
Abstract
FAK is a non-receptor tyrosine kinase contributing to migration and proliferation downstream of integrin and/or growth factor receptor signaling of normal and malignant cells. In addition to well-characterized tyrosine phosphorylations, FAK is phosphorylated on several serines, whose role is not yet clarified. We observed that phosphorylated FAK on serine 732 (P-FAKSer732) is present at variable levels in vitro, in several melanoma, ovarian and thyroid tumor cell lines and in vivo, in tumor cells present in fresh ovarian cancer ascites. In vitro P-FAKSer732 was barely detectable during interphase while its levels strongly increased in mitotic cells upon activation of the EGFR/MEK/ERK axis in an integrin-independent manner. P-FAKSer732 presence was crucial for the maintenance of the proliferation rate and its levels were inversely related to the levels of acetylated α-tubulin. P-FAKSer732 localized at the microtubules (MTs) of the spindle, biochemically associated with MTs and contributed to MT depolymerization. The lack of the phosphorylation on Ser732 as well as the inhibition of CDK5 activity by roscovitine impaired mitotic spindle assembly and correct chromosome alignment during mitosis. We also identified, for the first time, that the EGF-dependent EGFR activation led to increased P-FAKSer732 and polymerized MTs. Our data shed light on the multifunctional roles of FAK in neoplastic cells, being involved not only in integrin-dependent migratory signaling but also in integrin-independent MT dynamics and mitosis control. These findings provide a new potential target for inhibiting the growth of tumor cells in which the EGFR/MEK/ERK/CDK5 pathway is active.
Similar content being viewed by others


Main
FAK, a ubiquitously expressed non-receptor tyrosine protein kinase, has a key role in regulating signals at sites of cell extracellular matrix adhesion through integrins and at activated growth factor receptors.1 Integrin-dependent activated FAK is recruited to focal adhesions (FAs) and promotes dynamic actin and adhesion changes at the membrane and signaling to proliferation and to survival pathways. FAK expression is required for many normal cellular functions in development and angiogenesis,2, 3 and its expression is upregulated in a variety of late-stage cancers.4 Through different molecular connections, FAK can influence cell movement regulating cytoskeleton remodeling, FA turnover and membrane protrusions.1 FAK also seems to affect cell proliferation by regulating cell cycle progression from G1 to S phases by increasing the expression of cyclin D1.5, 6 For all these reasons, FAK expression and activation has been linked to cell transformation and metastasis.
The overall molecular structure of FAK is also consistent with its function as a protein scaffold. The amino-terminal tyrosine (Tyr) 397 residue of FAK, whose autophosphorylation leads to full FAK activation, creates a high-affinity binding site recognized by the SH2 domain of the Src family.7 Src binding promotes FAK phosphorylation within the kinase domain activation loop at Tyr576 and Tyr577, which are necessary for maximal FAK-associated activity.1 The carboxyl-terminal domain FAK-related non-kinase (FRNK) is a truncated isoform of FAK expressed in selected cellular types.8 FRNK lacks the Tyr397, thus acting as a dominant negative isoform of FAK. FRNK inhibits both integrin- and growth factor-induced FAK activation and blocks FAK-mediated cell migration and proliferation.9
The COOH-terminal domain of FAK undergoes several serine (Ser) phosphorylation events whose role is not well understood. In neocortical neurons, phosphorylation of FAK at Ser732 (P-FAKSer732), by the post-mitotic cyclin-dependent kinase (CDK) 5, regulates microtubule (MT) stability and migration by pulling the proximal region of the nucleus into the leading process, likely in the direction of the centrosome.10, 11 In endothelial cells (ECs), P-FAKSer732, phosphorylated by Rho-associated protein kinase (ROCK),12 has been found co-localized with γ-tubulin at the centrosomes; deletion of FAK in primary ECs causes an increase in centrosome numbers, and multipolar and disorganized spindles.13
P-FAKSer732 has been characterized mainly in normal cells.10, 12, 13 Here, we aimed to analyze in detail the presence and the role of P-FAKSer732 in tumor cells.
Results
P-FAKSer732 is detected in tumor cells and is not localized at the FAs during migration
The presence of P-FAKSer732 was analyzed together with that of -Tyr576, the site responsible for maximal FAK activity, in melanoma, ovarian and thyroid cancer cells. Basically, in all cell lines the expression of FAK was also associated with the presence of P-FAKSer732 and -Tyr576, although at variable levels (Figure 1a). FAK was mainly phosphorylated on Ser732 in Me#21 and #25. High expression of FAK, P-FAKSer732 and -Tyr576 was also observed in Me#20, a primary tumor and in Me#31, a visceral metastases. All these melanoma cell lines, which also express the highest levels of FAK, belong to a subset of melanomas lacking the melanocyte transcription factor MITF and melanoma differentiation antigens.14 Three out of eight ovarian carcinomas and one out of three thyroid cell lines expressed FAK, P-FAKSer732 and -Tyr576 at high levels. In the lysates from tumor cells contained in fresh ascites from advanced stage ovarian cancer (Supplementary Table S1), FAK was also found phosphorylated on Ser732 (Figure 1b), indicating that this phosphorylation is not restricted to cells cultured in vitro.

P-FAKSer732 is detected in tumor cells and is not localized at the FAs during migration. (a) Western blot analysis on total cell lysates from melanoma (n=14), ovarian (n=9) and thyroid (n=3) cancer cell lines. (b) Western blot analysis on total cell lysates from cells of ovarian cancer ascites (n=8). The presence in the ascites of tumor cells was evaluated by using the epithelial adhesion marker e-cadherin (cadh). (c) Wound-healing assay performed on control- (CO) or FAK- (FAK) silenced (Me)#28 cells. Left panel: western blotting showing the level of FAK silencing and the wound-healing repair capability at 4, 24 and 30 h from the scratch. Asterisks indicate statistically significant values (P≤0.01). Error bar, S.D. Right panel: representative images showing the wounds at the starting point (t=0 h), after 24 h and the wounds closure at the end of the experiment (t=30 h). Immunoblottings were performed with Abs against the proteins reported on the right. β-actin was used as control of gel loading. (d) IF performed on starved Me#28 cells induced to migrate though a wound. Cells were stained for P-FAKSer732, -Tyr 397, -Tyr 576, FAK (green) and α-tubulin (red). Nuclei were stained with DAPI (blue). Merged images are reported on the upper panels. (e) IP on Me#28 lysates with normal rabbit serum (CO) or anti-P-FAKSer732 (S732). Immunoprecipitated samples were analyzed by western blotting. Immunoblottings were performed with Abs against the proteins reported on the right
The melanoma Me#28 cell line was further characterized, and, for selected experiments, the ovarian carcinoma OAW42 and the thyroid carcinoma NIM-1 cell lines were also analyzed.
