
Abstract
A disintegrin and metalloprotease 17 (ADAM17) is a sheddase with important substrates including tumor necrosis factor-α (TNF-α) and its receptors, the p75 neurotrophin receptor (p75NTR), and members of the epidermal growth factor family. The rationale of this study was to inhibit ADAM17-induced shedding of soluble TNF-α in order to reduce detrimental inflammation after spinal cord injury (SCI). However, using the specific ADAM17 blocker BMS-561392 in neuronal and glial cell cultures, we show that proper functioning of ADAM17 is vital for oligodendrocyte and microglia survival in a p44 MAPK-dependent manner. In contrast, genetic ablation of ADAM17 specifically increases microglial death. Surprisingly, although blocking ADAM17 in vivo does not substantially change the ratio between membrane-bound and soluble TNF-α, it increases expression of the pro-apoptotic marker Bax and microglial apoptosis while impairing functional recovery after SCI. These data suggest that ADAM17 is a key survival factor for microglial cells after SCI.
Similar content being viewed by others


Main
Tumor necrosis factor-α (TNF-α) converting enzyme (TACE), also known as ADAM17,1 is a sheddase involved in multiple cell signaling pathways.2 Its first discovered biological function was the shedding of the membrane-bound form of TNF-α (mTNF-α)1 and has since had many other factors reported as substrates.3 It is also required for the processing of both TNF-α receptors (TNFRs),4 p75 neurotrophin receptor (p75NTR),5 and ligands of the epidermal growth factor receptor (EGFR) family.6, 7 First indications for a functional role of ADAM17 in neurodegenerative diseases have been found in ischemia8 and Alzheimer’s disease.9 However, its role in traumatic injuries of the central nervous system (CNS) is unclear.
CNS trauma, either in the form of brain injury or spinal cord injury (SCI), is characterized by an excessive post-traumatic inflammatory response leading to secondary injury processes and limited functional recovery. The composition and magnitude of these inflammatory processes vary among the different organs (the brain and spinal cord) as well as among the different phases after SCI.10 Several studies have demonstrated an upregulation of pro-inflammatory cytokines, including TNF-α within hours after injury.11 This increase in TNF-α levels has been linked to apoptosis, enhanced vascular permeability, and impaired glutamate metabolism and clearance.12 TNF-α is produced as a type II transmembrane protein (pro-TNF-α or mTNF-α) arranged in stable homotrimers. The soluble form of TNF-α (sTNF-α) is released after proteolytic cleavage of the membrane-bound form by ADAM17.1 The mTNF-α form decreases inflammation, whereas sTNF-α promotes strong inflammatory responses during infection.13 Recently, it was shown that mice lacking ADAM17 on lymphocytes are protected from sterile and bacterial sepsis due to loss of TNF-α shedding.14, 15 Therefore, ADAM17 blockers have been used in rheumatoid arthritis and multiple sclerosis models to reduce the production of sTNF-α in order to decrease inflammation.13 Some ADAM17 inhibitors have reached phase II of clinical trials for the treatment of breast cancer, but until now there is little information available about the functional role of ADAM17 and its inhibitors during CNS injury.
In the present study, we have investigated the role of ADAM17 using the specific ADAM17 blocker BMS-561392 in cultures of neuronal and glial cells in vitro as well as in a mouse model of T-cut hemisection SCI in vivo. We show that ADAM17-induced signaling is vital for the survival of cultured immature and mature oligodendrocytes, microglia, and astrocytes. Furthermore, blocking ADAM17 impairs functional recovery, increasing lesion size, astrogliosis, and microglial apoptosis after SCI.
Results
ADAM17 inhibition increases apoptosis of microglia and oligodendrocytes in vitro
To understand the cellular effects of ADAM17 inhibition, we used the main types of cells present in the CNS: oligodendrocytes, neurons, astrocytes, and microglia. First, we evaluated effects of ADAM17 modulation on cell survival during 48 h in two different cell lines of immature (Figures 1a–d) and mature oligodendrocytes (Figures 1e and f). Enzymatically active recombinant soluble ADAM17 (rADAM17) did not influence survival. In contrast, both immature and mature oligodendrocytes were strongly affected by the ADAM17 specific inhibitor BMS-561392. Undifferentiated oligodendrocytes were more susceptible to ADAM17 inhibition (Figures 1b and d), showing a concentration-dependent reduction in survival ranging from 10% with low concentration (0.3 mM), 45% with medium concentration (1.3 mM), and up to 89.5–93.5% with the highest concentration of the inhibitor (2.7 mM). A similar effect was found using 100 μM of the non-specific inhibitor TAPI-1, where viability was decreased about 10–20%; no significant changes were observed with 10 μM TAPI-1 (Figures 1b and d). However, mature oligodendrocytes were significantly affected only by the highest concentration of the ADAM17 inhibitor (2.7 mM) and TAPI-1 (100 μM), leading to a reduction of 80 or 25%, respectively, of oligodendrocyte survival (Figure 1f). Furthermore, we analyzed the effects of BMS-561392, TAPI-1, and rADAM17 treatment on primary cortical neurons in the presence (Figures 2a and b) or absence (Supplementary Figure S1) of B-27. Whereas a low concentration of rADAM17 (1 μM) promotes cell survival in the presence of B-27, a high concentration of rADAM17 (10 μM) decreases cell survival (Figure 2a). Deprivation of B-27 induced a reduction in cell survival of 30–35%, but rADAM17 did not have any effect on neuronal survival under these conditions (Supplementary Figure S1A). Similarly, inhibition of ADAM17 with different concentrations of BMS-561392 did not influence neuronal survival, neither in the presence (Figure 2b) nor in the absence (Supplementary Figure S1B) of B-27. TAPI-1 had a concentration-dependent effect on survival, inducing an increase (10 μM) or decrease (100 μM) in the presence or absence of B-27 (Figure 2b; Supplementary Figure S1B). Surprisingly, in astrocytes, both rADAM17 (10 μM) and BMS-561392 (2.7 mM) increased viability, while TAPI-1 significantly reduced viability (Figures 2c and d). Finally, in cultured BV-2 cells (i.e., henceforth ‘microglial cells’), inhibition of ADAM17 using the medium (1.3 mM) and high concentration (2.7 mM) of BMS-561392 decreased viability by 50 and 94%, respectively (Figure 2f), whereas the low concentration (0.3 mM) slightly increased viability compared with the control condition. TAPI-1 did not influence microglial survival (Figure 2f), whereas rADAM17 promoted survival in a dose-dependent manner (Figure 2e). In addition, treatment with a medium or high concentration (1.3 and 2.7 mM) of BMS-561392 significantly increased the number of activated caspase-3-positive microglial cells (Supplementary Figure S2A), while no significant changes were observed in astrocytes (Supplementary Figure S2B) and cultures of immature oligodendrocytes (Supplementary Figures S2C and D). However, blocking of ADAM17 with the high concentration (2.7 mM) of BMS-561392 induced a twofold increase in the number of activated caspase-3-positive mature oligodendrocytes (Supplementary Figure S2E).

