
Abstract
Angiogenesis has a key role in the tumor progression and metastasis; targeting endothelial cell proliferation has emerged as a promising therapeutic strategy for the prevention of cancer. Previous studies have revealed a complex association between the process of angiogenesis and autophagy and its outcome on tumorigenesis. Autophagy, also known as type-II cell death, has been identified as an alternative way of cell killing in apoptotic-resistant cancer cells. However, its involvement in chemoresistance and tumor promotion is also well known. In this study, we used a derivate of natural product magnolol (Ery5), a potent autophagy inducer, to study the association between the autophagy and angiogenesis in both in vitro and in vivo model system. We found that the robust autophagy triggered by Ery5, inhibited angiogenesis and caused cell death independent of the apoptosis in human umbilical cord vein endothelial cells and PC-3 cells. Ery5 induced autophagy effectively inhibited cell proliferation, migration, invasion and tube formation. We further demonstrated that Ery5-mediated autophagy and subsequent inhibition of angiogenesis was reversed when autophagy was inhibited through 3-methyl adenine and knocking down of key autophagy proteins ATG7 and microtubule-associated protein light chain 3. While evaluating the negative regulation of autophagy on angiogenesis, it was interesting to find that angiogenic environment produced by the treatment of VEGF and CoCl2 remarkably downregulated the autophagy and autophagic cell death induced by Ery5. These studies, while disclosing the vital role of autophagy in the regulation of angiogenesis, also suggest that the potent modulators of autophagy can lead to the development of effective therapeutics in apoptosis-resistant cancer.
Similar content being viewed by others


Main
Cancer progression is a complex process involving different cellular and physiological events. Several processes driving these events are intricately interconnected leading to tumorigenesis. Angiogenesis is one of the critical processes involved in the establishment of the tumor and metastasis. Several pro-angiogenic growth factors are secreted into the tumor microenvironment that facilitates the formation of new blood vessels from pre-existing vasculature.1 It is a process involving cell proliferation, migration, matrix degradation, tube formation and vessel maturation.1, 2 A number of therapeutic agents have been developed to inhibit these processes to achieve reduced blood supply to target tissues and is considered as one of the promising strategies in the treatment of cancer. Several angiogenesis inhibitors such as sorafenib, sunitinib, pazopanib, vandetanib, axitinib, cediranib and bevacizumab are either in final stages of their development or have been approved as anticancer drugs by FDA.3
During tumor growth, low oxygen supply in tumor tissue creates hypoxia that stimulates the angiogenesis through the upregulation of HIF-1α and VEGF.4 Tumor hypoxia and overexpression of HIF1 have been associated with therapeutic resistance to anticancer agents, increase in invasion, metastasis and poor outcome in malignancies.5 Hypoxia has also been shown to activate the process of autophagy, a lysosomal degradation pathway, which enables the degradation of proteins, carbohydrates and lipids allowing the cell to adapt its metabolism and meet its energy needs under stress.6 Autophagy also known as type-II programmed cell death is characterized by massive degradation of cellular content by the action of lysosomal hydrolases.7, 8 Autophagy has important role in various cellular processes and contributes to many normal and pathological processes. The role of autophagy in the regulation of cancer is contentious and depends upon tumor type, stage and genetic context.9 On one hand autophagy has been reported to inhibit the cell proliferation, an alternative to apoptotic death,10, 11 whereas it is also called responsible for its protective role against death in cancer cells.12, 13, 14 A defective autophagy due to the disruption or suppression of autophagy-related gene BECN1 was shown to promote tumorigenesis,15 whereas its overexpression was found to have the inhibitory effect.16 BECN1 gene has been found deleted in various cancers, for example, 40–75% of human breast, prostate and ovarian cancers have defective BECN1, suggesting that the defective autophagy may be linked with the tumor development.16 Several tumor suppressors genes have also been found associated with the onset of autophagy. Biallelic deletion of Atg4C (autophagy-related gene), a tumor suppressor gene, in mice results in defective autophagy under stressful conditions and is responsible for the formation of chemical-induced fibrosarcomas.17 Ultraviolet radiation resistance-associated gene (UVRAG) is another tumor suppressor protein that is also an important regulator of autophagy. UVRAG interact with beclin1, leading to autophagy and thus inhibit cancer progression.18 Bif1 is another important protein that interacts with beclin1 through UVRAG and participates in induction of autophagy by acting as a positive regulator of PI3KC3; loss of Bif1 resulted in reduced activity of PI3KC3 and knockout of Bif1 promoted spontaneous tumorigenesis in mice.19 Autophagy and angiogenesis have been shown to have complex relationship in different types of cancers. Many studies have revealed that autophagy inhibits angiogenic vasculature,20, 21 whereas other studies suggested that autophagy promotes cancer and its inhibition also prevents angiogenesis.22, 23
Target-based natural product anticancer agents are also capable of inducing autophagy in cancer cells by different mechanisms, like ROS generation, targeting PI3K/AKT or RAS signaling pathways with concomitant anti-angiogenic potential.24, 25, 26, 27, 28 The use of natural compounds capable of triggering autophagy-mediated inhibition of angiogenesis and cell death in apoptosis-resistant cancer cells can be a promising strategy to target highly malignant cancers. Magnolol is a natural compound that has been studied to have anticancer potential by showing antiproliferative activity against different cancer cell lines through the induction of apoptosis or autophagy.29, 30
In continuation of our recent study showing that a derivative of natural product magnolol (Ery5) induces robust autophagic cell death in human leukemia HL-60 cells,29 here for the first time we investigated the negative regulation of angiogenesis by Ery5-induced autophagy. We used well-studied cell model in angiogenesis, human umbilical vein endothelial cell (HUVEC) along with highly aggressive tumorigenic and metastatic human prostate cancer cell line PC-3.31 It was found that autophagy triggered by Ery5 was capable of inhibiting angiogenesis and induction of cancer cell death independent of apoptosis, when observed in both in vitro and in vivo systems.