In tumor cells, FAK regulates cell motility by directing cell migration. During migration, phosphorylated FAK at tyrosine residues localizes at the FAs and/or at the front and the rear of the cells.1 Indeed, transient knockdown of FAK through specific siRNA in Me#28 cells induced a statistically significant decrease of wound repair compared with cells transfected with a non-silencing siRNA both at 24 h and at 30 h (Figure 1c), when the latter had completely repaired the wound. Immunofluorescence (IF) was used to analyze the localization of FAK phosphorylated isoforms in Me#28 cells migrating into the wound. P-FAKSer732 localized in the cytoplasm and in a perinuclear region but not at FAs (Figure 1d), whereas both P-FAKTyr397 and -Tyr576 were detected in the cytoplasm and at FAs of the leading edge. In addition, western blot analysis of Me#28 samples following immunoprecipitation (IP) with anti-P-FAKSer732 antibody (Ab) indicated that the principal form in the immunoprecipitate consisted of FAK phosphorylated on Ser732 (Figure 1e), whereas the dominant form in the unbound fraction contained mainly P-FAKTyr576.
These results demonstrate that FAK exists as two distinct pools: the first associated with FAs and the second, phosphorylated on Ser732, localized in the cytoplasm in a perinuclear region.
FAK is phosphorylated at Ser732 independently from integrin engagement
To investigate whether the Ser732 phosphorylation was consequent to that at Tyr397, Me#28 cells were stably transfected with FRNK construct and the FRNK-expressing clone F6 was selected by western blotting (Figure 2a). Fetal calf serum (FCS) stimulation of Me#28 cells transfected with FRNK construct (clone F6) led to increased phosphorylation of FAK only at Ser732, whereas mock-transfected cells showed also increased P-FAKTyr397 and -Tyr576 (Figure 2b).

FAK is phosphorylated at Ser732 independently from integrin engagement. (a) Western blot analysis on total cell lysates of the clones obtained from FRNK-transfected (Me)#28. The clone F6, expressing the highest levels of FRNK protein, was selected for the experiment reported on panel b. (b) Western blotting on total lysates from Mock- or FRNK-transfected (clone F6) Me#28 cells. β-actin was used as control of gel loading. (c) Left panel: western blotting performed on lysates from starved Me#28 cells grown on plastic (−) or fibronectin (Fn) (+). β-actin was used as control of gel loading. Right panel: densitometric analysis reporting the levels of FAK phosphorylated isoforms respect to total FAK expression of cells grown on Fn. The mean values of five experiments are reported
To assess whether the phosphorylation at Ser732 was dependent on integrin engagement and activation, Me#28 cells were grown in absence of FCS for 24 h on plastic or fibronectin. Western blotting on cell lysates showed that P-FAKSer732 was not increased in cells grown onto fibronectin (Figure 2c), while P-FAKTyr397 and -Tyr576 increased, as expected, in cells grown on fibronectin indicating that P-FAKSer732 is not a downstream effector of integrin activation.
These results indicate that phosphorylation at Ser732 is independent of FAK autophosphorylation at Tyr397, while the latter is necessary for phosphorylation at Tyr576 upon integrin engagement, as already demonstrated.15
P-FAKSer732 accumulates in mitotic cells and co-localizes with MTs of the mitotic spindle
Then, we asked whether proliferative stimuli could induce P-FAKSer732. Indeed, FCS stimulation of starved Me#28, OAW42 and NIM-1 cell lines, induced, in all of them, increased levels of P-FAKSer732 (Figure 3a) that highly accumulated, as shown by IF in Figure 3b, in mitotic cells. In cells in interphase, P-FAKSer732 did not accumulate and was located in the cytoplasm, as already shown in Figure 1e.

P-FAKSer732 accumulates in mitotic cells and co-localizes with MTs of the mitotic spindle. (a) Western blotting on total lysates from starved or FCS-stimulated cells. (b) IF performed on fixed cells upon FCS stimulation with anti-P-FAKSer732 Ab (green). Nuclei were stained with DAPI (blue). (c) Confocal IF performed on fixed Me#28 cells with anti-P-FAKSer732 and ac-α- (left and middle panels) or γ-tubulin Abs (right panel). Images of metaphases (left and right panels, section 14 and 12, respectively) and late anaphase (section 11, middle panel) are shown. Bars, 10 μm. (d) IF performed on Me#28 cells upon extraction of free tubulin with PEM buffer (see Materials and Methods). The white arrows indicate the mitotic phases: 1, prophase; 2 anaphase; 3, telophase; 4, cytokinesis. The immunostaining was performed with anti-P-FAKSer732 (red) and ac-α-tubulin (green). Nuclei were stained with DAPI (blue). Bars, 60 μm. (e) IP on lysates from Me#28 cells with anti-P-FAKSer732 (S732) and -dynein intermediate chain Abs. Immunoprecipitated fractions were analyzed by western blotting. Normal rabbit or mouse sera were used as negative control (CO). For immunoblottings, Abs are reported on the right
Confocal IF was performed to better investigate on P-FAKSer732 localization in dividing Me#28 cells. P-FAKSer732 co-localized with α-tubulin at the mitotic spindle during metaphase (Figure 3c, left panel). During late anaphase, P-FAKSer732 co-localized with α-tubulin at the spindle, likely at the sites where MTs elongate and shrink (Figure 3c, middle panel), and accumulated in the middle of the spindle, indicating an interaction with the cortical actin. Anti-P-FAKSer732 also diffusely stained the cell cytoplasm, but no co-localization with γ-tubulin at the centrosome was observed (Figure 3c, right panel).
Furthermore, P-FAKSer732 co-localized with ac-α-tubulin of the spindle in all mitotic phases upon extraction of soluble tubulin (Figure 3d).
The MT motor protein dynein regulates MT dynamics and cortical pulling forces at the spindle poles.16 To biochemically analyze the possible association between FAK, observed mainly at the MTs of the spindle, and dynein, an IP was performed on Me#28 lysates with anti-P-FAKSer732 and -dynein intermediate light chain Abs. Anti-P-FAKSer732 co-immunoprecipitated with both ac-α-tubulin and dynein (Figure 3e, left panel). Accordingly, anti-dynein Ab co-immunoprecipitated with FAK phosphorylated on Ser732 as well as with ac-α-tubulin (Figure 3e, right panel).
