1. Furthermore, siRNA for RIPK3 strongly suppressed 24S-OHC-induce cell death. These results suggest that it is mainly necroptosis that is responsible for 24S-OHC-induced cell death in the absence of caspase-8 activity.
Here we have presented a novel mechanism of cell death that includes both apoptosis and necroptosis induced by an oxidation product of cholesterol. The esterification of oxysterol by ACAT1 is a common initial event in both of these cell death modalities. From the clinical viewpoint, it is noteworthy that 24S-OHC is predominantly produced in the brain and may be responsible for neuronal cell death in neurodegenerative diseases. We have therefore provided the basis for potential therapeutic strategies in which ACAT1 inhibitors are used to treat neurodegenerative diseases.
Materials and Methods
Materials
ACAT inhibitor F12511 and anti-ACAT1 antibody (DM102) were the generous gifts of Dr. Ta-Yuan Chang (Dartmouth Medical School, Hanover, NH, USA). ACAT1 inhibitor K-604 was kindly provided by Kowa Company, Ltd (Tokyo, Japan). Dulbecco’s modified Eagle’s medium/nutrient mixture Ham’s F-12 (DMEM/F-12) was obtained from Invitrogen (Carlsbad, CA, USA). RPMI-1640 was obtained from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum was from Hyclone (Thermo Scientific, Logan, UT, USA). 24S-OHC was from Biomol International (Plymouth Meeting, PA, USA), this having been dissolved in ethanol and stored at −20 °C. Pan-caspase inhibitor ZVAD was from Medical and Biological Laboratories (Nagoya, Japan). TNFα, CHX, and N,O-bis(trimethylsilyl)-trifluoroacetamide were purchased from Wako (Osaka, Japan). Nec-1 and Nile red were from Sigma-Aldrich, these, respectively, being dissolved in DMSO and stored at −20 °C under light-shielded conditions. The following antibodies were from commercial sources: caspase-8 (No. M058-3), Medical and Biological Laboratories; caspase-3 (No. 9662), Cell Signaling (Beverly, MA, USA); RIPK1 (No. 551041), BD Biosciences (San Jose, CA, USA); and β-actin (No. A5441), Sigma. All other chemicals, of analytical grade, were obtained from Sigma-Aldrich or Wako.
Cell lines and cell culture
SH-SY5Y human neuroblastoma cells, Jurkat human T cell leukemia cells, and caspase-8-deficient Jurkat cells (clone I 9.2) were purchased from American Type Culture Collection (Manassas, VA, USA). Cells were routinely maintained in DMEM/F-12 (SH-SY5Y) or RPMI-1640 (Jurkat) medium containing 10% heat-inactivated fetal bovine serum and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin; Invitrogen). For treatment, SH-SY5Y cells and Jurkat cells were seeded at a density of 3.0 × 105 cells/ml. Cells were grown at 37 °C under an atmosphere of 95% air and 5% CO2.
Cell treatment
To examine the toxicity of 24S-OHC in SH-SY5Y cells and Jurkat cells, cells were treated with 50 μM 24S-OHC for the indicated periods. To evaluate the effects of ZVAD or ACAT inhibitor, the cells were pretreated with 20 μM ZVAD for 1 h or with 5 μM F12511 or 2 μM K-604 for 15 min before further treatments.
Determination of cell viability
For determination of cell viability in Jurkat cells, WST-8 assay was performed using Cell Counting Kit-8 according to the manufacturer’s instructions (Dojindo, Kumamoto, Japan). Flow cytometry using propidium iodide staining was conducted as described previously.10 Briefly, control and treated cells were stained with propidium iodide, followed by analysis using a BD FACSAriaTM II cell sorter (Becton, Dickinson and Company, Franklin Lakes, NJ USA) with a 488 nm argon laser. For determination of cell viability in SH-SY5Y cells, LDH activity assay was performed as described previously.39 Data are expressed as percentage of total LDH activity, after subtraction of background determined from the serum medium alone.
Immunoblotting
Whole cell lysates were prepared in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 2 mM EDTA) to which protease inhibitor cocktail (nacalai tesque, Kyoto, Japan) had been added. Nuclei and unlysed cellular debris were removed by centrifugation at 16 000 × g for 10 min. The protein samples were subjected to SDS-PAGE and immunoblotting by using appropriate antibodies.