ADAM17 inhibition decreases viability of undifferentiated and differentiated oligodendrocytes. (a, c) Undifferentiated HOG and MO3.13 cells were treated for 48 h with selected concentrations of rADAM17. Cell survival was measured using an MTT assay. (b, d) Undifferentiated HOG and MO3.13 cells were incubated for 48 h with the specific ADAM17 inhibitor BMS- 561392 and the non-specific inhibitor TAPI-1. Cell viability was significantly reduced after ADAM17 inhibition. The values are expressed as percentage of the control. The results are the mean of three independent experiments, (n=12–17 wells per condition) (*P<0.05; **P<0.01; ***P<0.001). (e, f) MO3.13 cells were differentiated using PMA for 72 h and treated for 48 h with rADAM17, BMS-561392, or TAPI-1. Cell viability was measured using the MTT assay. There was no significant effect after rADAM17 treatment, but a significant decrease in cell viability was observed after ADAM17 inhibition (BMS-561392 (2.7 mM) and TAPI-1 (100 μM)). The results are the mean of three independent experiments and are represented as a percentage of the control condition (n=12–18 wells per condition) (*P<0.05; **P<0.01; ***P<0.001)

ADAM17 has a concentration-dependent effect on the survival of cortical neurons, astrocytes, and microglia. (a) Cortical neurons were incubated with rADAM17 (1 or 10 μM) for 48 h in the presence of B-27. The lower concentration (1 μM) significantly promotes cell survival while the higher concentration (10 μM) decreases survival. (b) Cortical neurons were treated with three selected concentrations of the ADAM17 inhibitor BMS-561392 and two concentrations of the non-specific inhibitor of ADAM17, TAPI-1. While BMS-561392 did not have an effect on survival, TAPI-1 concentration dependently increased (10 μM) or decreased (100 μM) survival. The values are expressed as percentage of control with B-27 and represent three independent experiments (n=15–21 wells per condition) (*P<0.05; **P<0.01). (c) The astrocytic cell line, CCF, was treated for 48 h with rADAM17. Afterwards, cell survival was measured using an MTT assay. Viability was increased after treatment with 10 μM of rADAM17 (n=15 wells per condition) (**P<0.01). (d) CCF cells were treated for 48 h with BMS-561392 or TAPI-1. CCF cell survival was increased by BMS-561392 (2.7 mM) while TAPI-1 (100 μM) decreased survival (n=15 wells per condition) (*P<0.05; **P<0.01). (e) The microglial cell line BV-2 was treated for 48 h with rADAM17. Cell survival was measured using an MTT assay. Microglial survival was significantly increased after rADAM17 treatment (n=15 wells per condition) (1 and 10 μM; *P<0.05; **P<0.01). (f) Inhibition of ADAM17 by BMS-561392 for 48 h decreased BV-2 cell survival. The results are the mean of three independent experiments and are represented as a percentage of the control condition (n=15 wells per condition) (*P<0.05; ***P<0.001)
ADAM17 inhibition increases membrane TNFR-1 and pro-NGF expression in microglia
As a next step, we investigated the signaling pathways potentially affected by ADAM17 inhibition that are involved in microglia and oligodendrocyte apoptosis. These include (1) TNF-α signaling via TNFR-1 and TNFR-2, (2) unprocessed nerve growth factor precursor (pro-NGF), which induces apoptosis by binding to p75NTR, and (3) EGF receptor (EGFR) signaling via MAPK activation/inhibition.16, 17, 18 First, we analyzed the expression of TNFR-1 and TNFR-2 in the membrane of microglia and oligodendrocytes. The membrane expression of TNFR-1 and TNFR-2 was not significantly changed by BMS-561392 for oligodendrocytes (Figures 3a and b). However, in microglia, a significant increase in membrane expression of TNFR-1 and TNFR-2 was found using BMS-561392 in a concentration of 2.7 and 1.3 mM, respectively (Figures 3c and d), together with a significant reduction in TNFR-2 expression in the presence of TAPI-1 (100 μM) in comparison with the control condition (Figure 3d). TNF receptor-associated death domain (TRADD) is a specific mediator of TNFR-1 activation inducing recruitment of other adaptor proteins, such as TNF receptor associated factor 2 (TRAF2) in a first step,19 followed by receptor-interacting protein 1 (RIP1) and associated protein with death domain (FADD),20 eventually leading to apoptosis.21 Surprisingly, neither the ADAM17 inhibitor nor TAPI-1 significantly influenced TRADD expression on oligodendrocytes or microglia (Figure 3e and Figure 3g). Second, as the microglial cell line BV-2 does not express p75NTR,22 we analyzed the expression of p75NTR in oligodendrocytes and astrocytes (data for astrocytes not shown). No significant changes were found in p75NTR expression (Figure 3f). We also analyzed the expression of pro-NGF and NGF in oligodendrocytes and microglia cells. As for p75NTR, no significant changes were detected in pro-NGF expression in oligodendrocytes (Figure 3h). However, a significant increase in pro-NGF expression was found in the presence of the low concentration of BMS-561392 (0.3 mM) in microglial cells (Figure 3i). The levels of NGF were below the detection limit under the experimental conditions used in this study.