Therefore, P-FAKSer732 associates with polymerized MTs of the spindle during all mitotic stages and is biochemically linked to dynein arguing for the notion of a role during mitosis.
FAK contributes to cell proliferation when it is phosphorylated at Ser732
To assess a role of P-FAKSer732 in proliferation, Me#28 cells were treated with a specific siRNA against FAK and growth capability was assayed. The growth rate of melanoma cells decreased 25% after 48 h and up to 40% after 72 h (Figure 4a). Furthermore, phosphorylation on Ser10 of Histone H3 (herein named PHH3), a marker of mitosis,17 was 30% lower in FAK-silenced cells in comparison with cells transfected with a control siRNA (Figure 4b), suggesting a role of FAK in the control of the mitosis.

FAK contributes to cell proliferation when is phosphorylated at Ser732. (a) Proliferation assay performed on cells silenced with control- (siCO) or FAK- (siFAK) siRNA. Representative growth curves of one of three independent experiments are shown; each point represents the mean of three replicates. Error bars, S.D. Asterisks indicate statistical significant values (P<0.0001). (b) Mitotic index performed on control- (siCO) or FAK- (siFAK) silenced (Me)#28 cell. The level of mitosis is reported as the percentage of PHH3-positive cells measured by FACS. Asterisk, P≤0.05. (c) Proliferation rate of control- (siCO)- or FAK- (siFAK)-silenced Me#28 cells transfected with GFP alone, wt GFP- or Mut GFP-FAK vectors. The experiment was done in triplicate. Asterisks indicate statistical significant values (P<0.001). Error bars, S.D. (d) Western blotting on cell lysates of the transfectants described above, treated with control (siCO) or FAK (siFAK) siRNA. Endogenous FAK, thin arrow, ectopic FAK, thick arrow. For immunoblottings, Abs are reported on the right. β-actin was used as control of gel loading
To specifically assess a role of P-FAKSer732 in cell proliferation, we transiently transfected, in control or FAK-silenced Me#28 cells, a GFP construct alone, or containing the wild-type (wt) GFP-FAK or a mutated FAK on Ser732→Ala10 (herein named Mut GFP-FAK). Control silenced cells transfected with Mut GFP-FAK displayed a 53% growth rate reduction in comparison with those transfected with wt GFP-FAK (Figure 4c). Growth reduction reached 70% when Mut GFP-FAK was transfected into FAK-silenced cells. The re-expression of wt GFP-FAK in FAK-silenced cells was not able to return proliferation to that observed in control-siRNA-treated cells although a 30% increase was observed respect to siFAK-treated cells transfected with GFP only. These data argue for the hypothesis that a delicate balance between total FAK and P-FAKSer732 is necessary to maintain the correct growth rate.
The above reported results were further confirmed by western blotting of cell lysates from the transfectants described above. Both wt and Mut GFP-FAK-transfected cells showed a decrease of PHH3 (Figure 4d), indicating a role in the entry in mitosis. Conversely, expression of the Mut GFP-FAK increased the amount of polymerized MTs, evaluated with anti-acetylated α-tubulin (ac-α-tubulin) Ab.
These data strongly indicate that P-FAKSer732 is associated to an increased growth potential and, together with the results shown above, also argue for the hypothesis of its involvement in the regulation of the MT dynamics and in the mitotic spindle assembly.
P-FAKSer732 contributes to MT depolymerization
To investigate whether the effect on MTs, analyzed as ac-α-tubulin, could only be due to P-FAKSer732, NIM-1 cells, that express low levels of FAK (Figure 1a), were transfected with the construct expressing wt and Mut GFP-FAK. Western blot analysis of cell lysates showed that cells transfected with Mut GFP-FAK contained about 2.5-fold more ac-α-tubulin compared with cells transfected with wt GFP-FAK (Figure 5a). Conversely, iper-expression of FAK, as in wt GFP-FAK-transfected cells, was associated to 50% decrease of ac-α-tubulin respect to GFP-transfected cells.

P-FAKSer732 contributes to MT depolymerization. (a) Upper panel: western blotting on cell lysates from NIM-1 cells transfected with GFP alone, wt or Mut GFP-FAK vectors. Endogenous FAK, thin arrow, ectopic FAK, thick arrow. Lower panel: densitometric analysis performed on the western blotting shown in the upper panel. The results of an experiment of three performed are shown. (b) MT re-growth assay was performed on Me#28 and OAW42 cells transfected with control (siCO) or FAK- (siFAK) siRNA. For immunoblottings, Abs are reported on the right. β-actin was used as control of gel loading
The fact that the amount of polymerized MTs was inversely related to the amount of P-FAKSer732 argues for the hypothesis that FAK can modulate MT dynamics. To test this possibility, we performed a MT re-growth assay on Me#28 and OAW42 cells upon FAK silencing. Silenced cells were treated overnight (o.n.) with nocodazole to force MT depolymerization, evaluated as the absence of ac-α-tubulin, and then MT re-growth was analyzed after nocodazole washout. FAK-silenced cells showed a higher amount of ac-α-tubulin indicating higher levels of polymerized MTs and, after nocodazole treatment, ac-α-tubulin was slightly detected. Of note, the knockdown of FAK induced a better rescue of MT re-growth after nocodazole removal compared with cells transfected with control siRNA (Figure 5b).
These data strongly indicate that FAK phosphorylated at Ser732 has a role in MT depolymerization.
Lack of Ser732 phosphorylation impairs spindle assembly and mitosis
To specifically evaluate the role of P-FAKSer732 during mitosis, wt and Mut GFP-FAK were transiently transfected in Me#28 and NIM-1 cells. In interphase, wt (Figure 6a, left panel) and Mut GFP-FAK (Figure 6b, left panel) were mainly localized at the sites of the FAs, and no differences were observed between the two transfectants. In mitotic cells, Mut GFP-FAK localized in randomly positioned spindle showing a diffused staining along the entire spindle and the chromosomes appeared significantly unaligned (Figure 6b, right panel). wt GFP-FAK-transfected cells showed a regular spindle with wt GFP-FAK mainly localized on polar MTs (Figure 6a, right panel). In agreement with these results, the number of mitosis was six- and threefold lower in Mut GFP-FAK-transfected Me#28 and NIM-1 cells (Figure 6c).