Nile red staining
To confirm the presence of lipid droplets in 24S-OHC-treated cells, cells were fixed by 4% paraformaldehyde phosphate buffer solution at room temperature for 30 min. After washing out fixing solution, the cells were incubated with PBS, including 50 ng/ml Nile red for 5 min in the dark. Fluorescence images of Nile red staining were obtained using a fluorescence microscope (OLYMPUS IX71, Tokyo, Japan).
Knockdown of ACAT1 and RIPK3 by small interfering RNA
Stealth siRNA targeting human ACAT1 (5′-UAGAACUCCAAUCUGGAAGAUCAUG-3′) and Stealth RNAi negative control (NC) were obtained from Invitrogen. The siRNA targeting human RIPK34 was obtained from Hokkaido System Science (Sapporo, Japan). SH-SY5Y cells grown in six-well plates were transfected with siRNA, at a concentration of 50 or 75 pmol/well by Lipofectamine RNAiMAX (Invitrogen) for 48 h (ACAT1) or by 60 pmol/well by HiPerFect (Qiagen, Hilden, Germany) for 72 h (RIPK3) before further experiments. To confirm knockdown efficiency for ACAT1, whole cell lysates were analyzed by immunoblotting using DM102 antibody.
Cholesterol and 24S-OHC measurement by GC–MS
The levels of cholesterol and 24S-OHC were measured by GC–MS using a previously reported method.40 Briefly, the cells in 1 ml PBS were divided equally into two portions, one for total sterol (with saponification) and the other for free sterol (without saponification). Each cell suspension was added to 500 μl of 100 nM internal standards and 100 μM butylated hydroxytoluene in methanol. Saponification was carried out by adding 500 μl of 1 M KOH in methanol followed by incubation on a shaker at 40 °C for 30 min. Lipids were extracted by adding 2 ml of 10% acetic acid in water and 5 ml of chloroform/ethyl acetate (4:1, v/v), followed by vortexing for 1 min and centrifugation at 3000 × g for 10 min at 4 °C. After removal of the water layer, the chloroform/ethyl acetate layer was evaporated to dryness under nitrogen. As silylating agent, 30 μl of N,O-bis(trimethylsilyl)-trifluoroacetamide was added to the dried residue. The solution was mixed vigorously by vortexing and was incubated for 60 min at 60 °C to obtain trimethylsilyl esters and ethers. After mixing with 50 μl of acetone, an aliquot of this sample was injected into a gas chromatograph (GC 6890 N; Agilent Technologies, Palo Alto, CA, USA) that was equipped with a quadrupole mass spectrometer (5973 Network; Agilent Technologies). A fused silica capillary column (HP-5MS, 5% phenylmethyl siloxane, 30 × 0.25 mm2; Agilent Technologies) was used. Helium was used as carrier gas at a flow rate of 1.2 ml/min. Temperature programming was carried out to bring the temperature from 60 °C to 280 °C at 10 °C/min. The injector temperature was set to 250 °C, and the temperatures of the transfer lines to the mass detector and ion source were 250 °C and 230 °C, respectively. Electron energy was set to 70 eV. Cholesterol and 24S-OHC were identified based on their retention times and mass patterns; ions having m/z 458 for cholesterol and 413 for 24S-OHC were selected for the quantification. Cholesterol and 24S-OHC were identified quantitatively using cholesterol-d7 and 27-OHC-d6 (Avanti Polar Lipids, Alabaster, AL, USA) as internal standards, respectively.
Lipid fractionation by HPLC
Cells were lysed in 1 ml of 0.2 M NaOH and were neutralized by 64 μl of 3 M HCl and 45 μl of 1 M KH2PO4.41 Lysate was transferred to glass tubes and to the lysate in each tube three times its volume of Folch solvent (chloroform/methanol, 2 : 1, v/v) and 1 ml H2O were then added. After centrifugation at 3500 r.p.m. (2330 × g) for 10 min, the chloroform phase was transferred into fresh tubes. The extracts were evaporated under a stream of N2, the dry residue was then dissolved in 100 μl eluent without water, and this was injected to HPLC. HPLC was carried out on an Inertsil ODS-3 column (5 μm, 4.6 × 250 mm; GL Science, Tokyo, Japan) as described previously.42 The HPLC apparatus consisted of a pump (PU-980 Intelligent HPLC Pump; Nihon Bunko, Tokyo, Japan), a detector (UV-970 Intelligent UV/VIS Detector, Nihon Bunko), and a calculator (EPC-500; Eicom, Kyoto, Japan). The flow rate of eluent (acetonitrile/2-propanol/H2O, 43/53/4) was 1.0 ml/min.