Results
Ery5 inhibits cell proliferation and attenuates angiogenesis in HUVECs and PC-3 cells
Ery5 inhibited cell proliferation in HUVECs with IC50 value 10 μM after 24 h treatment (Figure 1a). Recently, we reported the cell growth inhibitory effect of Ery5 in different cell lines including PC-3.29, 32 In PC-3, Ery5 treatment significantly inhibited colony formation at 1 μM concentration and was more prominent with increasing doses of Ery5 (Figures 1b and c). While examining the anti-angiogenic effect in HUVECs and PC-3 cells, it was found that Ery5 significantly inhibited the tube formation in HUVECs in a dose-dependent manner. Interestingly, the inhibitory potential was better than known anti-angiogenic inhibitor ‘sunitinib’ at the concentration of 20 μM after 24 h (Figure 1d). The inhibitory effect of Ery5 on tube formation was more prominent than that on the cell viability when observed under lower concentrations (Figure 1d, Supplementary Figures S2A and S2B). It was observed that the cell growth inhibition by Ery5 treatment at the concentration of 5 μM was only 7–8%, whereas inhibition of tube formation was about 35%; same was the case with 10 and 20 μM treatments. Anti-angiogenic activity of Ery5 was further confirmed by its inhibitory effect on cell migration in HUVECs and PC-3 cells in a wound-healing experiment (Figures 1e and f), the inhibitory effect on migration increased with increasing time periods (Figures 1e and f). To further evaluate the anti-angiogenic potential of Ery5, an ex vivo microvessel sprout formation was determined by aortic ring assay that mimics several stages of angiogenesis. It was observed that in the absence of Ery5, sprouts emerged from the aortic ring and grew outward after 4 days in culture with VEGF; however, Ery5 treatment resulted in a significant dose-dependent decrease in sprout length and density induced by VEGF (Figure 1g).


Ery5 diminishes angiogenesis in HUVECs and PC-3 cells. (a) Ery5-attenuated HUVEC proliferation. HUVECs were plated in 96-well plates, treated with various concentrations of Ery5 for 24 h. MTT was added 3 h before the termination of the experiment; the procedure of the assay is discussed in Materials and Methods. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (b and c) Ery5-inhibited colony formation in PC-3 cells. PC-3 cells were treated with different concentrations of Ery5 for 5 days; the procedure of the assay is described in Materials and Methods. (d) Ery5-inhibited tube formation in HUVECs. For tube formation assay, 50 μl of extracellular matrix was transferred to each well of a precooled 96-well tissue culture plate. Then the plate was incubated at 37 °C for at least 1 h to allow the matrix solution to solidify. HUVECs were added to each well and were allowed to attach overnight. Cells were treated with 1 μM of sunitinib and 5, 10 and 20 μM of Ery5 for 24 h. The tube formation was observed under an inverted light microscope at × 10 magnification using an inverted microscope equipped with digital camera (Olympus Imaging Corp.). Ares of formed tubes were measured as described in Materials and Methods. (e and f) Ery5-inhibited migration in HUVECs and PC-3 cells. HUVECs and PC-3 cells were plated in six-well plates. A wound was given with a sterile tip. Ery5 (10 μM) was added for 6, 12 and 24 h. The procedure for the assay is discussed in Materials and Methods. (g) Ery5-attenuated angiogenesis in ex vivo model. Ex vivo angiogenesis inhibition was analyzed through aortic arch ring formation assay. The procedure of the experiment is described in Materials and Methods. (h and i) Ery5 attenuated the expression of angiogenic proteins in PC-3 and HUVECs. Cells (1 × 106) were plated in 60 mm dishes, cells were allowed to attach and grow and were incubated with 10 μM of Ery5 for 6, 12 and 24 h. Cells were lysed with RIPA buffer, proteins were separated on SDS-PAGE, transferred to PVDF membranes and probed for different antibodies as described in Materials and Methods
Ery5 significantly inhibited the expression of angiogenic proteins
Ery5 treatment showed strong inhibitory effect on the expression of angiogenic proteins in HUVECs and PC-3 cells. Essential angiogenic proteins like, VEGFR2, HIFα, HIFβ, c-MYC, pAKT and p90RSK were significantly inhibited by Ery5 treatment in both HUVECs and PC-3 cells. The expression of the receptor of vascular endothelial growth factor receptor 2 (VEGFR2), an important target of anti-angiogenic drugs, was completely diminished after 6 h treatment of Ery5 in HUVECs and PC-3 cells (Figures 1h and i). Expression of other important angiogenic proteins, such as HIFα, HIFβ and c-MYC also showed significant downregulation in 6 h, which was further attenuated or completely abrogated after 12- and 24-h Ery5 treatments (Figures 1h and i). As evident, PI3K and STAT pathways have important role in the progression of angiogenesis; treatment of Ery5 effectively reduced the expression of pAKT, p90RSK and pSTAT3 in HUVECs and PC-3 cells starting from 6 h (Figures 1h and i). Interestingly, we found that the expression of PHD2, an anti-angiogenic factor negatively regulating HIFα and HIFβ,33 was significantly elevated in Ery5-treated HUVECs and PC-3 cells (Figure 1i).
Ery5 induced robust autophagy in PC-3 and HUVECs
Ery5-treated HUVECs and PC-3 cells underwent through robust induction of autophagy. Ery5-induced autophagy was demonstrated by acridine orange staining (AOS) of PC-3 cells, which measures autophagic acidic vesicular organelles (Figure 2a). Autophagy initiation was further confirmed by using microtubule-associated protein light chain 3 (LC3)-specific immunofluorescence assay, where LC3 puncta per cell were counted and shown as bar diagram in Figure 2b. Autophagy induction was further confirmed by western blot analysis of key autophagic proteins. It was found that Ery5 enhanced the conversion of LC3-I into LC3-II; a significant increase in the expression of LC3-II was observed after 6 h treatment that was further increased through 12- and 24-h treatments (Figure 2c). Ery5 treatment also decreased the level of p62, an important regulator of autophagy. Studies have shown that p62/SQSTM1 is recruited to autophagosomal membrane through interaction with LC3 and thus, the lysosomal degradation of autophagosome leads to decrease in p62 levels during autophagy.34 Further, a significant increase in the expression of autophagic protein ATG7 was observed in 6-h treatment, which improved with the increased treatment periods (Figure 2c). Simultaneously, similar to PC-3 cells, robust autophagy induction by Ery5 was also observed in HUVECs (Figure 2d).