Lack of Ser732 phosphorylation impairs spindle assembly and mitosis. Me#28 and NIM-1 cells transiently transfected with wt (a) or Mut GFP-FAK (b) vectors. IF was performed with anti-α-tubulin (red) Ab on fixed cells. Nuclei were stained with DAPI (blue). Inserts on the bottom report green/red merge showing the localization in the spindle of the exogenous wt or Mut GFP-FAK. Bars, 10 μm. (c) Number of mitosis in transfected cells. Error bars, S.D. Asterisks indicate P≤0.05
The MEK/ERK pathway is involved in the phosphorylation at Ser732 associated with polymerized MTs
Several solid tumors have a hyper-activated MEK/ERK pathway and ERK was already proposed to be a regulator of MT dynamics and mitosis.18, 19 Thus, a possible involvement of MEK/ERK and Rho/ROCK pathways, which generates P-FAKSer732 in ECs,12 was analyzed upon treatment with UO126 and Y27632, which are inhibitors of MEK and ROCK, respectively. In FCS-stimulated Me#28 cells, UO126 inhibited P-FAKSer732 nearly 90% and Y27632 reduced P-FAKSer732 up to 30% (Figure 7a). As FAK can be an up- and a downstream effector of both MEK/ERK and Rho/ROCK pathways,20 the effects of FAK expression on these pathways was analyzed. Transient FAK silencing in Me#28 cells induced 35% reduction of ERK phosphorylation (Figure 7b), confirming that FAK expression and/or activation could regulate ERK phosphorylation. Y27632 treatment in control siRNA-transfected cells slightly decreased ERK phosphorylation and reached a 70% decrease in FAK-silenced cells arguing for the notion that ROCK further induced the decrease of ERK phosphorylation when FAK decreased. As shown above, UO126 completely inhibited P-FAKSer732. These results strongly indicate that MEK/ERK pathway is responsible for P-FAKSer732.

The MEK/ERK pathway is involved in the phosphorylation at Ser732 associated with polymerized MTs. (a) Me#28 cells treated with UO126 or Y27632 inhibitors for 24 h. Upper panel: western blotting performed on lysates from untreated or treated cells. Bottom panel: densitometric analysis of the western blotting shown in the upper panel, reporting the percentage of P-FAKSer732 respect to total FAK upon inhibitor treatments. Error bars, S.D. (b) Upper panel: western blotting performed on lysates from Me#28 cells transfected with control- (CO) or FAK- (FAK) siRNA. Bottom panel: densitometric analysis of the western blotting shown in the upper panel, reporting the percentage of P-ERK respect to total ERK upon inhibitor treatments. β-Actin was used as control of gel loading. (c) Western blotting on lysates from starved Me#28 and OAW42 cells stimulated with FCS and treated with increasing concentration of UO126 (1–20 μM). Total ERK was used as control of gel loading. (d) Western blotting of soluble (S) or polymerized MT (MT) fractions (see Materials and Methods) from Me#28 cells left untreated or treated with UO126 (20 μM)
Obserivng the relationship among P-FAKSer732, MEK/ERK activation and polymerized tubulin (Figures 4 and 5), we assessed whether MEK/ERK inhibitor was able to modulate the amount of polymerized tubulin. UO126 treatment on Me#28 and OAW42 cells increased the amount of ac-α-tubulin and decreased the amount of P-FAKSer732 in a dose-dependent manner, indicating an inverse correlation between FAK, phosphorylated on Ser732 by MEK/ERK and MT polymerization (Figure 7c).
The biochemical association of P-FAKSer732 with polymerized MTs was then investigated by western blotting on cell extracts upon separation of soluble and polymerized α-tubulin fractions. P-FAKSer732 was found in both soluble and MT fractions, but UO126 treatment decreased the levels of P-FASer732 in the MT fraction only (Figure 7d), indicating that a part of P-FAKSer732 is associated with ac-α-tubulin and that de novo phosphorylation on Ser732 of FAK occurs at the level of MTs. Ac-α-tubulin of MT fraction increased in UO126-treated cells.
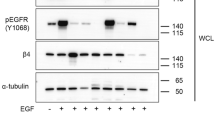
EGF-dependent EGFR/MEK/ERK/CDK5 pathway induces P-FAKSer732, thus contributing to mitosis
MEK/ERK pathway is one of the downstream effector of EGFR21 and CDK5 phosphorylates FAK on Ser732 in neural cells.11 To test the hypothesis that P-FAKSer732 is induced by EGFR/CDK5 pathway activation, starved Me#28, NIM-1 and OAW42 cells were stimulated with EGF. In all the three cell lines, EGF stimulation increased P-ERK together with P-FAKSer732 and P-CDK5Tyr15, the active kinase isoform (Figure 8a). To test whether CDK5 was downstream of EGFR/MEK/ERK signaling cascade, ERK2 siRNA-treated Me#28 cells were stimulated with EGF. EGF stimulation strongly decreased P-FAKSer732 and P-CDK5Tyr15 of ERK2-silenced cells but not of control siRNA-treated cells (Figure 8b). To confirm the presence of the axis MEK/ERK/CDK5/P-FAKSer732, starved Me#28, OAW42 and NIM-1 cells were treated with increasing concentrations of roscovitine, a potent CDK5 inhibitor.22 Roscovitine treatment decreased the amount of P-FAKSer732 in a dose-dependent manner in all cell lines (Figure 8c), but did not affect the level of P-FAKTyr397 and of P-ERK. Moreover, roscovitine treatment increased the amount of ac-α-tubulin in a dose-dependent manner.

EGF-dependent EGFR/MEK/ERK/CDK5 pathway induces P-FAKSer732, thus contributing to mitosis. (a) Western blotting on lysates from starved Me#28, NIM-1 and OAW42 cells stimulated with EGF (20 ng/ml) for up to 20 min. For immunoblottings, Abs are reported on the right. β-Actin was used as control of gel loading. (b) Western blotting on cell lysates from Me#28 cells silenced with control (−) or ERK2 (+) siRNA, starved and then stimulated with EGF (20 ng/ml) for 15 min. Abs are reported on the right. α-Tubulin was used as control of gel loading. (c) Western blotting on lysates from starved Me#28, OAW42 and NIM-1 cells untreated or treated with roscovitive (2–20 μM) and stimulated with FCS. Abs are reported on the right. β-Actin was used as control of gel loading. (d) Confocal IF performed on fixed NIM-1 cells untreated or treated with roscovitive (10 μM) and stained with anti-P-FAKSer732 (green) and α-tubulin (red) Abs. Scale bars, 20 μm
IF on roscovitine-treated NIM-1 cells showed impaired spindle (Figure 8d), according to the data reported in Figure 6. P-FAKSer732 staining was lower and diffused with respect to that observed on FCS-treated cells, and no co-localization with acetylated α-tubulin was observed.