RNA isolation and real-time PCR
Total RNA isolation and real-time PCR were conducted as described previously.15 The following primers were used: human ACAT1 forward, 5′-GCAAGGCGCTCTCTCTTAGATG-3′; reverse, 5′-AGGAGGGCAATAAACATGTGATATATT-3′; and human ACAT2 forward, 5′-TGGCTCAACGCCTTTGC-3′; reverse, 5′-CCAGTCCCGGTAGAACATCCT-3′.
Statistical analysis
Data are reported as mean±S.D. of at least three independent experiments unless otherwise indicated. The statistical significance of the difference between the determinations was calculated by an analysis of variance using Tukey’s test for multiple comparisons. The difference was considered significant when the P value was <0.05.
Abbreviations
- ACAT:
-
acyl-CoA:cholesterol acyltransferase
- AD:
-
Alzheimer’s disease
- CHX:
-
cycloheximide
- CYP46A1:
-
cholesterol 24-hydroxylase
- GC–MS:
-
gas chromatography–mass spectrometry
- HPLC:
-
high-performance liquid chromatography
- LDH:
-
lactate dehydrogenase
- Nec-1:
-
Necrostatin-1
- RIPK:
-
receptor-interacting protein kinase
- TNFα:
-
tumor necrosis factor α
- ZVAD:
-
ZVAD-fmk
- 24S-OHC:
-
24(S)-hydroxycholesterol
References
Christofferson DE, Yuan J . Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 2010; 22: 263–268.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1: 112–119.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC et al. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem 2007; 103: 2004–2014.
Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res 2010; 88: 1569–1576.
Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W . Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine 1997; 9: 801–808.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Wilson NS, Dixit V, Ashkenazi A . Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol 2009; 10: 348–355.
Yamanaka K, Saito Y, Yamamori T, Urano Y, Noguchi N . 24(S)-hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J Biol Chem 2011; 286: 24666–24673.
Dietschy JM, Turley SD . Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 2004; 45: 1375–1397.
Russell DW, Halford RW, Ramirez DM, Shah R, Kotti T . Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem 2009; 78: 1017–1040.
Björkhem I, Lütjohann D, Diczfalusy U, Ståhle L, Ahlborg G, Wahren J . Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res 1998; 39: 1594–1600.
Jeitner TM, Voloshyna I, Reiss AB . Oxysterol derivatives of cholesterol in neurodegenerative disorders. Curr Med Chem 2010; 18: 1515–1525.
Urano Y, Ochiai S, Noguchi N . Suppression of amyloid-beta production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J 2013; 27: 4305–4315.
Shafaati M, Solomon A, Kivipelto M, Björkhem I, Leoni V . Levels of ApoE in cerebrospinal fluid are correlated with Tau and 24S-hydroxycholesterol in patients with cognitive disorders. Neurosci Lett 2007; 425: 78–82.
Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, Oehring RD et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 2000; 41: 195–198.
Brown J 3rd, Theisler C, Silberman S, Magnuson D, Gottardi-Littell N, Lee JM et al. Differential expression of cholesterol hydroxylases in Alzheimer's disease. J Biol Chem 2004; 279: 34674–34681.
Kölsch H, Lütjohann D, Tulke A, Björkhem I, Rao ML . The neurotoxic effect of 24-hydroxycholesterol on SH-SY5Y human neuroblastoma cells. Brain Res 1998; 818: 171–175.
Walther TC, Farese RV Jr . Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 2012; 81: 687–714.
Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol 2003; 5: 781–792.
Chang TY, Li BL, Chang CC, Urano Y . Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab 2009; 297: E1–E9.
Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci USA 2010; 107: 3081–3086.
Bonilla E, Prelle A . Application of nile blue and nile red, two fluorescent probes, for detection of lipid droplets in human skeletal muscle. J Histochem Cytochem 1987; 35: 619–621.
Chang CC, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R et al. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J Biol Chem 2000; 275: 28083–28092.
Ikenoya M, Yoshinaka Y, Kobayashi H, Kawamine K, Shibuya K, Sato F et al. A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 2007; 191: 290–297.
Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther 2011; 339: 487–498.
He MX, He YW . A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ 2013; 20: 188–197.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43: 449–463.
Cheng D, Chang CC, Qu X, Chang TY . Activation of acyl-coenzyme A:cholesterol acyltransferase by cholesterol or by oxysterol in a cell-free system. J Biol Chem 1995; 270: 685–695.