Blocking of ADAM17 using a specific inhibitor increases TNFR-1 and TNFR-2 expression on microglial membranes. (a, b). Membranes of undifferentiated HOG were treated for 3 h with TAPI-1 or BMS-561392, after which the cells were lysed and membrane fractions were collected. Electrophoretic analysis revealed no significant changes in TNFR-1 and TNFR-2 expression (n=6 control; n=3-4 ADAM17 inhibitor BMS-561392). (c, d) Electrophoretic analysis of BV-2 cell membranes showed a significant increase of >50% in TNFR-1 expression in the cell membrane after 2.7 mM of BMS-561392 and in TNFR-2 expression using 1.3 mM of BMS-561392. TAPI-1 significantly reduced TNFR-2 expression (n=6 control; n=3-4 ADAM17 inhibitor BMS-561392). Representative blots in Figure 3c show the control condition for the 2.7 mM of BMS-561392, while in 3D the control condition for 1.3 mM of BMS-561392 is shown to facilitate readability. (e, g) TRADD expression was unchanged after treatment in both oligodendrocytes and microglia cells. (f) p75NTR expression was not affected in oligodendrocytes after ADAM17 inhibition. (h, i) Oligodendrocytes did not show differences in pro-NGF levels after inhibition of ADAM17 neither with the specific nor the non-specific inhibitor (mean of three experiments; n=3 per condition), while low inhibition of ADAM17 (0.3 mM) significantly increased pro-NGF expression of cultured BV-2 cells (mean of four independent experiments; n=4 per condition). Data are presented as a mean percentage of the control condition (set as 100%) (*P<0.05)
ADAM17 inhibition modulates p44 MAPK phosphorylation in a concentration-dependent manner
MAPKs are serine/threonine protein kinases involved in diverse cellular signaling pathways in many different cell types. Among these, extracellular signal-regulated kinase (ERK) is one of the most important MAPKs regulating cell death and survival.18, 23 We therefore investigated the modulation of phosphorylation of ERK 1/2 or p44/42 MAPK by BMS-561392 and TAPI-1, in oligodendrocytes and microglia. In both cell types, levels of p42 MAPK phosphorylation were unchanged. On the contrary, levels of p44 MAPK phosphorylation were significantly decreased with the highest concentration of BMS-561392 (2.7 mM) in oligodendrocytes (Figure 4a), whereas in microglial cells phosphorylation of p44 MAPK was significantly increased by the lower concentration of BMS-561392 (0.3 mM) and significantly diminished below detection by the high concentration of the inhibitor (2.7 mM) (Figure 4b).

Inhibition of ADAM17 modulates phosphorylation of p44 MAPK. (a) Levels of p44 MAPK phosphorylation were decreased after ADAM17 inhibition with medium and high concentrations of BMS-561392, whereas p42 MAPK phosphorylation was not affected in immature HOG oligodendrocytes. (b) Microglial BV-2 cells were treated with different concentrations of BMS-561392 and TAPI-1. MAPK p44 phosphorylation was significantly increased using 0.3 mM of the inhibitor, while higher concentrations decreased or even abolished p44 MAPK phosphorylation. MAPK p42 phosphorylation was not affected by BMS-561392 or TAPI-1 treatment. The results are the mean of three independent experiments and are represented as a percentage of the control condition (n=3 per condition)
Recombinant ADAM17 transiently improves functional recovery during the acute phase after SCI in vivo while inhibition of ADAM17 impairs recovery
To investigate the role of ADAM17 in functional recovery after SCI in vivo, we applied rADAM17 (0.3 mM) or vehicle solution locally immediately after injury by placing a soaked gelfoam on top of the lesion (Figure 5a). Under these conditions, rADAM17 led to a slight but significant transient increase in the BMS score during the first 3 days after injury compared with the vehicle group (Figure 5b). Histological analysis of spinal cord cryosections from mice treated with rADAM17 16 days post-SCI did not show significant differences in lesion size compared with vehicle (control) mice (Figures 5c–e) or astrogliosis, as determined by GFAP intensity analysis (Figures 5f and g, 5g1 and 2). In addition, we assessed whether treatment with rADAM17 influences microglia/macrophages activation/infiltration by Iba1 expression analysis. Similarly, no significant differences were found between the two treatment groups (Figures 5h–j).

Local application of rADAM17 transiently improves functional outcome during the acute phase after SCI. (a) Immediately after injury (SCI) at day 0, a gelfoam soaked with 0.3 mM of rADAM17 or vehicle solution, was placed on top of the lesion site. The animals were scored during 15 days. (b) One single administration of rADAM17 (0.3 mM) slightly but significantly improves the BMS score (i.e., locomotor recovery) in the acute phase after SCI (until 3 days after injury; n=7-8 mice per condition) (*P<0.05). (c) rADAM17-treated mice did not show differences in lesion size compared with vehicle-treated mice. (d, e) Representative pictures of the lesion area in mice treated with vehicle solution or rADAM17. (f) There were no significant differences in astrogliosis analyzed 100 μm cranial or caudal to the lesion site between the two treatment groups. (g) Representative picture showing the quantified regions in a control mouse. G1–G2. Higher magnification of the areas indicated by white boxes in g showing GFAP immunoreactivity cranial and caudal to the lesion epicenter. (h) No significant differences were found in Iba1 expression rostro-caudal to the lesion epicenter between vehicle and rADAM17-treated mice. (i–j) Representative pictures showing perilesional Iba1 expression. Scale bars =100 μm for d, e, g, G1, G2, i, and j (n=3 mice per condition)
To further investigate the role of ADAM17 after SCI, we treated mice with daily injections of the specific ADAM17 inhibitor BMS-561392 (2.2 mM/day for 15 days) or PBS (control mice) for 15 days (Figure 6a). Mice treated with the inhibitor had a significantly reduced locomotion recovery compared with the control group, as assayed by the BMS score (Figure 6b). Histological analysis of spinal cord cryosections from mice treated with BMS-561392 16 days post-SCI showed a significant increased lesion size compared with PBS (control) mice (Figures 6c–e) and significantly increased astrogliosis caudal to the lesion site, as determined by GFAP intensity analysis (Figures 6f and g, 6g1 and 2). The area of demyelination (MBP immunoreactivity) and T-cell numbers showed no differences between both groups (data not shown). Furthermore, we investigated the influence of ADAM17 inhibition in microglia/macrophages activation/infiltration. Mice treated with BMS-561392 displayed a significantly reduced expression of Iba1, especially close to the lesion center and caudally (Figures 6h–j).

Blocking of ADAM17 impairs locomotion recovery after SCI. (a) Scheme of the treatment approach used after SCI. From the day of injury (day 0) until day 15 post SCI, mice were daily injected subcutaneously (s.c.) with 2.2 mM of the ADAM17 inhibitor or vehicle solution. (b) Treatment with the specific ADAM17 inhibitor (2.2 mM) significantly reduces spontaneous recovery (n=7–11 mice per condition; *P<0.05) as shown by a reduced BMS score. (c) Quantification of the lesion size after T-cut hemisection injury. ADAM17 inhibitor-treated mice showed an increase in lesion size after 15 days of treatment (*P<0.05). (d, e) Representative pictures of the lesion area in mice treated with ADAM17 inhibitor or control mice; the asterisk indicates the delineated area. (f) Quantification of astrogliosis 100 μm cranial and caudal to the lesion epicenter. There was a significant increase in gliosis (i.e., GFAP immunoreactivity) caudal to the lesion site in mice treated with the ADAM17 inhibitor (*P<0.05). (g) Representative picture showing the quantified regions. G1-G2. Higher magnification of the areas indicated by white boxes in g showing GFAP immunoreactivity cranial and caudal to the lesion epicenter. (h) Quantification of Iba1 immunoreactivity from 500 μm cranial to 500 μm caudal to the lesion epicenter. A significant decrease in Iba1 expression rostro-caudal to the lesion epicenter was found after ADAM17 inhibition (***P<0.001). (i, j) Representative pictures showing perilesional Iba1 expression. Scale bars=100 μm for d, e, g, G1, G2, i, and j (n=4–7 mice per condition)
ADAM17 inhibition increases apoptosis of microglia/macrophages in the spinal cord
ADAM17 is responsible for the cleavage of mTNF-α in the sTNF-α form.1 However, in our model, ADAM17 inhibition did not significantly alter the ratio between mTNF-α and sTNF-α (Supplementary Figures S3A and B). Inhibition of TNF-α after SCI leads to decreased apoptosis in the spinal cord.24 Therefore, we investigated the expression of the anti-apoptotic marker B-cell lymphoma-2 (Bcl-2) and the pro-apoptotic marker Bcl-2-associated X protein (Bax). Inhibition of ADAM17 resulted in a slight but non-significant decrease in levels of Bcl-2 (Supplementary Figures S3C and D) and a significant increase in Bax expression (Supplementary Figures S3E and F).