These data strongly indicate that EGFR/MEK/ERK/CDK5 pathway induces the phosphorylation of FAK at Ser732 independently from the FAK autophosphorylation, thus contributing to spindle formation and mitosis of tumor cells.
Discussion
We present here a deep analysis on the role of FAK phosphorylated on Ser 732 in tumor cells. We have found that the phosphorylation of FAK at Ser732 occurs independently from integrin activation, is not localized at the sites of FAs and accumulates in mitotic cells strongly suggesting that, regardless of FAK involvement in migration, P-FAKSer732 has a role in mitosis, thus regulating the proliferation of tumor cells. P-FAKSer732 appears to control mitotic spindle assembly and correct chromosome segregation. Hence, FAK can exert in tumor cells a dual role: one, fully described elsewhere, integrin-dependent associated to phosphorylation of Tyr397 and -576 with a role in migration, and the other, characterized here for the first time, integrin-independent associated to phosphorylation on Ser732 upon activation of the EGFR/MEK/ERK/CDK5 pathway, regulating MT dynamics.
Although P-FAKSer732 was already observed in neural and in few non-neural cells, that is, Cos, HeLa cells and ECs, phosphorylated by CDK5,10, 13 our results clearly increase the knowledge about the role of FAK phosphorylated on Ser732 focusing in tumor cells. While in migrating neural cells P-FAKSer732 was shown to be responsible for the MT assembly to generate forces for nuclear movement, in ECs P-FAKSer732 associated to the centrosomes was related to centrosome function during mitosis. On the contrary, we found P-FAKSer732 localized at the mitotic spindle and not at the centrosomes indicating that P-FAKSer732 exerts, in the analyzed tumor cells, a role at the level of the spindle MTs. Indeed, we also gave evidences of an inverse relationship between P-FAKSer732 and ac-α-tubulin, and the MT re-growth assay clearly showed that the lack of FAK expression delayed MT polymerization, strongly indicating for the first time a role of FAK in MT dynamics.
Dynein is a minus-end MT motor protein that contribute to the generation of forces necessary to keep the spindle pole at the correct distance from the equator.23 On the other hand, different actin structures and actin-associated proteins contribute to spindle orientation through the anchorage of astral MTs.24 The localization of P- FAKSer732 at both MTs of the spindle and cortical ring as well as the association with dynein suggest a role of P-FAKSer732 as molecular cue for correct MT dynamics, spindle assembly and/or correct chromosome segregation. Further studies will be required to elucidate the involvement of FAK during mitotic spindle assembly and positioning.
In solid tumors, one of the hallmarks is the chromosome instability caused by wrong chromosome segregation. The alteration of MT dynamics is one of the causes of the chromosomal instability.25 In this context, P-FAKSer732 seems to participate to MT depolymerization, thus maintaining normal spindle assembly and function. However, as already observed for MCAK, the mitotic centromere-associated kinesin,26 the phosphorylation of FAK at the Ser732 could be finely orchestrated by kinases and phosphatases specifically activated in tumor cells. Therefore, future attention should be paid to analyze, in a wide panel of tumor biopsies, as well as in their normal counterparts, the relationship between the presence of P-FAKSer732 and the clinical outcome or sensitivity to conventional therapies. In addition, our data also suggests that the activation of the identified signaling pathway could be preferentially activated in different tumor subsets. Indeed, melanoma cell lines displaying the highest levels of P-FAKSer732 belong to a tumor subgroup lacking MITF and melanoma differentiation antigens such as MART-1 and gp100.14 The presence of P-FAKSer732 in ovarian carcinoma cells from ascites further supports a correlation between this particular FAK phosphorylation and malignant cells with high proliferative capability in vivo. These data indicate that the analysis of the involvement of FAK in the mitotic spindle assembly and function in tumor cells appear to be instrumental for developing specific inhibitors to this particular FAK function in malignant cells.
We also showed that P-FAKSer732 was generated by the activation of the EGFR/MEK//ERK/CDK5 signaling. In neural cells, CDK5 is considered a post-mitotic kinase whose activation is dependent on the expression of CDK activators p35 and p39.27 In tumors, p35-dependent activation of CDK5 appeared to occur following ERK activation, which in turn led to increased Egr1 transcriptional activity and upregulation of p35 expression.28 In pancreatic cancer, inhibition of CDK5 kinase activity was found to suppress ras signaling,29 indicating a possible relationship between MEK/ERK and CDK5 activations. As we have clearly shown here that the MEK/ERK pathway activation and the activated P-CDK5Tyr15 are associated with the generation of P-FAKSer732, at least in the analyzed tumor cells, the induction of p35 or p39 expression can be hypothesized. Several years ago, Harrison and Turley18 showed in H-ras-transformed cells that ERK associated with the MTs of the mitotic spindle, thus regulating their stability. Based on our results, we hypothesize that those observations were not due to ERK itself, but due to FAK upon phosphorylation at Ser732 caused likely by CDK5. Analysis of CDK5 expression and activation as well as p35 and p39 expression await further investigation in tumor cells especially when the ras/RAF/MEK/ERK is constitutively activated by mutations.
Although ROCK does not appear to phosphorylate FAK on Ser732, as suggested previously,12 we cannot exclude involvement of Rho/ROCK pathway in the activation of the signaling circuit MEK/ERK/FAK/ERK. This hypothesis is not weird, as the Rho/ROCK pathway regulates actin dynamics through cofillin30 and spindle orientation is regulated by cortical actin.24 Experiments are ongoing to better clarify the mechanism that links Rho/ROCK pathway and P-FAKSer732.
The link between EGFR activation and P-FAKSer732, never demonstrated before for tumor cells, might be considered an important step forward in understanding EGFR function in tumors. Further knowledge may be useful to design alternative therapeutic approach, especially in tumors that become resistant to anti-EGFR compounds.