Zhang Y, Yu C, Liu J, Spencer TA, Chang CC, Chang TY . Cholesterol is superior to 7-ketocholesterol or 7 alpha-hydroxycholesterol as an allosteric activator for acyl-coenzyme A:cholesterol acyltransferase 1. J Biol Chem 2003; 278: 11642–11647.
Freeman NE, Rusinol AE, Linton M, Hachey DL, Fazio S, Sinensky MS et al. Acyl-coenzyme A:cholesterol acyltransferase promotes oxidized LDL/oxysterol-induced apoptosis in macrophages. J Lipid Res 2005; 46: 1933–1943.
Fukunaga M, Nunomura S, Nishida S, Endo K, Gon Y, Hashimoto S et al. Mast cell death induced by 24(S),25-epoxycholesterol. Exp Cell Res 2010; 316: 3272–3281.
Bao L, Li Y, Deng SX, Landry D, Tabas I . Sitosterol-containing lipoproteins trigger free sterol-induced caspase-independent death in ACAT-competent macrophages. J Biol Chem 2006; 281: 33635–33649.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Bantel H, Engels IH, Voelter W, Schulze-Osthoff K, Wesselborg S . Mistletoe lectin activates caspase-8/FLICE independently of death receptor signaling and enhances anticancer drug-induced apoptosis. Cancer Res 1999; 59: 2083–2090.
von Haefen C, Wieder T, Essmann F, Schulze-Osthoff K, Dörken B, Daniel PT . Paclitaxel-induced apoptosis in BJAB cells proceeds via a death receptor-independent, caspases-3/-8-driven mitochondrial amplification loop. Oncogene 2003; 22: 2236–2247.
Xaus J, Comalada M, Valledor AF, Lloberas J, López-Soriano F, Argilés JM et al. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood 2000; 95: 3823–3831.
Saito Y, Nishio K, Ogawa Y, Kinumi T, Yoshida Y, Masuo Y et al. Molecular mechanisms of 6-hydroxydopamine-induced cytotoxicity in PC12 cells: involvement of hydrogen peroxide-dependent and -independent action. Free Radic Biol Med 2007; 42: 675–685.
Shichiri M, Yoshida Y, Ishida N, Hagihara Y, Iwahashi H, Tamai H et al. α-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic Biol Med 2011; 50: 1801–1811.
Urano Y, Watanabe H, Murphy SR, Shibuya Y, Geng Y, Peden AA et al. Transport of LDL-derived cholesterol from the NPC1 compartment to the ER involves the trans-Golgi network and the SNARE protein complex. Proc Natl Acad Sci USA 2008; 105: 16513–16518.
Urano Y, Hayashi I, Isoo N, Reid PC, Shibasaki Y, Noguchi N et al. Association of active gamma-secretase complex with lipid rafts. J Lipid Res 2005; 46: 904–912.
Acknowledgements
We thank Dr. Yoko Ogawa Akazawa (National Institute of Advanced Industrial Science and Technology, Osaka, Japan) for supporting GC–MS analysis. We thank Dr. Paul Huang in Dr. Ta-Yuan Chang’s lab (Dartmouth Medical School) for providing the ACAT1 siRNA sequence used in this study. We also thank Dr. Ta-Yuan Chang and Kowa Co. Ltd (Tokyo, Japan) for providing valuable reagents. This work was supported in part by the KAKENHI Grant-in-Aid for Young Scientists (B) 23700428, 25830041 to YU and for JSPS fellows 243567 to KY from JSPS and the MEXT-Supported Program for the Strategic Research Foundation at Private Universities in Japan for the years 2012–2016.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yamanaka, K., Urano, Y., Takabe, W. et al. Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA:cholesterol acyltransferase 1. Cell Death Dis 5, e990 (2014). https://doi.org/10.1038/cddis.2013.524
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.524
Keywords
This article is cited by
-
Dysregulation of cholesterol metabolism in cancer progression
Oncogene (2023)
-
Integrated stress response is involved in the 24(S)-hydroxycholesterol-induced unconventional cell death mechanism
Cell Death Discovery (2022)
-
Idarubicin combats abiraterone and enzalutamide resistance in prostate cells via targeting XPA protein
Cell Death & Disease (2022)
-
Sterol auto-oxidation adversely affects human motor neuron viability and is a neuropathological feature of amyotrophic lateral sclerosis
Scientific Reports (2021)
-
Cholesterol Metabolism in Neurodegenerative Diseases: Molecular Mechanisms and Therapeutic Targets
Molecular Neurobiology (2021)