Ery5 induces autophagy in PC-3 cells and HUVECs. (a) PC-3 cells were incubated with 10 μM of Ery5 for 6, 12 and 24 h. AO (1 μg/ml) was added 15 min before termination of the experiment; cells were collected, washed once with PBS and AO fluorescence was observed under fluorescence microscope. (b) Ery5-induced LC3 level in PC-3 cells. PC-3 cells were grown on cover slips and treated with 10 μM of Ery5 for 6, 12 and 24 h. The samples were processed for immunofluorescence as described in Materials and Methods. Autophagic puncta per cell were counted and are displayed in the form of bar diagram. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (c and d) Ery5 activates autophagy in PC-3 and HUVEC. PC-3 and HUVECs were seeded in 60 mm dishes and incubated with 10 μM of Ery5 for 6, 12 and 24 h. Cells were lysed with RIPA buffer, proteins were separated on SDS-PAGE and western blots for indicated proteins was done as described in Materials and Methods. (e) Ery5 induces G2 arrest in PC-3 cells. PC-3 cells were plated in six-well plate and incubated with 10 μM of Ery5 for indicated time periods. Cell cycle phase distribution was done as described in Materials and Methods. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with **P<0.01 and ***P<0.001
Ery5 treatment caused cell cycle arrest and autophagic cell death in PC-3 cells
While observing the mode of cell death caused by Ery5, a double staining of annexin V/propidium iodide (PI) was performed in PC-3 cells at various treatment periods. Interestingly, it was found that Ery5 was unable to cause any significant impact on the induction of apoptosis in PC-3 cells as observed through annexin V staining by flow cytometry (Supplementary Figure S1A). As mitochondrial dysfunction and decrease in the mitochondrial membrane potential are associated with the induction of apoptotic cell death,35 flow cytometric analysis of Ery5-treated PC-3 did not show any significant change in the mitochondrial membrane potential loss even up to 24 h (Supplementary Figure S1B). While exploring the role of Ery5 in the inhibition of cell growth, cell cycle analysis was performed in PC-3 cells. Cell cycle phase distribution analysis displayed prominent arrest in G2 phase that increased from 11% in control to 26 and 34% after 12 and 24 h treatment of Ery5, respectively (Figure 2e). As mitotic arrest-mediated cell death refers to the execution of a death pathway through different mechanisms including apoptosis, autophagy and necrosis,36 we assume that Ery5-induced mitotic arrest and subsequent autophagy has a role in the death of PC-3 cells.
Ery5-induced autophagy inhibits the angiogenesis
Induction of the autophagy has been controversially reported to have both pro-angiogenic and anti-angiogenic properties. As our earlier experiments depicted that Ery5 causes autophagy and suppress angiogenesis, we tried to examine the role of autophagy in the process of angiogenesis and consequential death in PC-3 cells. Cells were incubated with 3-methyl adenine (3-MA), a known inhibitor of autophagy, before the treatment of Ery5, which lead to partial reversal of Ery5-mediated cell death (Figure 3a). It was further analyzed that 3-MA was able to reverse the expression of pro-angiogenic factors like VEGFR2, HIFα, HIFβ and c-MYC in Ery5-treated cells (Figure 3b). To further confirm whether Ery5-mediated autophagy was responsible for the inhibition of angiogenesis, we silenced ATG7 and LC3B, two of the important regulators of autophagy activated by Ery5, by using specific small interfering RNAs (siRNAs). Our results showed that the cell death induced by Ery5 was significantly protected in ATG7- and LC3B-silenced PC-3 cells (Figures 3c and e). This led us to believe that angiogenesis inhibition might be reversed in ATG7- and LC3B-silenced PC-3 cells and the western blot results revealed that the inhibitory effect of Ery5 on angiogenic proteins in ATG7- and LC3B-silenced cells was significantly restored. ATG7-silenced sample displayed almost complete reversal of HIFα, HIFβ and VEGFR2 (Figure 3d). Similarly, in LC3B siRNA-transfected PC-3 cells, the expression of VEGFR2 and HIFβ was restored completely and expression of c-MYC was also restored to a certain level (Figure 3f). To unravel the effect of combined knockdown of LC3 and ATG7 on Ery5-mediated autophagic cell death, an impressive protection in cell death was observed when compared with mock PC-3 cells treated with Ery5 at different time points (Figure 3g). Double knockdown of ATG7 and LC3B improved the viability form 63 to 91%, 46 to 71% and 26 to 70% in 6, 12 and 24 h treatments, respectively (Figure 3g). Simultaneously, it was also found that the double knockdown of LC3B and ATG7 in Ery5-treated cells reversed the expression of key angiogenic proteins,VEGFR2, HIFα, HIFβ and c-MYC when compared with Ery5-only-treated cells (Figure 3h).

Autophagy inhibition reversed the expression of angiogenic protiens and improves cell death in PC-3 cells. (a) Inhibition of autophagy through 3-MA improves viability inhibited by Ery5 in PC-3 cells. PC-3 cells were plated in 96-well plates, and 3-MA (2 mM) was added 30 min before the addition of Ery5 for 6, 12 and 24 h. Cell viability was assessed through MTT assay as described in Materials and Methods. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (b) 3-MA reversed the expression of angiogenic proteins. PC-3 cells were pretreated with 3-MA (2 mM) before treatment with 10 μM of Ery5 for indicated time periods; cells were collected and lysed using RIPA buffer. Western blot analysis for indicated proteins was done as described in Materials and Methods. Quantification for LC3-II was done using image J software. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (c) Autophagy inhibition through ATG7 siRNA reversed the cell death induced by Ery5 for different time period. PC-3 cells were seeded in six-well plates, ATG7 was silenced through siRNA as described in Materials and Methods. Cell viability was determined by MTT assay as described in materials and methods. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (d) ATG7 inhibition caused reversal of Ery5-mediated inhibition of angiogenic proteins. Cells were seeded in six-well plates; ATG7 was silenced through siRNA as described in Materials and Methods. ATG7 siRNA-transfected and non-transfected cells were treated with 10 μM of Ery5 for 6, 12 and 24 h. Cells were lysed in RIPA buffer and proteins were separated on SDS-PAGE and transferred to PVDF membrane. Membranes were probed with different angiogenic and autophagic proteins as described in Materials and Methods. Quantification for LC3-II was done using image J software. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (e) LC3 silencing reversed the inhibitory effect of Ery5 on viability. LC3 was downregulated in PC-3 cells through siRNA as described in Materials and Methods. MTT assay was done to calculate cell viability as described in Materials and Methods. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (f) LC3 inhibition reversed the expression of angiogenic proteins. LC3 siRNA-transfected and non-transfected PC-3 cells were treated with 10 μM of Ery5 for indicated time periods. Cells were lysed and proteins were separated on SDS-PAGE, transferred to PVDF membranes and membranes were probed for different antibodies. β-actin was used as an internal control. (g) Cell death induced by Ery5 is autophagic. Combined silencing of ATG7 and LC3 improved the viability dramatically in PC-3 cells. siRNA-transfected and non-transfected cells were seeded in 96-well plates and treated with 10 μM of Ery5 for 6, 12 and 24 h. Viability was determined through MTT assay as described above. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (h) Combined knockdown of LC3 and ATG7 reduced the anti-angiogenic effect of Ery5. Cells were transfected with LC3 and ATG7 siRNA as described in Materials and Methods. Transfected and non-transfected cells were treated with 10 μM of Ery5 for indicated time periods. Western blot for indicated antibodies was done as described in Materials and Methods. Western blotting results revealed that combined silencing of LC3 and ATG7 reversed the expression of angiogenic proteins very significantly. β-actin was used as an internal control. (i) PC-3 cells (3 × 106) were seeded in 90 mm dishes and incubated with Ery5 (10 μM) for different time points. Cells were collected and lysed in non-denaturing lysis buffer and immunoprecipitated with LC3 antibody as described in Materials and Methods section. After immunoprecipitation, western blot for indicated proteins was done with immunoprecipitated samples as described in Materials and Methods. (j) The figure represents western blot of the input from the IP experiment. Western blot was done as described in Materials and Methods
Ery5 inhibits the interaction between autophagy and angiogenesis
It was interesting to examine how Ery5-mediated autophagy downregulates the angiogenesis. We tried to rule out whether interaction between the autophagy and angiogenic factors facilitates the cross talk during the regulation of two processes. The interaction of ‘LC3’ with angiogenic factor HIFα and VEGFR2 influencing the angiogenesis during the induction of autophagic flux has been reported earlier.37, 38 For this purpose, whole-cell extracts from Ery5-treated and -untreated cells were immunoprecipitated with an anti-LC3 antibody followed by the western blot analysis of HIFα and VEGFR2. As shown in Figure 3i, control cells showed strong association of HIFα and VEGFR2 with LC3-II, which disappeared with the increasing treatment time points of Ery5. The corresponding input from the samples also exhibited comparable expression of VEGFR2 and HIFα as obtained from immunoprecipitation (Figure 3j).