Materials and Methods
Antibodies and reagents
Abs against P-FAKSer732, -Tyr576 and -Tyr 397 (rabbit) were from Biosource (Invitrogen, San Francisco, CA, USA); anti-FAK (rabbit); anti-MAP kinase (ERK1/2; rabbit); anti-P-EGFRTyr1173 (mouse), anti-EGFR (rabbit), anti-carboxyl-terminal domain of FAK from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-α-tubulin (mouse) from Thermo Scientific (Fremont, CA, USA); anti-ac-α-tubulin (mouse) from Promega (Promega, Madison, WI, USA); anti-β-actin (rabbit), anti-dynein intermediate chain (mouse) from Sigma-Aldrich (St. Louis, MO, USA); anti-phosphorylated ERK1/2 kinase (rabbit), anti-P-histone H3 (Ser10) (rabbit) from Cell Signaling Technology (New England BioLabs, Beverly, MA, USA); anti-CDK5Tyr15 (rabbit) from Abcam (Abcam, Cambridge, UK) Fluorochrome-conjugated Alexa Fluor 488 and 546 secondary Abs were from Molecular Probes (Invitrogen). UO126 was from Promega; Y27632, nocodazole and roscovitine were from Sigma-Aldrich. Human recombinant EGF was from PeproTech, Inc. (Rocky Hill, NJ, USA). G418 and Lipofectamine 2000 were purchased from Invitrogen.
Cells
Human melanoma cell lines were obtained from surgical specimens of patients admitted to INT. These cells have been extensively characterized as reported elsewhere.14 Nine human ovarian cancer cell lines were used in this study: IGROV-1 kindly provided by Dr. Bénard (Paris, France);31 SKOV3 and CAOV4 from ATCC; OVCAR4 and OVCAR5 provided by Dr. Camalier (NCI-NIH, USA); 3507OVA and 413OVA by Dr. van den Berg-Bakker (Leiden, the Netherlands);32 COR by Dr. Balconi (M Negri, Milan, Italy). They were maintained in RPMI 1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10% FCS (Hyclone, Logan, UT, USA), 1% L-glutamine at 37 °C in a humidified atmosphere of 5% CO2 in air. OAW42 was provided by Dr. A. Ullrich, (Max Planck Institute of Biochemistry, Martinsried, Germany)33 and maintained in DMEM supplemented with 10% FCS and 1% L-glutamine in a humidified atmosphere of 5% CO2. The human thyroid cell lines NIM-1,34 TPC1 and BCPAP35 were maintained in DMEM supplemented with 10% FCS and 1% L-glutamine in a humidified atmosphere of 5% CO2 in air. Ascites from patients with stage III ovarian cancer were collected at the time of debulking surgery. Cytological analysis confirmed that the ascites mainly contained tumor cells, as also demonstrated by the expression of the epithelial marker E-cadherin. The features of ovarian carcinoma specimens are listed in Supplementary Table S1. UO126, Y27632 and roscovitine treatment were performed o.n. on starved cells before the relevant stimulation.
Stable transfection
Me#28 cells were placed on 96-well dishes and transfected after 24 h with 0.2 μg per well of the FRNK-containing vector (kindly provided by Dr. Ungefrorer, Hamburg) and pcDNA3.1 (Invitrogen), as mock-transfected vector, using Lipofectamine 2000 reagent following the manufacturer’s protocol. At 48 h after the transfection, G418-containing media was added and changed every 48–72 h thereafter to generate stable transfectants. After 2 weeks, isolated colonies were analyzed by western blotting for the presence of FRNK (41 kDa) in total cell lysates using the anti-carboxyl-terminal domain of FAK.
Cell growth on fibronectin
Fibronectin-coated 12-well coated plate was prepared with a 2 μg/ml fibronectin solution in RPMI and incubated (o.n.) at 37 °C. The next day, cells (0.8 × 105/well) were plated in serum-free condition on plastic or fibronectin for 20 min. Western blotting was performed on total lysates as described below.
Western blotting and IP
Cells were washed with ice-cold PBS containing Na3VO4 (0.1 mM) and lysated with NuPAGE LDS sample buffer (1 × ) (Invitrogen) under reducing conditions. Proteins were separated on precast 4–12% SDS-polyacrylamide gels (Invitrogen). Western blot was performed as described36 and analyzed using ChemiDoc XRS System and the Quantity One software (Bio-Rad, Hercules, CA, USA). The relevant values were always normalized with the values of the protein used as control of gel loading.
The separation of soluble and polymerized tubulin has been described elsewhere.37 In some experiments, cells were grown in standard medium o.n., washed twice in PEM buffer (100 mM PIPES pH 6.9, 1 mM EGTA, 2 mM MgCl2) and then cells were incubated in PEM buffer containing 0.2% Triton X-100 for 1 min at 37 °C and then rinsed and incubated in PEM buffer at 37 °C for 5 min. Cells were lysed as described above.
IP was performed essentially as described.36 Briefly, cells were lysed in cold lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 0.25% NaDoc, 1 mM EDTA pH 8, 1 mM Na3VO4, 1% PMSF, 1% NP-40 and protease inhibitors) for 30 min on ice. Cellular debris and nuclei were removed by centrifugation at 13 000 r.p.m./min for 30 min at 4 °C. Normal rabbit or mouse sera were used as negative control. Primary Ab was bound to beads conjugated with goat anti-mouse or -rabbit Ab (Dynabeads; Dynal ASA, Oslo, Norway) and incubated with cell extracts o.n. at 4 °C with rotation. Beads were washed once with cold lysis buffer, twice with PBS plus BSA and protease inhibitors, and once with PBS plus inhibitor (10 min/wash). Immunoprecitated proteins were separated on 4–12% SDS-PAGE under reducing conditions.
siRNA treatment and transient transfection
siRNA transfections were performed using Lipofectamine 2000 according to the manufacturer’s protocol. For FAK silencing, 40 pmol/ml small-interfering RNA (siRNA) duplex specific for FAK (Smart Pool, Thermo Scientific, Dharmacon, Inc. Chicago, IL, USA) or control siRNA (Qiagen-Xeragon, Germantown, MD, USA) were applied. Total cell lysates were prepared 48 h after transfection. Otherwise, 24 h after siRNAs treatment, transfected cells were placed on six-well dishes and transfected with 2 μg of GFP, wt GFP- and Mut GFP-FAK vectors10 using Lipofectamine 2000 reagent (Invitrogen), following the manufacturer’s protocol. In some experiments, cells were transfected with wt and Mut GFP-FAK vectors without FAK silencing.