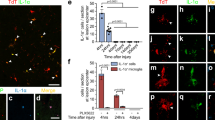
On the basis of our in vitro results, we performed double labeling for activated caspase-3 and CC-1 (a marker for mature oligodendrocytes) to identify apoptotic oligodendrocytes in vivo. The analysis showed a slight but non-significant increase in the number of apoptotic oligodendrocytes caudal to the lesion site (data not shown). In addition, we quantified apoptosis of genetically labeled oligodendrocytes in transgenic proteolipid protein-enhanced cyan fluorescent protein (PLP-eCFP) mice. Similarly, no change in the number of activated caspase-3/PLP-double-positive cells was observed (Figures 7a and b). Using double-labeling for activated caspase-3 and CD11b to identify apoptotic phagocytes in vivo, we found a significant increase in the number of apoptotic microglia/macrophages caudal to the lesion site (Figures 7c and d). Finally, we measured viability in primary oligodendrocyte cultures isolated from hypomorphic ADAM17ex/ex mice, which showed over 95% reduced expression of ADAM17 in all tissues investigated.25 As in our in vivo model, there was no difference in viability between wild-type and ADAM17-deficient oligodendrocytes (Figure 7e). In contrast, survival of ADAM17-deficient primary microglia was significantly reduced by nearly 40% compared with wild-type controls (Figure 7f).

ADAM17 inhibition in vivo or deficiency in vitro increases microglial apoptosis. (a) Representative pictures of PLP/activated caspase-3 double staining. Scale bar=20 μm. The arrows indicate apoptotic oligodendrocytes. (b) Quantification of the number of apoptotic oligodendrocytes (PLP/activated caspase-3 double-positive) in PLP-eCFP transgenic mice in a squared area of 225 × 225 μm around the lesion site. There is a slight but non-significant increase in the number of apoptotic oligodendrocytes caudal to the lesion site (225 μm; n=3-4 mice per condition). (c) Representative pictures of CD11b+/activated caspase-3 caudal to the lesion site. The arrows show apoptotic microglia in both groups. Scale bar=20 μm. (d) A significant increase in the number of apoptotic microglial (CD11b/activated caspase-3 double positive) cells caudal to the lesion site was found after inhibition of ADAM17 (n=4 mice per condition; *P<0.05). (e) Survival of primary immature oligodendrocyte cultures from ADAM17-deficient mice is not affected. (f) Primary microglial cultures from ADAM17-deficient mice have a reduced survival compared with microglial cultures from wild-type animals (n=26–41 wells (primary oligodendrocytes), n=27–28 wells (primary microglia)(***P<0.001)
Discussion
ADAM17 is involved in several cell signaling pathways, which are crucial for survival and apoptosis, including signaling via TNFR, p75NTR, and EGFR. In the present study, we have used the specific ADAM17 blocker BMS-561392 and ADAM17-deficient cell cultures to show that ADAM17 is a key survival factor for microglial cells in vitro and in vivo.
ADAM17 is best known for the shedding of TNF-α and its receptors.1, 4 After SCI, inhibition of both TNF-α forms with etanercept leads to a decrease in Bax and an increase in Bcl-2 expression and reduces apoptosis in the spinal cord.24 The two forms of TNF-α show distinct binding affinities for TNFR-1 and TNFR-2. TNFR-1 has been primarily associated with apoptosis through the recruitment of TRADD, whereas TNFR-2 lacks a death domain and is associated with the anti-apoptotic effects of TNF-α.26, 27 There is increasing evidence that both TNF-α forms may have complementary roles. For example, mTNF-α has a higher affinity for TNFR-2 and may therefore have a more important role in the regulation of cell survival,27 remyelination after experimental autoimmune encephalomyelitis,13 and reduction of the inflammatory response in atherosclerosis,28 whereas sTNF-α has a higher affinity for TNFR-1 and may be more important for apoptosis and inflammation.29, 30 Therefore, the rationale of the present study was to modify the ratio of mTNF-α and sTNF-α, using the specific ADAM17 inhibitor BMS-561392, in order to improve functional recovery after SCI.
It has been speculated previously that tissue inhibitor of metalloproteinases-3 (TIMP-3), which is the natural inhibitor of ADAM17, may contribute to increased death of immature oligodendrocytes via ADAM17 in a cerebral ischemia model.31 On the basis of these data, we expected that blocking ADAM17 would increase the levels of mTNF-α leading to a reduction of apoptotic oligodendrocytes. Surprisingly, in our SCI model, ADAM17 inhibition did not lead to differences in the levels of the two TNF-α forms in spinal cord homogenates. Our findings are supported by corresponding data in ADAM17-deficient leukocytes and leukocytes from gene-targeted mice expressing non-cleavable TNF-α.32, 33 The authors suggested that mTNF-α levels may be regulated by a feedback mechanism independent of shedding.32, 33 A similar mechanism may explain our findings. Consistently, the number of activated caspase-3+/CC-1+ oligodendrocytes and activated caspase-3+/PLP+ oligodendrocytes was not influenced by ADAM17 inhibition in vivo, indicating that oligodendrocytes are less affected by the inhibitor in our in vivo model. Interestingly, survival of primary oligodendrocytes cultures derived from ADAM17-deficient mice (ADAM17ex/ex) was also unchanged. In contrast, in in vitro cultures of two different oligodendrocytic cell lines (HOG and MO3.13), cell survival as measured using MTT assay was significantly reduced by ADAM17 inhibition. Meanwhile, the number of activated caspase-3-positive immature oligodendrocytes was not affected by ADAM17 inhibition, whereas blocking of ADAM17 significantly increased the number of activated caspase-3-positive mature oligodendrocytes in vitro. It is important to note that the MTT assay rather measures cell viability than cell death; thus, viability can be decreased without actual cell death as a final result, which might explain why MTT values are decreased in immature neurons while no changes in caspase-3 cell numbers were found in this population. Furthermore, susceptibility to cell death may be related to a different activation state of the cell or even the timelines for the progression of apoptosis might vary between different cell types or different cellular activation or maturation states.34, 35 For example, for cerebellar granule neurons, it was shown that effects on survival by certain cytokines (such as TNF-α) were dependent on the maturation state of these cells.36 It is tempting to speculate that a similar mechanism occurs in immature versus mature oligodendrocytes.