Angiogenic environment counteracts Ery5-induced autophagy in PC-3 cells
So far, our results demonstrated that intervention in the Ery5-mediated autophagy counteracts the autophagic cell death and its anti-angiogenic effect. It was interesting to investigate the consequence of pro-angiogenic environment on the Ery5-induced autophagy, and its outcome on cell death. As VEGF and CoCl2 have been widely used for the induction of angiogenesis,39, 40, 41, 42 PC-3 cells were incubated with VEGF and CoCl2 before Ery5 treatment. The results confirmed that protective effect of VEGF and CoCl2 treatments on Ery5-mediated autophagic cell death was significant (Figure 4a). This protective effect of angiogenesis inducers was augmented with increasing time points. In case of cells pretreated with VEGF, the viability of the cells was found increased from 66 to 81%, 46 to 67% and 27 to 43% after 6, 12 and 24 h treatments, respectively (Figure 4a). Similarly, effect on Ery5-mediated autophagic cell death was significantly reduced in case of CoCl2 pretreatment, and protective effect was noticeably better than VEGF after 24 h (Figure 4b). Although there are contentious reports regarding the association between autophagy and angiogenesis, our study showed that autophagy and angiogenesis are inversely regulated. Some studies had recently reported that the anti-angiogenesis agents promote autophagy.20 In our experiments, cells pretreated with pro-angiogenic VEGF were subsequently treated with Ery5 for various time periods to investigate its effect on autophagy. It was found that contrary to the Ery5-induced increasing expression of LC3-II from 6 through 24 h treatments, cells pretreated with VEGF for the same time durations were either unable to express or showed a very reduced expression of LC3-II (Figure 4c). As predicted, VEGF pretreatment also reversed the inhibitory effect of Ery5 on various pro-angiogenic factors such as VEGFR2, HIFα, HIFβ and c-MYC (Figure 3c). Similarly, CoCl2 treatment also inhibited Ery5-induced autophagy, where effect was even more prominent when compared with VEGF. CoCl2-pretreated cells displayed reduced expression of LC3-II in comparison with that of the cells treated with Ery5 only (Figure 4d). Pertinently, the expression levels of pro-angiogenic HIFα, HIFβ, c-MYC and VEGFR2 were retained in CoCl2-pretreated cells compared with cells treated with Ery5 only (Figure 4d).

Angiogenesis induction reverses Ery5-induced autophagic cell death in PC-3 cells. (a and b)VEGF and Cocl2 improve viability in PC-3 cells. PC-3 cells (6 × 103 per well) were plated in 96-well plates. Cells were allowed to attach for 24 h, VEGF (20 ng/ml) and Cocl2 (100 μM/ml) were treated 30 min before treatment of Ery5 for 6, 12 and 24 h. MTT dye was added 3 h before termination of the experiment and MTT crystal were dissolved in 150 μl of DMSO. OD was measured at 570 nm. Percent viability was calculated as described in Materials and Methods section. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with *P<0.05 and ***P<0.001. (c and d) Angiogenesis induction through VEGF and Colc2 inhibited autophagy in PC-3 cells. PC-3 cells (1 × 106) were seeded in 60 mm dishes, cells were allowed to attach followed by treatment with VEGF (20 ng/ml) and Cocl2 (100 μM/ml) 30 min before treatment of Ery5 for indicated time periods. After treatment for various indicated time periods, cells were detached and lysed in RIPA buffer; proteins were separated on SDS-PAGE and transferred to PVDF membranes. Membranes were probed with different antibodies. The detailed procedure is described in Materials and Methods. Quantification for LC3-II was done using image J software. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with **P<0.01 and ***P<0.001
Ery5-triggered autophagy inhibits angiogenesis in vivo
Matrigel plug assay was performed to validate the anti-angiogenic effect of Ery5 in vivo. We observed a deep red appearance in matrigel containing VEGF (Figure 5a) indicating the formation of new vasculature in matrigel. In contrast, mice receiving the treatment of Ery5 (50 mg/kg) matrigel plug showed less vasculature, whereas no vasculature was found in matrigel plug at 100 mg/kg, which interestingly was comparable to the group of mice treated with 2.5 mg/kg sunitinib (Figure 5a). Anti-angiogenic effect was also confirmed by estimating the hemoglobin levels that were significantly reduced in matrigel plugs of mice treated with Ery5 (Figure 5b). Further, protein analysis from matrigel plugs displayed that Ery5-induced autophagy could inhibit angiogenesis in vivo also. The expression of LC3-II was increased in protein samples extracted from matrigel plugs treated with Ery5. Contrary to this, expression of VEGFR2 was reduced by Ery5 treatment in vivo and the inhibition was comparable to that of positive control sunitinib (Figure 5c).