For ERK2 silencing, 40 pmol/ml siRNA duplex specific for MAPK1(Smart Pool, Thermo Scientific, Dharmacon, Inc.) or control siRNA (Qiagen-Xeragon) were applied. Treatment with EGF (20 ng/ml) was carried out 24 h after transfection in a serum-free medium. Total cell lysates were prepared 48 h after transfection.
Wound-healing assay
Confluent cultures of Me#28 transfected with FAK siRNA or control siRNA were wounded using a sterile 200-μl pipette tip. Wound closure was assessed 4, 24 and 30 h later with an inverted microscope with a × 10, 0.75 NA PanFluor objective (Nikon, Tokyo, Japan). Images were acquired with ACT-1 software (Nikon) at a resolution of 1280 × 1024 pixels. The wound closure was measured on 10 different micrographs captured in three wells for each condition. Data are expressed as percentage of wound closure of the original wound width.
Immunofluorescence
Cells (2 × 104) were seeded on 8-well glass chamber slides (Nalge Nunc International, Rochester, NY, USA). Before immunostaining, cells were washed with cold PBS, fixed with 2% paraformaldehyde for 20 min and permeabilized for 10 min in PBS/Tween 0.1%. To dilute out of the cell-free tubulin heterodimers cells grown on 8-well glass chamber slides were washed twice in PEM buffer.37 Then cells were incubated in PEM buffer containing 0.2% Triton X-100 for 1 min at 37 °C and then rinsed and incubated in PEM buffer at 37 °C for 5 min. Cells were then fixed in methanol/acetone (1 : 1). Samples were mounted with Prolong Gold Antifade reagent with DAPI (Invitrogen) and analyzed using an Eclipse TE2000-S microscope with a × 40, 0.75 NA PanFluor objective (Nikon). Images were acquired with ACT-1 software (Nikon). Confocal microscopy was carried out using a Microradiance 2000 microscopy (Bio-Rad Laboratories, Hercules, CA, USA) equipped with Ar (488 nm) and HeNe (543 nm) lasers. Images were obtained using a × 40 oil immersion lens 1.4 NA and analyzed using Lasersharp 2000 software. z-stacks of 24 (metaphase) and 19 (anaphase) sections were acquired. Images were processed using ImageJ and Adobe Photoshop.
Cell proliferation and mitotic index
Cells (1 × 103 cells/well) were plated into 48-well plates. At 0, 24, 48 and 72 h, the number of cells was evaluated by Countess automated cell counter (Invitrogen).
For evaluation of the mitotic index, cells were transiently transfected with siRNA. After 48 h, trypsinized cells were fixed by adding 4.5 ml of ice-cold 70% ethanol and incubated at 4 °C. After two washes with PBS/0.03% BSA, cells were incubated with anti-PHH3 (1 : 40) diluted in PBS/0.03% BSA for 1 h at 37 °C followed by incubation with Alexa Fluor 488 goat-anti-rabbit Ab (1 : 1000) in PBS/0.03% BSA for 1 h at room temperature. The mitotic index was reported as the percentage of PHH3-positive cells determined by FACS using the FACSCalibur (Becton Dickinson, San Jose, CA, USA) with CELLQuest software.
MT re-growth assay
Cells (8 × 104/well) seeded on six-well plates, were transfected with FAK or control siRNA and grown for 24 h. MTs were depolymerized treating the cells with 5 μg/ml nocodazole in complete medium at 37 °C o.n. Cells were washed with complete medium once and incubated at 37 °C. For Me#28 and OAW42 cells, the MT re-growth was allowed for 30 min and 3 h, respectively. Total cell lysates were analyzed by western blotting.
Statistical analyses
GraphPad Prism software (GraphPad Software, San Diego, CA, USA) was used to analyze all data. Differences between mean values were determined by Student's t-test and 2way ANOVA test.
Abbreviations
- Ab:
-
antibody
- ac-α-tubulin:
-
acetylated α-tubulin
- CDK:
-
cyclin-dependent kinase
- EC:
-
endothelial cell
- FA:
-
focal adhesion
- FRNK:
-
FAK-related non kinase
- IF:
-
immunofluorescence
- IP:
-
immunoprecipitation
- MAb:
-
monoclonal antibody
- MT:
-
microtubule
- Mut:
-
mutated
- o.n.:
-
overnight
- ROCK:
-
Rho-associated protein kinase
- Ser:
-
serine
- Tyr:
-
tyrosine
- wt:
-
wild type
References
Schaller MD . Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci 2010; 123: 1007–1013.
Corsi JM, Houbron C, Billuart P, Brunet I, Bouvree K, Eichmann A et al. Autophosphorylation-independent and -dependent functions of focal adhesion kinase during development. J Biol Chem 2009; 284: 34769–34776.
Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R . Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol 2006; 172: 151–162.
Gabarra-Niecko V, Schaller MD, Dunty JM . FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev 2003; 22: 359–374.
Zhao J, Pestell R, Guan JL . Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol Biol Cell 2001; 12: 4066–4077.
Wang X, Urvalek AM, Liu J, Zhao J . Activation of KLF8 transcription by focal adhesion kinase in human ovarian epithelial and cancer cells. J Biol Chem 2008; 283: 13934–13942.
Calalb MB, Polte TR, Hanks SK . Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol 1995; 15: 954–963.
Schaller MD, Borgman CA, Parsons JT . Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol Cell Biol 1993; 13: 785–791.
Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ . Structural basis for the autoinhibition of focal adhesion kinase. Cell 2007; 129: 1177–1187.
Xie Z, Sanada K, Samuels BA, Shih H, Tsai LH . Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 2003; 114: 469–482.
Xie Z, Tsai LH . Cdk5 phosphorylation of FAK regulates centrosome-associated miocrotubules and neuronal migration. Cell Cycle 2004; 3: 108–110.
Le Boeuf F, Houle F, Sussman M, Huot J . Phosphorylation of focal adhesion kinase (FAK) on Ser732 is induced by rho-dependent kinase and is essential for proline-rich tyrosine kinase-2-mediated phosphorylation of FAK on Tyr407 in response to vascular endothelial growth factor. Mol Biol Cell 2006; 17: 3508–3520.
Park AY, Shen TL, Chien S, Guan JL . Role of focal adhesion kinase Ser-732 phosphorylation in centrosome function during mitosis. J Biol Chem 2009; 284: 9418–9425.