As ADAM17 controls ectodomain shedding of various substrates, we studied selected factors of key pathways associated with both apoptosis and ADAM17, namely NGF, p75NTR, TNF-α, and EGFR/MAPK signaling.16, 17, 18 We found a concentration-dependent modulation of p44 MAPK phosphorylation in microglia cultures. The low concentration of the inhibitor slightly increased p44 MAPK phosphorylation, whereas the high concentration reduced it below detection levels, indicating a non-linear effect of the inhibitor as sometimes also seen with other enzyme-inhibitors. The high concentration also reduced p44 MAPK phosphorylation in undifferentiated oligodendrocytes (HOG). ADAM17 and MAPK signaling appear to be closely linked. Among the MAPK signaling pathways, p44 MAPK and p38 signaling are necessary for ADAM17 activation/trafficking,37, 38 but at the same time ADAM17 controls EGFR and MAPK activation via the cleavage of EGFR ligands.6 ADAM17 interacts with its natural inhibitor TIMP3, which is abolished after ADAM17 phosphorylation by MAP kinases.39 In our study, other signaling pathways associated with apoptosis, such as TNFRs, p75NTR, and pro-NGF signaling, were not affected by ADAM17 inhibition in cultures of oligodendrocytes and microglia. To our knowledge, we show for the first time that ADAM17 inhibition leads to a concentration-dependent modulation of p44 MAPK phosphorylation in microglia and oligodendrocyte cultures.
Another interesting finding was that ADAM17 inhibition significantly increased the number of apoptotic microglia/macrophages in the injured spinal cord by approximately 40%, combined with a reduced histological and functional outcome after SCI. The inhibition of ADAM17 promoted astrogliosis and increased lesion size after SCI. This is consistent with our in vitro results, which showed that ADAM17 inhibition promotes survival of astrocytes (CCF). In line with the increased microglia apoptosis, we also found a significant reduction of Iba1+ microglia/macrophages after ADAM17 inhibition. Microglial cells have a dual role after CNS injury, participating as phagocytes to remove tissue debris and dead cells, as well as exacerbating tissue damage through the release of pro-inflammatory factors.40 After SCI, peripheral monocytes may contribute to recovery by infiltrating the injury site and producing IL-10.41 As in our SCI model a reduced histological and clinical outcome after ADAM17 inhibition is combined with increased microglia apoptosis, it is tempting to speculate that a beneficial subtype of microglia cells is protected by ADAM17. Consistently, it was demonstrated that ADAM17-mediated signaling promotes survival and anti-apoptotic pathways of colonic epithelial cells after injury by activation of the EGFR-MAPK-ERK axis.25 Thus, these data indicate that ADAM17 signaling activates pro-inflammatory as well as anti-inflammatory pathways in a cell-type specific manner.
Survival of primary neurons was not influenced by ADAM17 inhibition but significantly supported by rADAM17. These findings may explain the slight but significant improvement in locomotor performance by rADAM17 in the very early phase after SCI (first 3 days). Unfortunately, no histological changes were found between the two groups. This minor effect on behavior might be explained by the half-life of ADAM17 (over 24 h),42 suggesting that rADAM17 treatment might need repeated local applications to have a sustained effect over time after SCI. Moreover, it has been previously shown that soluble rADAM17 is unable to cleave mTNF-α on endothelial cells;43 therefore, our data suggest that rADAM17 may have additional functions such as the modulation of cell-adhesion molecules as suggested by its integrin domain.44, 45 Our in vitro data using cell cultures of oligodendrocytic cell lines (HOG and MO3.13), astrocytes (CCF), and microglia (BV-2) indicated cell-type specific actions of ADAM17 inhibition. Whereas only mature oligodendrocytes showed increased caspase-3 staining, blocking of ADAM17 significantly increased the number of activated caspase-3 microglial cells. It is a known phenomenon that the same agent may induce apoptosis in one cell line but not in other cells, although the result of the treatment is the same (cell death). This might be due to a divergent mechanism independent of caspase-3,35 a different activation state of the cell, different cell types, or different cellular activation states,34, 35 as already mentioned. For example, it has been shown after postnatal excitotoxicity in the brain that caspase-3 in glial cells (mainly astrocytes and oligodendrocytes) works via different spatial and temporal pathways than in neurons.46 To make things even more complicated, there are studies showing distinct cell types from caspase-3-knockout mice that undergo cell death via an incomplete form of apoptosis.47 Our in vivo data also show this divergence in cell-type specific cell death, the number of activated caspase-3+/CC-1+ oligodendrocytes and activated caspase-3+/PLP+ oligodendrocytes was not influenced by ADAM17 inhibition, indicating that oligodendrocytes were less affected by the inhibitor in our in vivo model. Consistently, survival of primary oligodendrocytes cultures derived from ADAM17-deficient mice ADAM17ex/ex was unchanged, whereas survival of primary microglia from these ADAM17ex/ex mice was reduced by more than 40%.
In summary, these findings support the concept that ADAM17 has a key role in protecting potentially beneficial microglia and in supporting functional recovery after SCI.
Materials and Methods
ADAM17 inhibitor
The ADAM17 inhibitor, BMS-561392, was kindly provided by Bristol-Myers Squibb, Princeton, NJ, USA. The drug was prepared as described before.9 In brief, the drug was reconstituted in freshly prepared PBS (pH 3.5). For the in vitro study, three concentrations were used: 0.3 mM, 1.3 mM, and 2.7 mM. All the in vivo experiments were performed using 2.2 mM (210 μg/day).9 rADAM17 (R&D Systems, Abingdon, UK) was diluted to a final concentration of 1 μM, 10 μM (in vitro experiments), or 0.3 mM (in vivo experiments) in ultrapure water.
In vitro experiments
Cell lines
In this study, we used the following cell lines: the immortalized murine BV-2 cell line,48 a human astrocytoma cell line (CCF),49 and two undifferentiated oligodendrocyte cell lines, i.e., human oligodendroglioma (HOG) and human glial (oligodendrocytic) hybrid cell line (MO3.13).50 To increase readability, these cell lines were referred to as microglia, astrocytes, and oligodendrocytes in some parts of the text.