Ery5 inhibits angiogenesis and induces autophagy in vivo. (a) C57/BL6J mice (4- to 6-week old) were obtained from central animal facility of Indian Institute of Integrative Medicine, Jammu. Matrigel was embedded in mice as described in Materials and Methods. Animals were treated with Ery5 50 mg/kg, 100 mg/kg and sunitinib 2.5 mg/kg. Animals were killed on 10th day of experiment, matrigel plug was extracted and photographs were taken as described in Materials and Methods. (b) Hemoglobin content was measured from tissue samples as described in Materials and Methods. There was significant decrease in hemoglobin content in samples treated with Ery5, which represent its anti-angiogenic activity. Data are mean±S.D. of three similar experiments; statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with ***P<0.001. (c) Tissue samples from mice (matrigel) were homogenized in a homogenizer, followed by lysing in RIPA buffer. Protein estimation was done by the Bradford method, 60 μg protein was loaded in each well. Western blot for indicated antibodies was done as described in Materials and Methods. β-actin was used as an internal control
Discussion
Cancer progression is a complex phenomenon influenced by genetic, epigenetic and factors associated with tumor microenvironment. Angiogenesis is known to be one of the key processes involved in tumor growth, migration and metastasis. Drugs targeting devascularization in angiogenesis are being developed as promising candidates to inhibit tumor growth.43 However, autophagy triggered by anti-angiogenic agents has controversially shown to be involved in both cell survival and cell death.44, 45 The complex relationship between the autophagy and angiogenesis is poorly studied and there are several conflicting reports about the role of autophagy in the process of angiogenesis and tumor progression. In this study, we demonstrated that autophagy can negatively regulate angiogenesis and cause cell death independent of apoptosis. In one of our recent studies, we showed that Ery5, a derivate of natural product Magnalol, induced autophagy-mediated cell death in cancer cells.29 As Ery5 induces robust autophagy in cells, we tried to employ Ery5 to explore the association between the autophagy and angiogenesis. It was interesting to find that Ery5-initiated autophagy can restrain tumor growth by inhibiting angiogenesis, leading to cell death. Ery5-induced robust autophagy in HUVECs and PC-3 cells was observed by AOS and activation of key autophagic proteins. Unlike most chemotherapeutic agents that inflict cell death in cancer cells by targeting mitochondria, Ery5 treatment could not disrupt mitochondrial integrity, as no change was detected in the mitochondrial membrane potential. Further, FACS analysis of Annexin/PI staining indicated that Ery5-induced cell death was predominantly autophagic in nature rather than apoptotic. To confirm whether the cell death caused by Ery5 was the consequence of autophagy, we pretreated cells with pharmacological inhibitors of autophgay and RNAi targeting translation of essential autophagy-driving proteins.46, 47 Interestingly, it was found that inhibition of autophagy by 3-MA enabled partial protection of Ery5-induced cell death, whereas in case of ATG7 knockdown by siRNA, the protective effect on cell death was more pronounced, corroborating that Ery5-arbitrated cell death was autophagic in nature.
Next, we tried to explore the outcome of Ery5-triggered autophagy on angiogenesis. It was found that Ery5-induced autophagy was able to inhibit angiogenesis both in in vitro and in vivo systems. Ery5 treatment inhibited cell migration of PC-3 and HUVECs and also repressed microvessel growth in ex vivo aortic ring assay. Further, immunoblot analysis revealed that Ery5 treatment either downregulated or completely abrogated the expression of important pro-angiogenic proteins in HUVECs and PC-3 cells. Interestingly, during matrigel plug assay in mice, vascularization inhibition by Ery5 treatment was found comparable to that of sunitinib.
Pertinently, autophagy inhibition through 3-MA not only protected cell death but also reversed the inhibitory effect of Ery5 on various angiogenic protein, such as VEGFR2, HIFα, HIFβ and c-MYC. Similarly, when ATG7 expression was knocked down by siRNA, a significant decline in the inhibitory effect of Ery5 on angiogenesis was observed. ATG7 knockdown considerably restored the decreased expression of VEGFR2, HIFα and HIFβ in Ery5-treated cells. To further evaluate the role of autophagy in the inhibition of angiogenesis, expression of another important autophagic marker LC3B was silenced using siRNA. It was observed that inhibition of autophagy through LC3B silencing also decreased the inhibitory effect of Ery5 on the expression profile of angiogenic proteins similar to that of ATG7-silenced samples. Interestingly, double knockdown of ATG7 and LC3B in PC-3 cells showed a dramatic improvement in the cell viability in Ery5-treated cells, with concurrent restoration of the expression of VEGFR2, HIFα, HIFβ and c-MYC. To confirm the negative regulation of angiogenesis by autophagy in vivo, it was imperative to compare the protein expression of tissue samples isolated from the mice matrigel angiogenesis model. Compared with untreated, samples isolated from Ery5-treated mice, generation of autophagy and suppression of angiogenesis was confirmed by observing elevated expression of ‘LC3-II’ with concurrent downregulation of angiogenic marker ‘VEGFR2’.
Although it was demonstrated that autophagy negatively regulates angiogenesis, it was imperative to examine the role of angiogenic environment on autophagy. Several reports have earlier mentioned that angiogenic inhibitors downregulate the autophagy.22 To explore whether angiogenic environment affects the Ery5-triggered autophagy and autophagic cell death, angiogenesis was induced through VEGF and CoCl2 treatment in PC-3 cells. It was found that angiogenic environment inflicted marked reduction in the Ery5-triggered autophagy and autophagic cell death. A protein–protein interaction of LC3 with VEGFR2 and HIF through co-immunoprecipitation also confirmed the cross talk between two processes. This led us to believe that there is a mechanism of cross-regulation between the two processes.
In conclusion, our study tried to elucidate the association between the two important processes involved in the tumor biology by using natural product small molecule. We showed that the robust autophagy can inhibit the tumor progression by downregulating the angiogenesis and promoting the type-II cell death in apoptosis-resistant cancers. As the induction of autophagic cell death is an alternative to apoptosis in the treatment of cancer, a number of chemopreventive dietary compounds have been found to initiate autophagy in cancer cells.48 Intervention and modulation of autophagy by using natural products can be a promising strategy to find new drug-like molecules having significant clinical prospect for the treatment of cancer.
Materials and Methods
Cell culture, growth conditions and treatments
Human prostate carcinoma cells PC-3 and HUVECs were obtained from European Collection of Cell Cultures. PC-3 cells were grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin (100 units/ml), streptomycin (100 μg/ml), L-glutamine (0.3 mg/ml), sodium pyruvate (550 mg/ml) and NaHCO3 (2 mg/ml), whereas HUVECs were grown in culture media obtained from Millipore, Bangalore, India. Cells were grown in CO2 incubator (Thermocon Electron Corporation, Waltham, MA, USA) at 37 °C in an atmosphere of 95% air and 5% CO2 with 98% humidity. Ery5, VEGF and Cocl2 were dissolved in DMSO and was delivered to cell cultures in complete medium.