Sensi M, Catani M, Castellano G, Nicolini G, Alciato F, Tragni G et al. Human cutaneous melanomas lacking MITF and melanocyte differentiation antigens express a functional Axl receptor kinase. J Invest Dermatol 2011; 131: 2448–2457.
Hanks SK, Calalb MB, Harper MC, Patel SK . Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci USA 1992; 89: 8487–8491.
Laan L, Pavin N, Husson J, Romet-Lemonne G, van DM, Lopez MP et al. Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell 2012; 148: 502–514.
Juan G, Traganos F, James WM, Ray JM, Roberge M, Sauve DM et al. Histone H3 phosphorylation and expression of cyclins A and B1 measured in individual cells during their progression through G2 and mitosis. Cytometry 1998; 32: 71–77.
Harrison RE, Turley EA . Active erk regulates microtubule stability in H-ras-transformed cells. Neoplasia 2001; 3: 385–394.
Roberts EC, Shapiro PS, Nahreini TS, Pages G, Pouyssegur J, Ahn NG . Distinct cell cycle timing requirements for extracellular signal-regulated kinase and phosphoinositide 3-kinase signaling pathways in somatic cell mitosis. Mol Cell Biol 2002; 22: 7226–7241.
Mitra SK, Hanson DA, Schlaepfer DD . Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 2005; 6: 56–68.
Tarcic G, Yarden Y . MAP Kinase activation by receptor tyrosine kinases: in control of cell migration. Methods Mol Biol 2010; 661: 125–135.
Lopes JP, Agostinho P . Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol 2011; 94: 49–63.
Kotak S, Busso C, Gonczy P . Cortical dynein is critical for proper spindle positioning in human cells. J Cell Biol 2012; 199: 97–110.
Stevermann L, Liakopoulos D . Molecular mechanisms in spindle positioning: structures and new concepts. Curr Opin Cell Biol 2012; 24: 816–824.
Thompson SL, Compton DA . Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci USA 2011; 108: 17974–17978.
Sanhaji M, Friel CT, Wordeman L, Louwen F, Yuan J . Mitotic centromere-associated kinesin (MCAK): a potential cancer drug target. Oncotarget 2011; 2: 935–947.
Liebl J, Furst R, Vollmar AM, Zahler S . Twice switched at birth: cell cycle-independent roles of the ‘neuron-specific’ cyclin-dependent kinase 5 (Cdk5) in non-neuronal cells. Cell Signal 2011; 23: 1698–1707.
Harada T, Morooka T, Ogawa S, Nishida E . ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol 2001; 3: 453–459.
Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, Ball DW et al. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res 2010; 70: 4460–4469.
Geneste O, Copeland JW, Treisman R . LIM kinase and Diaphanous cooperate to regulate serum response factor and actin dynamics. J Cell Biol 2002; 157: 831–838.
Bénard J, De Silva J, De Blois MC, Boyer P, Duvilland P, Chiric E et al. Characterization of a human ovarian adenocarcinoma line, IGROV1, in tissue culture and in nude mice. Cancer Res 1985; 45: 4970–4979.
van den Berg-Bakker CA, Hagemeijer A, Franken-Postma EM, Smit VT, Kuppen PJ, van Ravenswaay C et al. Establishment and characterization of 7 ovarian carcinoma cell lines and one granulosa tumor cell line: growth features and cytogenetics. Int J Cancer 1993; 53: 613–620.
Ramakrishna V, Negri DRM, Brusic V, Fontanelli R, Canevari S, Bolis G et al. Generation and phenotypic characterization of new human ovarian cancer lines with the identification of antigens potentially recognizable by HLA-restricted cytotoxic T cells. Int J Cancer 1997; 73: 143–150.
Tohyama K, Yoshida Y, Ohashi K, Sano E, Kobayashi H, Endo K et al. Production of multiple growth factors by a newly established human thyroid carcinoma cell line. Jpn J Cancer Res 1992; 83: 153–158.
Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab 2008; 93: 4331–4341.
De Santis G, Miotti S, Mazzi M, Canevari S, Tomassetti A . E-cadherin directly contributes to PI3K/AKT activation by engaging the PI3K-p85 regulatory subunit to adherens junctions of ovarian carcinoma cells. Oncogene 2009; 28: 1206–1217.
Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell 2005; 7: 51–63.
Acknowledgements
We thank Ms. Gabriella Nicolini for her technical assistance. We also thank Dr. Ungefrorer for kindly providing FRNK construct, Professor Tsai for the wt and Mut GFP-FAK vector constructs, Dr. Paola Defilippi for her fruitful discussion, the gynecologic clinical staff and the biorepository personnel at the Fondazione IRCCS Istituto Nazionale dei Tumori for the ovarian carcinoma specimens and Dr. Marina Bagnoli for their management. This work was supported by Italian Ministry of Health, (grants ‘Tumori Femminili’ to SC and ‘Programma integrato in Oncologia, to AT) and Associazione Italiana per la Ricerca sul Cancro (grant IG13055 to AT and grant IG11608 to AA). We thank Dr. Moore for the English editing of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Raschellá
Author Contributions
KR performed research, analyzed and interpreted data and wrote manuscript (including first draft). MS interpreted and discussed data; AA discussed data and obtained funding; SC interpreted and discussed data and obtained funding; AT designed study, interpreted data, wrote manuscript and obtained funding. All authors approved the final version of the manuscript.
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Rea, K., Sensi, M., Anichini, A. et al. EGFR/MEK/ERK/CDK5-dependent integrin-independent FAK phosphorylated on serine 732 contributes to microtubule depolymerization and mitosis in tumor cells. Cell Death Dis 4, e815 (2013). https://doi.org/10.1038/cddis.2013.353
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.353
Keywords
This article is cited by
-
PRC2 mediated KLF2 down regulation: a therapeutic and diagnostic axis during tumor progression
Cancer Cell International (2023)
-
CDK5-mediated phosphorylation and stabilization of TPX2 promotes hepatocellular tumorigenesis
Journal of Experimental & Clinical Cancer Research (2019)
-
CDK5-dependent phosphorylation and nuclear translocation of TRIM59 promotes macroH2A1 ubiquitination and tumorigenicity
Nature Communications (2019)
-
A mitotic CDK5-PP4 phospho-signaling cascade primes 53BP1 for DNA repair in G1
Nature Communications (2019)
-
Simultaneous E-cadherin and PLEKHA7 expression negatively affects E-cadherin/EGFR mediated ovarian cancer cell growth
Journal of Experimental & Clinical Cancer Research (2018)