Cell cytotoxicity assay
Primary cortical neuronal cells were prepared from embryonic day 15 (E15) C57BL/6 mice, and primary oligodendrocyte and microglia cultures were prepared from postnatal day 3 ADAM17 wild-type (wt/wt) and ADAM17-deficient (ADAM17ex/ex) mice.25 BV-2, CCF, HOG, and MO3.13 cell lines were used to study survival of microglia, astrocytes, and oligodendrocytes, respectively. All cells were seeded on poly-D-lysine-coated 96-wells at a density of 5 × 104 cells/well. To study the effect of ADAM17 on survival, cells were grown under optimal conditions and treated with different concentrations of BMS-561392, TAPI-1, or rADAM17 to measure cell viability. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) solution (1 mg/ml) was added for 4 h. The cells were lysed in a mixture of dimethyl sulfoxide (DMSO) and glycine (0.1 M), and the absorption was measured at 550 nm using a microplate reader (Bio-Rad, Nazareth, Belgium) (see Supplementary Materials and methods for details).
Western blot analysis of primary cell lines
The above described cell lines were used to perform western blotting against different protein targets. In brief, when the cells reached 80–90% confluence, different concentrations of BMS-561392 or TAPI-1 were added to the cells for 3 h. The cells were then washed with PBS and lysed on ice using Procarta lysis buffer. The lysed product was centrifuged for 30 min at 14 000 r.p.m. at 4 °C. The supernatant was aliquoted and stored at −20 °C until measurement. To determine membranous TNFR-1 and TNFR-2 expression, the Pierce cell surface protein isolation kit (Thermo Fisher Scientific, Erembodegem, Belgium) was used according to the manufacturer’s instructions. Total protein levels were determined using the BCA Protein Assay Kit. Between 5 and 20 μg of protein was electrophoretically separated and transferred to PVDF membranes. The primary antibodies were incubated for 1 h at room temperature (RT) or overnight at 4 °C (Supplementary Table S2). Afterwards, the membranes were washed with PBS and then incubated for 1 h at RT with the corresponding secondary antibody (Supplementary Table S2). The membranes were re-blotted using an antibody against β-actin as a loading control. The signal was detected using the ECL plus western blotting detection (GE Healthcare, Diegem, Belgium). Densitometric analysis was performed using ImageJ (US National Institutes of Health; http://rsb.info.nih.gov/nih-image/). The results are expressed as a percentage of the control group (set as 100%). To minimize bias due to differences in densitometric measurements between experiments, each control condition per experiment was set at 100%, therefore lacking an error (S.E.M.) bar (Figures 3 and 4).
In vivo experiments
Animals
All experiments were performed using either 10-week-old C57BL/6 mice (Harlan, The Netherlands), ADAM17ex/ex and ADAM17wt/wt mice generated from ADAM17wt/ex mice (kindly provided by Dr. S Rose-John, Christian Albrechts University, Kiel, Germany),25 or transgenic PLP-eCFP mice51 (kindly provided by Dr. F Kirchhoff, University of Saarland, Germany). Mice were housed in a conventional animal facility at Hasselt University under standardized conditions (i.e., temperature-controlled room (20±3 °C), a 12 h light-dark cycle, food and water ad libitum); all evaluations were performed by blinded investigators. All experiments were approved by the local ethical committee of Hasselt University and were performed according to the guidelines described in Directive 2010/63/EU on the protection of animals used for scientific purposes.
Spinal cord injury T-cut hemisection injury
The T-cut hemisection injury was performed as described previously.52, 53 In brief, mice were randomized, anesthetized, and underwent a partial laminectomy at thoracic level T8. The mice were subjected to a bilateral dorsal T-cut hemisection using iridectomy scissors to transect the left and right dorsal funiculus, the dorsal horns, and the ventral funiculus. This injury procedure leads to a complete transection of the dorsomedial and ventral corticospinal tract and induces impairment of several other descending and ascending tracts. The muscles were sutured and the back skin closed with wound clips. After surgery, the mice were placed in a recovery chamber until they were well awake and could be returned to their home cage. The animals’ bladders were manually voided daily until the mice were capable to urinate independently.
Experimental groups
The treatment protocols are summarized in the Figures 5a and 6a. To investigate the effect of rADAM17, the lesion site was covered with a piece of gelfoam soaked with 0.3 mM of rADAM17 or vehicle solution (ultrapure water) as control (Figure 5a). In a second approach, to investigate the effect of ADAM17 inhibition, mice were distributed over two groups; one group of mice was subcutaneously injected daily for 15 days with PBS pH 3.5 and the other group with BMS-561392 (2.2 mM/day) (Figure 6a).
Locomotion tests
Animals were scored daily for functional recovery after SCI using the Basso mouse scale (BMS)54 starting one day after injury. The BMS is a 10-point scale (9=normal locomotion; 0=complete hind limb paralysis) that is based on hind limb movements made in an open field. The analysis was done using the mean of the left and right hind limb scores for each animal.
Immunofluorescence analysis of the spinal cord
Animals were transcardially perfused 16 days after surgery with Ringer’s solution only, for animals treated with rADAM17, or Ringer’s solution followed by 4% paraformaldehyde (PFA), for mice treated with the ADAM17 inhibitor BMS-561392. Spinal cords were isolated and used to prepare cryosections (10 μm thickness) for measurement of lesion size and astrogliosis (glial fibrillary acidic protein (GFAP) immunoreactivity), inflammatory infiltrate (number of CD4-positive T cells; Iba1 immunoreactivity for microglia/macrophages), and demyelination (myelin basic protein (MBP) immunoreactivity) as described before.53 Cryosections of mice treated with rADAM17 (or vehicle) were post-fixed using 100% acetone for measurement of lesion size and astrogliosis or with 100% methanol for infiltration of microglia/macrophages. Images were obtained with a Zeiss LSM510 meta confocal microscope or with a Nikon 80i Eclipse fluorescence microscope (see Supplementary Materials and methods for details).
Western blotting of spinal cord tissue
Animals were transcardially perfused with Ringer’s solution 16 days post surgery. Spinal cord tissue was dissected out (from 0.5 cm cranial to 0.5 cm caudal from the lesion site) and homogenized using Procarta lysis buffer (Panomics, Vignate-Milano, Italy). Homogenized tissue was centrifuged at 10 000 r.p.m. for 10 min at 4 °C. The supernatants were collected and stored at −80 °C until further use. Total protein levels were determined using the BCA Protein Assay Kit (Thermo Fisher Scientific, Erembodegem, Belgium) according to the manufacturer’s instructions. A total amount of 75 μg (for measurement of Bax and Bcl-2 levels) or 40 μg of total protein (to measure TNF-α expression) were separated electrophoretically and transferred to PVDF membranes. The membranes were blocked with 5% nonfat dry milk in PBS-Tween 0.05% for 1 h. Primary antibodies were incubated overnight (4 °C) or for 1 h at RT (Supplementary Table S2). The membranes were washed in PBS-Tween 0.05% and incubated with the secondary antibody for 1 h at RT (Supplementary Table S2). The membranes were further analyzed as described above.