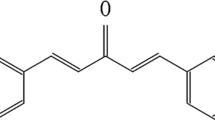
Chemical structure of Ery5
The synthesis of Ery5 from magnolol has been described earlier.32
Reagents and chemicals
RPMI-1640(R8758), 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (M2128), penicillin (P3032), streptomycin (S9137), L-glutamine (G3126), pyruvic acid (107360), ribonuclease A (R6513) and protease inhibitor cocktail (P8340) were purchased from Sigma Chemical Co., St. Louis, MO, USA. FBS was obtained from GIBCO Invitrogen Corporation (number 16000-044, lot number 1237517, Carlsbad, CA, USA). AnnexinV-FITC apoptosis detection kit (sc4252), β-actin (sc47778) and siRNA transfection reagent (sc29528) were purchased from Santa Cruz Biotechnology Incorporation, Texas, TX, USA. pAKT (s473) (s4051), pAKT, T308 (s2965), LC3 (s2775), p62(s5114), LC3B siRNA (s6212), ATG7 siRNA (s6604) and anti mouse IgG (s7076) were acquired from Cell Signaling Technology, Danvers, MA, USA. Electrophoresis reagents, protein markers were acquired from Bio-Rad, Hercules, CA, USA; hyper film and ECL plus reagents were purchased from Amersham Biosciences, Buckinghamshire, UK.
Cell proliferation assay
MTT assay was done to check cell viability.49 Briefly, cells were seeded in 96-well plates. MTT dye was added 3 h before the termination of experiment. Optical density (OD) was acquired at 570 nm.
Colony formation assay
PC-3 cells (1 × 103) were seeded in six-well plate. The cells were allowed to attach and fresh media was added the next day; 1, 5, 10 and 20 μM of Ery5 was delivered to cells and were incubated at 37 °C for 5 days. The cells were then fixed with 4% formaldehyde for 10 min, followed by incubation with 0.5% crystal violet for 30 min. Photographs were taken with a digital camera.
In vitro angiogenesis assay (wound-healing assay)
HUVECs were seeded in six-well Bio-Coat cell culture plates (BD Bioscience, San Jose, CA, USA) at a concentration of 1 × 106 cells per ml and maintained in EndoGRO-LS Complete Media Kit (Millipore, catalog number-SCME001), whereas PC-3 were seeded in six-well plate and maintained in RPMI media. Scratches were given with a 200-μl sterile tip. Ery5 for different time periods was delivered to cells in fresh culture medium. The wound closure was recorded by photography at 0, 6, 12 and 24 h after injury using an inverted microscope equipped with digital camera (Olympus Imaging Corp., Center Valley, PA, USA).
Tube formation assay
Tube formation assay was performed according to the kit manufacturer’s protocol (Millipore, catalog number-ECM 625). Briefly, matrix and diluents buffer were thawed at 4 °C overnight. Extracellular matrix was diluted with diluent buffer before use. The solution was kept on ice to avoid solidification. About 50 μl of this solution was transferred to each well of a precooled 96-well tissue culture plate. Then the plate was incubated at 37 °C for at least 1 h to allow the matrix solution to solidify. The endothelial cells were collected and suspended in endothelial cell growth media (EndoGRO-LS Complete Media Kit; Millipore, catalog number-SCME001). About 5 × 103–1 × 104 cells per well were seeded in matrix-coated plate, followed by overnight incubation at 37 °C. After formation of cellular network, 5, 10 and 20 μM of Ery5 was added for 24 h, sunitinib 1 μM (24 h) was used as a positive control. The tube formation was observed under an inverted light microscope at × 10 magnification using an inverted microscope equipped with digital camera (Olympus Imaging Corp.). Areas of tubes formed were measured using NIH image J software (Bethesda, MA, USA) as described earlier.50
Flow cytometric analysis of apoptosis and necrosis
PC-3 cells (0.5 × 106) were treated with 10 μM of Ery5 for different time periods. Cells were double stained with annexin V/PI kit by using the manufacturer’s protocol. The FACS analysis for apoptosis and necrosis was done as described earlier.51
Cell cycle analysis
Cells were treated with Ery5 (10 μM) for 6, 12 and 24 h, collected and washed once with PBS. Pellet was then fixed in 70% ethanol overnight, followed by one washing with PBS and incubation with RNAse A for 90 min at 37 °C. Cells were then stained with PI and acquired in FACS as described earlier.29
Measurement of mitochondrial membrane potential (ψm)
Mitochondrial membrane potential was measured by using rhodamine-123 (RH-123). PC-3 cells were treated with Ery5 (10 μM), for 6, 12 and 24 h and RH-123 (200 nM) was added 30 min before termination of the experiment. FACS analysis for loss of the mitochondrial membrane potential (Δψm) was done as described earlier.29
Microscopic detection of autophagy after AOS
Induction of autophagy was analyzed by staining cells with acridine orange (AO) as described earlier.29 Briefly, 0.5 × 106 cells were seeded in six-well plate and treated with Ery5 (10 μM) for 6, 12 and 24 h. Cells were incubated with 1 μg/ml AO for 15 min before the termination of the experiment and were washed with PBS before analysis on fluorescence microscope.
Immunofluorescence confocal microscopy for LC3 detection
After treatment with Ery5 for different time periods, PC-3 cells were fixed in 4% paraformaldehyde for 10 min at room temperature and permeabilized using 0.5% Triton-X in PBS for 5 min. The cells were blocked with 10% goat serum for 20 min at room temperature. Autophagy was detected with a rabbit LC3B antibody (Sigma Chemical Co.) diluted 1 : 100 in 0.1% Triton X-100 in PBS for 1 h at room temperature and Alexa Fluor 555-conjugated secondary antibody (Invitrogen, Carlsbad, CA, USA) diluted 1 : 500 in PBS for 1 h at room temperature. Cells were then washed three times in PBS and stained with 4′,6-diamidino-2-phenylindole (1 μg/ml in PBS). The cover slips were mounted over glass slides and cells were imaged by a laser scanning confocal microscope (Olympus Fluoview FV1000) at × 40.
Western blot analysis
Treated and untreated cells were centrifuged at 400 × g at 4 °C, washed in PBS and cell pellets were lysed in RIPA buffer for preparation of whole-cell lysate as described earlier.49 Equal amount of protein (60 μg) was loaded into each well for SDS-PAGE. Blots were incubated with different primary antibodies, and chemiluminiscence was captured on hyperfilm after incubating the blots in ECL plus solution.
Immunoprecipitation
PC-3 cells (3 × 106) were seeded in 90 mm dishes, followed by incubation with 10 μM Ery5 for indicated time points. Cells were lysed in denaturing lysis buffer (RIPA buffer). The DNA was denatured by passing through syringes. Lysates were exposed to LC3 antibody and lysate antibody mixture was kept overnight under constant agitation at 4 °C. Protein agarose A bead was added to the lysate antibody mixture and was incubated for overnight under constant agitation at 4 °C. Lysates were washed thrice with lysis buffer each time centrifuged at 12 000 × g at 4 °C. After three washings with lysis buffer, 50 μl of 2 × loading buffer was added to each sample and samples were run for SDS-PAGE for 3 h. Western blot analysis of immunoprecipitates were further performed as described above.
Small interfering RNA
Human LC3- and ATG7-specific siRNA were transfected into PC-3 by using manufacturer’s protocol. Briefly, cells were incubated in transfection media containing transfection reagent and siRNA for 8 h, followed by addition of complete media for 48 h. Knocking down of the expression of the respective proteins was checked by western blotting.
Aortic ring assay
Sprague–Dawley rats were obtained from central animal facility of Indian Institute Of Integrative Medicine, Jammu, India. Rats were killed by cervical dislocation and surface sterilized with 70% ethanol, dissected and aorta was removed under aseptic conditions as described earlier.52 Dissected aortae were transferred to petri dishes containing opti MEM, excess of fat tissue was removed by forceps, aortae were then embedded in matrigel with or without VEGF (30 nM/ml), followed by treatment with Ery5 (5, 10 and 15 μM) for 4 days. Photographs were taken using DP-72 camera.
In vivo matrigel angiogenesis assay
C57/BL6J mice (4- to 6-week old) were obtained from central animal facility of Indian Institute of Integrative Medicine. Female (20–25 gm) C57/BL6J mice were housed and cared under standard conditions, with 12 : 12 h light and dark cycle. Animal studies were performed in accordance with experimental protocols approved by the animal ethics committee of Indian Institute of Integrative Medicine. Animals were injected subcutaneously into the right flanks with 0.5 ml ice-cold matrigel (BD Bioscience) supplemented with VEGF-A (150 ng/ml) and different concentration of Ery5 (50 and 100 mg/kg BW) and sunitinib (2.5 mg/kg BW); control mice were injected with matrigel without VEGF. At the end of each study on day 10, animals were killed to remove matrigel plugs, and photographs showing the extent of vascularization were taken by using Sony digital camera (Tokyo, Japan). The neovascularization of matrigel plugs was quantified by using 4 ml Drabkin’s reagent by adding well homogenated 20 ml of neovascularized matrigel. After thorough mixing, the absorbance was measured by spectrophotometer at a wavelength of 540 nm to estimate hemoglobin. The hemoglobin estimation was calculated using formula Hb (g/dl)=absorbance of sample/absorbance of standard × concentration of standard.53
Statistical analysis
Statistical analysis was done by using the Bonferroni method and P-value<0.05 was considered to be significant with *P<0.05, **P<0.01 and ***P<0.001.
Abbreviations
- AO:
-
acridine orange
- ATG:
-
autophagy-related gene
- LC3:
-
microtubule-associated protein light chain 3
- MTT:
-
3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide
- PI:
-
propidium iodide
- RH-123:
-
rhodamine-123
- siRNA:
-
small interfering RNA
- HUVECs:
-
human umbilical cord vein endothelial cells
- VEGFR2:
-
vascular endothelial growth factor receptor 2
- 3-MA:
-
3-methyl adenine
References
Carmeliet P, Jain RK . Angiogenesis in cancer and other diseases. Nature 2000; 407: 249–257.
Stetler-Stevenson WG . Matrix metalloproteinases in angiogenesis: a moving target for therapeutic intervention. J Clin Invest 1999; 103: 1237–1241.
Ichihara E, Kiura K, Tanimoto M . Targeting angiogenesis in cancer therapy. Acta Med Okayama 2011; 65: 353–362.
Semenza GL . HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 2010; 20: 51–56.
Höckel M, Vaupe IP . Biological consequences of tumor hypoxia. Semin Oncol 2001; 28: 36–41.
Xie Z, Klionsky D . Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9: 1102–1109.
Shintani T, Klionsky DJ . Autophagy in health and disease: a double edged sword. Science 2004; 306: 990–995.
Kondo Y, Kanzawa T, Sawaya R, Kondo S . The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5: 726–734.
Kimmelman AC . The dynamic nature of autophagy in cancer. Genes Dev 2011; 25: 1999–2010.
Levine B, Yuan J . Autophagy in cell death: an innocent convict? J Clin Invest 2005; 115: 2679–2688.
Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi Z et al. PARP and RIP-1 are required for autophagy induced by 11'-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy 2011; 7: 598–612.
Rouschop KM, Wouters BG . Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr Mol Med 2009; 9: 417–424.
Codogno P, Meijer AJ . Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005; 12: 1509–1518.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–752.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112: 1809–1820.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672–676.
Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C . Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 2007; 282: 18573–18583.
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8: 688–699.
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9: 1142–1151.
Nguyen TM, Subramanian IV, Kelekar A, Ramakrishnan S . Kringle 5 of human plasminogen, an angiogenesis inhibitor, induces both autophagy and apoptotic death in endothelial cells. Blood 2007; 109: 4793–4802.
Ramakrishnan S, Nguyen TM, Subramanian IV, Kelekar A . Autophagy and angiogenesis inhibition. Autophagy 2007; 3: 512–515.
Du J, Teng RJ, Guan T, Eis A, Kaul S, Konduri GG et al. Role of autophagy in angiogenesis in aortic endothelial cells. Am J Physiol Cell Physiol 2012; 302: 383–391.
Shen W, Tian C, Chen H, Yang Y, Zhu D, Gao P et al. Oxidative stress mediates chemerin-induced autophagy in endothelial cells. Free Radic Biol Med 2013; 55: 73–82.
Zhang X, Chen LX, Ouyang L, Cheng Y, Liu B . Plant natural compounds: targeting pathways as anti-cancer therapeutic agents. Cell Prolif 2012; 45: 466–476.
Bhandarkar SS, Arbiser JL . Curcumin as an inhibitor of angiogenesis. Adv Exp Med Biol 2007; 595: 185–195.