Statistical analysis
Statistical analyses were performed with Prism 5.0 (GraphPad Software, La Jolla, CA, USA) and PASW statistics 18 (SPSS; IBM, Brussel, Belgium) software. Behavioral data (BMS) were analyzed using a two-way ANOVA as previously described.54 Immunohistochemical and western blotting analyses were performed using a Mann–Whitney U-test for comparison between two groups. For semi-quantitative protein detection (western blotting) and cell cytotoxicity assay with more than two groups, values were compared using one-way ANOVA followed by Bonferroni post hoc test. All data are given as mean±S.E.M.
Abbreviations
- ADAM17:
-
a disintegrin and metalloprotease 17
- Bax:
-
Bcl-2-associated X protein
- Bcl-2:
-
B-cell lymphoma-2
- BMS:
-
Basso Mouse Scale
- CNS:
-
central nervous system
- EGFR:
-
epidermal growth factor receptor
- ERK:
-
extracellular signal-regulated kinase
- FADD:
-
FAS-associated protein with death domain
- GFAP:
-
glial fibrillary acidic protein
- Iba1:
-
ionized calcium-binding adapter molecule 1
- MBP:
-
myelin basic protein
- mTNF-α:
-
membrane-bound form of tumor necrosis factor-alpha
- NGF:
-
nerve growth factor
- p75NTR:
-
p75 neurotrophin receptor
- PLP:
-
proteolipid protein
- pro-NGF:
-
nerve growth factor precursor
- rADAM17:
-
recombinant ADAM17
- RIP1:
-
receptor-interacting protein 1
- SCI:
-
spinal cord injury
- sTNF-α:
-
soluble form of tumor necrosis factor-alpha
- TNF-α:
-
tumor necrosis factor-alpha
- TACE:
-
tumor necrosis factor-alpha converting enzyme
- TNFRs:
-
tumor necrosis factor-alpha receptors
- TNFR-1:
-
tumor necrosis factor-alpha receptor 1
- TNFR-2:
-
tumor necrosis factor-alpha 2
- TRADD:
-
TNF receptor-associated death domain
- TRAF2:
-
TNF receptor-associated factor 2
References
Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997; 385: 729–733.
Seals DF, Courtneidge SA . The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Development 2003; 17: 7–30.
Scheller J, Chalaris A, Garbers C, Rose-John S . ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 2011; 32: 380–387.
Yang P, Baker KA, Hagg T . The ADAMs family: coordinators of nervous system development, plasticity and repair. Progress Neurobiol 2006; 79: 73–94.
Weskamp G, Schlondorff J, Lum L, Becherer JD, Kim TW, Saftig P et al. Evidence for a critical role of the tumor necrosis factor alpha convertase (TACE) in ectodomain shedding of the p75 neurotrophin receptor (p75NTR). J Biol Chem 2004; 279: 4241–4249.
Murthy A, Defamie V, Smookler DS, Di Grappa MA, Horiuchi K, Federici M et al. Ectodomain shedding of EGFR ligands and TNFR1 dictates hepatocyte apoptosis during fulminant hepatitis in mice. J Clin Invest 2010; 120: 2731–2744.
La Marca R, Cerri F, Horiuchi K, Bachi A, Feltri ML, Wrabetz L et al. TACE (ADAM17) inhibits Schwann cell myelination. Nature Neurosci 2011; 14: 857–865.
Wang X, Feuerstein GZ, Xu L, Wang H, Schumacher WA, Ogletree ML et al. Inhibition of tumor necrosis factor-alpha-converting enzyme by a selective antagonist protects brain from focal ischemic injury in rats. Mol Pharmacol 2004; 65: 890–896.
Kim ML, Zhang B, Mills IP, Milla ME, Brunden KR, Lee VM . Effects of TNFalpha-converting enzyme inhibition on amyloid beta production and APP processing in vitro and in vivo. J Neurosci 2008; 28: 12052–12061.
Vidal PM, Lemmens E, Dooley D, Hendrix S . The role of ‘anti-inflammatory’ cytokines in axon regeneration. Cytokine Growth Factor Rev 2012; 24: 1–12.
Pineau I, Lacroix S . Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comparative Neurol 2007; 500: 267–285.
Oyinbo CA . Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta Neurobiologiae Experimentalis 2011; 71: 281–299.
Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE et al. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011; 134 (Pt 9): 2736–2754.
Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y et al. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol 2007; 179: 2686–2689.
Long C, Wang Y, Herrera AH, Horiuchi K, Walcheck B . In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli-mediated peritonitis. J Leukocyte Biol 2010; 87: 1097–1101.
Ware CF, VanArsdale S, VanArsdale TL . Apoptosis mediated by the TNF-related cytokine and receptor families. J Cell Biochem 1996; 60: 47–55.
Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM et al. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 2002; 36: 375–386.
Zhan YH, Liu J, Qu XJ, Hou KZ, Wang KF, Liu YP et al. beta-Elemene Induces Apoptosis in Human Renal-cell Carcinoma 786-0 Cells through Inhibition of MAPK/ERK and PI3K/Akt/mTOR Signalling Pathways. Asian Pac J Cancer Prev 2012; 13: 2739–2744.
Kim JY, Lee JY, Kim DG, Koo GB, Yu JW, Kim YS . TRADD is critical for resistance to TRAIL-induced cell death through NF-kappaB activation. FEBS Lett 2011; 585: 2144–2150.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Hsu H, Shu HB, Pan MG, Goeddel DV . TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996; 84: 299–308.
Zhang J, Geula C, Lu C, Koziel H, Hatcher LM, Roisen FJ . Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp Neurol 2003; 183: 469–481.
Lee JH, Lee SW, Choi SH, Kim SH, Kim WJ, Jung JY . p38 MAP kinase and ERK play an important role in nitric oxide-induced apoptosis of the mouse embryonic stem cells. Toxicol in vitro 2012; 27: 492–498.
Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muia C, Bramanti P et al. Immunomodulatory effects of etanercept in an experimental model of spinal cord injury. J Pharmacol Exp Therapeut 2006; 316: 1006–1016.
Chalaris A, Adam N, Sina C, Rosenstiel P, Lehmann-Koch J, Schirmacher P et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J Exp Med 2010; 207: 1617–1624.
McCoy MK, Tansey MG . TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation 2008; 5: 45.
Wajant H, Pfizenmaier K, Scheurich P . Tumor necrosis factor signaling. Cell Death Differentiation 2003; 10: 45–65.