Lau DH, Xue L, Young LJ, Burke PA, Cheung AT . Paclitaxel (Taxol): an inhibitor of angiogenesisin a highly vascularized transgenic breast cancer. Cancer Biother Radiopharm 1999; 14: 31–36.
Igura K, Ohta T, Kuroda Y, Kaji K . Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett 2001; 171: 11–16.
Meade-Tollin LC, Wijeratne EM, Cooper D, Guild M, Jon E, Fritz A et al. Ponicidin and oridonin are responsible for the antiangiogenic activity of Rabdosiarubescens, a constituent of the herbal supplement PC SPES. J Nat Prod 2004; 67: 2–4.
Kumar S, Kumar A, Pathania AS, Guru SK, Jada S, Sharma PR et al. Tiron and trolox potentiate the autophagic cell death induced by magnolol analog Ery5 by activation of Bax in HL-60 cells. Apoptosis 2013; 18: 605–617.
Rasul Azhar, Yu Bo, Khan Muhammad, Zhang K, Iqbal F, Ma T et al. Magnolol, a natural compound, induces apoptosis Of Sgc-7901 human gastric adenocarcinoma cells via the mitochondrial and pi3k/akt signaling pathways. Int J Oncol 2012; 40: 1153–1161.
Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW . Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 1979; 17: 16–23.
Jada S, Doma MR, Singh PP, Kumar S, Malik F, Sharma A et al. Design and synthesis of novel magnolol derivatives as potential antimicrobial and antiproliferative compounds. Eur J Med Chem 2012; 51: 35–41.
Rebecca J, AppelhoffYa-Min Tian, Raju R, Turley H, Harris AL, Pugh CW et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 2004; 279: 38458–38465.
Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T . Monitoringautophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452: 181–197.
Düssmann H, Rehm M, Kögel D, Prehn JH . Outer mitochondrial membrane permeabilization during apoptosis triggers caspase-independent mitochondrial and caspase-dependent plasma membrane potential depolarization: a single-cell analysis. J Cell Sci 2003; 116: 525–536.
Ricci MS, Zong WX . Chemotherapeutic approaches for targeting cell death pathways. Oncologist 2006; 11: 342–357.
Tafani M, Schito L, Anwar T, Indelicato M, Sale P, Di Vito M et al. Induction of autophagic cell death by a novel molecule is increased by hypoxia. Autophagy 2008; 4: 1042–1053.
Liu H, Yu S, Zhang H, Xu J . Angiogenesis impairment in diabetes: role of methylglyoxal-induced receptor for advanced glycationend products, autophagy and vascular endothelial growth factor receptor 2. PLoS One 2012; 7: e46720.
Zhang ZG, Zhang L, Jiang Q, Zhang R, Zhang R, Davies K et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest 2000; 106: 829–838.
Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA . Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 2004; 56: 549–580.
Loboda A, Jazwa A, Wegiel B, Jozkowicz A, Dulak J . Heme oxygenase-1-dependent and -independent regulation of angiogenic genes expression: effect of cobalt protoporphyrin and cobalt chloride on VEGF and IL-8 synthesis in human microvascular endothelial cells. Cell Mol Biol 2005; 51: 347–355.
Liu S, Wu P, Ye D, Huang Y, Zhou X, Li Y et al. Effects of lipoxinA(4) on CoCl(2)-induced angiogenesis and its possible mechanisms in human umbilical vein endothelial cells. Pharmacology 2009; 84: 17–23.
Folkman J . Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 2007; 6: 273–286.
Janku F, McConkey DJ, Hong DS, Kurzrock R . Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8: 528–539.
Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 2012; 72: 1773–1783.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Shen HM, Codogno P . Autophagic cell death: loch ness monster or endangered species? Autophagy 2011; 7: 457–465.
Hannigan AM, Gorski SM . Macroautophagy: the key ingredient to a healthy diet? Autophagy 2009; 5: 140–151.
Malik F, Kumar A, Bhushan S, Khan S, Bhatia A, Suri KA et al. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic cell death of human myeloid leukemia HL-60 cells by a dietary compound withaferin A with concomitant protection by N-acetyl cysteine. Apoptosis 2007; 12: 2115–2133.
Kondo T, Ohta T, Igura K, Hara Y, Kaji K . Tea catechins inhibit angiogenesis in vitro, measured by human endothelial cell growth, migration and tube formation, through inhibition of VEGF receptor binding. Cancer Lett 2002; 180: 139–144.
Kumar A, Malik F, Bhushan S, Shah BA, Taneja SC, Pal HC et al. A novel parthenin analog exhibits anti-cancer activity: activation of apoptotic signaling events through robust NO formation in human leukemia HL-60 cells. Chem Biol Interact 2011; 193: 204–215.
Baker M, Robinson SD, Lechertier T, Barber PR, Tavora B, D'Amico G et al. Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc 2011; 7: 89–104.
Song Y, Dai F, Zhai D, Dong Y, Zhang J, Lu B et al. Usnic acid inhibits breast tumor angiogenesis and growth by suppressing VEGFR2-mediated AKT and ERK1/2 signaling pathways. Angiogenesis 2012; 15: 421–432.
Acknowledgements
We are thankful to the Council of Scientific and Industrial Research, India for financial assistance to carry out this research work. We are also thankful to Director III M, Ram Vishwakarma, for his generous support during these studies. We appreciate the technical assistance provided by Dr. Praduman R Sharma and Mr. Ashok Kumar during microscopy. We are grateful to the University Grants Commission, India for providing research fellowship to SK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by GM Fimia
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Kumar, S., Guru, S., Pathania, A. et al. Autophagy triggered by magnolol derivative negatively regulates angiogenesis. Cell Death Dis 4, e889 (2013). https://doi.org/10.1038/cddis.2013.399
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.399
Keywords
This article is cited by
-
Angiogenesis-on-a-chip coupled with single-cell RNA sequencing reveals spatially differential activations of autophagy along angiogenic sprouts
Nature Communications (2024)
-
FOXA1 prevents nutrients deprivation induced autophagic cell death through inducing loss of imprinting of IGF2 in lung adenocarcinoma
Cell Death & Disease (2022)
-
The P-type ATPase transporter ATP7A promotes angiogenesis by limiting autophagic degradation of VEGFR2
Nature Communications (2021)
-
Small extracellular vesicles secreted by human iPSC-derived MSC enhance angiogenesis through inhibiting STAT3-dependent autophagy in ischemic stroke
Stem Cell Research & Therapy (2020)
-
Arginine deaminase from Pseudomonas aeruginosa PS2: purification, biochemical characterization and in-vitro evaluation of anticancer activity
3 Biotech (2020)