Canault M, Peiretti F, Mueller C, Kopp F, Morange P, Rihs S et al. Exclusive expression of transmembrane TNF-alpha in mice reduces the inflammatory response in early lipid lesions of aortic sinus. Atherosclerosis 2004; 172: 211–218.
Grell M . Tumor necrosis factor (TNF) receptors in cellular signaling of soluble and membrane-expressed TNF. J Inflammation 1995; 47: 8–17.
Holtmann MH, Neurath MF . Differential TNF-signaling in chronic inflammatory disorders. Curr Mol Med 2004; 4: 439–444.
Yang Y, Jalal FY, Thompson JF, Walker EJ, Candelario-Jalil E, Li L et al. Tissue inhibitor of metalloproteinases-3 mediates the death of immature oligodendrocytes via TNF-alpha/TACE in focal cerebral ischemia in mice. J Neuroinflammation 2011; 8: 108.
Ruuls SR, Hoek RM, Ngo VN, McNeil T, Lucian LA, Janatpour MJ et al. Membrane-bound TNF supports secondary lymphoid organ structure but is subservient to secreted TNF in driving autoimmune inflammation. Immunity 2001; 15: 533–543.
Bell JH, Herrera AH, Li Y, Walcheck B . Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J Leukocyte Biol 2007; 82: 173–176.
Jessel R, Haertel S, Socaciu C, Tykhonova S, Diehl HA . Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. J Cell Mol Med 2002; 6: 82–92.
Green DR . Apoptotic pathways: the roads to ruin. Cell 1998; 94: 695–698.
De Luca A, Weller M, Frei K, Fontana A . Maturation-dependent modulation of apoptosis in cultured cerebellar granule neurons by cytokines and neurotrophins. Eur J Neurosci 1996; 8: 1994–2005.
Soond SM, Everson B, Riches DW, Murphy G . ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J Cell Sci 2005; 118 (Pt 11): 2371–2380.
Diaz-Rodriguez E, Montero JC, Esparis-Ogando A, Yuste L, Pandiella A . Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol Biol Cell 2002; 13: 2031–2044.
Xu P, Liu J, Sakaki-Yumoto M, Derynck R . TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci signaling 2012; 5: ra34.
David S, Kroner A . Repertoire of microglial and macrophage responses after spinal cord injury. Nature Rev Neurosci 2011; 12: 388–399.
Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 2009; 6: e1000113.
Schlondorff J, Becherer JD, Blobel CP . Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem J 2000; 347 (Pt 1): 131–138.
Canault M, Leroyer AS, Peiretti F, Leseche G, Tedgui A, Bonardo B et al. Microparticles of human atherosclerotic plaques enhance the shedding of the tumor necrosis factor-alpha converting enzyme/ADAM17 substrates, tumor necrosis factor and tumor necrosis factor receptor-1. Am J Pathol 2007; 171: 1713–1723.
Trad A, Riese M, Shomali M, Hedeman N, Effenberger T, Grotzinger J et al. The disintegrin domain of ADAM17 antagonises fibroblastcarcinoma cell interactions. Int J Oncol 2013; 42: 1793–1800.
Gooz P, Dang Y, Higashiyama S, Twal WO, Haycraft CJ, Gooz M . A disintegrin and metalloenzyme (ADAM) 17 activation is regulated by alpha5beta1 integrin in kidney mesangial cells. PloS ONE 2012; 7: e33350.
Villapol S, Acarin L, Faiz M, Castellano B, Gonzalez B . Distinct spatial and temporal activation of caspase pathways in neurons and glial cells after excitotoxic damage to the immature rat brain. J Neurosci Res 2007; 85: 3545–3556.
Porter AG, Janicke RU . Emerging roles of caspase-3 in apoptosis. Cell Death Differ 1999; 6: 99–104.
Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F . Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunology 1990; 27: 229–237.
Hashimura T, Tubbs RR, Connelly R, Caulfield MJ, Trindade CS, McMahon JT et al. Characterization of two cell lines with distinct phenotypes and genotypes established from a patient with renal cell carcinoma. Cancer Res 1989; 49 (24 Pt 1): 7064–7071.
Buntinx M, Moreels M, Vandenabeele F, Lambrichts I, Raus J, Steels P et al. Cytokine-induced cell death in human oligodendroglial cell lines: I. Synergistic effects of IFN-gamma and TNF-alpha on apoptosis. J Neurosci Res 2004; 76: 834–845.
Steffens H, Nadrigny F, Kirchhoff F . In vivo two-photon imaging of neurons and glia in the mouse spinal cord. Cold Spring Harbor protocols 2012; 2012.
Tuszynski MH, Steward O . Concepts and methods for the study of axonal regeneration in the CNS. Neuron 2012; 74: 777–791.
Boato F, Hendrix S, Huelsenbeck SC, Hofmann F, Grosse G, Djalali S et al. C3 peptide enhances recovery from spinal cord injury by improved regenerative growth of descending fiber tracts. J Cell Sci 2010; 123 (Pt 10): 1652–1662.
Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG . Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 2006; 23: 635–659.
Acknowledgements
We thank Dr. F Kirchhoff and Dr. Anja Scheller (University of Saarland, Germany) to provide us with the PLP-eCFP mice. We thank Bristol-Myers Squibb for providing the specific ADAM17 inhibitor BMS-561392 to perform the study. This study was supported in part by grants from Deutsche Forschungsgemeinschaft (SPP1394), SFB877 project A1 to AC and SRJ, and from Research Foundation Flanders - FWO (G.0834.11N, G.0389.12N, G0A1413N) to SH and EL (1.2.703.10N), and to PV by the transnational University Limburg (tUL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Vidal, P., Lemmens, E., Avila, A. et al. ADAM17 is a survival factor for microglial cells in vitro and in vivo after spinal cord injury in mice. Cell Death Dis 4, e954 (2013). https://doi.org/10.1038/cddis.2013.466
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.466
Keywords
This article is cited by
-
Expression of SARS-CoV-2-related receptors in cells of the neurovascular unit: implications for HIV-1 infection
Journal of Neuroinflammation (2021)
-
Matrix metalloproteinases and ADAMs in stroke
Cellular and Molecular Life Sciences (2019)
-
Analysis of the Role of CX3CL1 (Fractalkine) and Its Receptor CX3CR1 in Traumatic Brain and Spinal Cord Injury: Insight into Recent Advances in Actions of Neurochemokine Agents
Molecular Neurobiology (2017)
-
Interleukin-25 is detrimental for recovery after spinal cord injury in mice
Journal of Neuroinflammation (2016)
-
MiR-200c regulates ROS-induced apoptosis in murine BV-2 cells by targeting FAP-1
Spinal Cord (2015)

{kind=link}
{kind=link}
{kind=link}