
Abstract
We previously demonstrated that overexpression of RanBP9 led to enhanced Aβ generation in a variety of cell lines and primary neuronal cultures, and subsequently, we confirmed increased amyloid plaque burden in a mouse model of Alzheimer’s disease (AD). In the present study, we found striking reduction of spinophilin protein levels when RanBP9 is overexpressed. At 12 months of age, we found spinophilin levels reduced by 70% (P<0.001) in the cortex of APΔE9/RanBP9 mice compared with that in wild-type (WT) controls. In the hippocampus, the spinophilin levels were reduced by 45% (P<0.01) in the APΔE9/RanBP9 mice. Spinophilin immunoreactivity was also reduced by 22% (P<0.01) and 12% (P<0.05) in the cortex of APΔE9/RanBP9 and APΔE9 mice, respectively. In the hippocampus, the reductions were 27% (P<0.001) and 14% (P<0.001) in the APΔE9/RanBP9 and APΔE9 mice, respectively. However, in the cerebellum, spinophilin levels were not altered in either APΔE9 or APΔE9/RanBP9 mice. Additionally, synaptosomal functional integrity was reduced under basal conditions by 39% (P<0.001) in the APΔE9/RanBP9 mice and ∼23% (P<0.001) in the APΔE9 mice compared with that in WT controls. Under ATP- and KCl-stimulated conditions, we observed higher mitochondrial activity in the WT and APΔE9 mice, but lower in the APΔE9/RanBP9 mice. Significantly, we confirmed the inverse relationship between RanBP9-N60 and spinophilin in the synaptosomes of Alzheimer’s brains. More importantly, both APΔE9 and APΔE9/RanBP9 mice showed impaired learning and memory skills compared to WT controls. These data suggest that RanBP9 might play a crucial role in the loss of spines and synapses in AD.
Similar content being viewed by others


Main
RanBP9 is a multi-modular scaffolding protein implicated in a variety of functions through integration of cell surface receptors with intracellular signaling targets.1, 2 Recently RanBP9 was found to be within the clusters of RNA transcript pairs associated with markers of Alzheimer’s disease (AD) progression, suggesting that RanBP9 might contribute to the pathogenesis of AD.3 In fact even before this discovery, we showed for the first time that RanBP9 increased Aβ generation by four-fold in varieties of cell cultures.4 We initially identified RanBP9 as a binding partner to low-density lipoprotein receptor-related protein (LRP).5 We subsequently showed that RanBP9 reduced cell surface levels of APP, LRP, and β1-integrin, resulting in strongly enhanced Aβ generation and focal adhesion disruption.4, 6 Surprisingly, RanBP9-N60, a 60-kDa proteolytically derived species of RanBP9 that binds to APP, LRP and BACE-1 stronger than that of the full-length protein, was increased six-fold in the brains of patients with AD,7 and RanBP9 holoprotein was increased four-fold in the brains of J20 transgenic mice.8
There is now compelling evidence that the neurodegenerative process in AD begins in synapses. For example, the extent of synaptic loss that begins prior to the appearance of tangles, plaques or neuronal loss is an excellent correlate of dementia.9, 10, 11 Several transgenic mouse models generated to understand the molecular mechanisms of AD also reproduce loss of dendrites and spines.12, 13, 14, 15 In spite of a large body of evidence accumulating on the loss of synapses, understanding of the molecular mechanisms by which the structure and function of synapses are altered in AD remains largely unknown.
Several protein interactions with RanBP9 suggest a critical role for RanBP9 in the brain synapses. RanBP9 interaction with plexin-A regulates semaphorin3A signaling, controlling axonal outgrowth.16 In addition, RanBP9 regulates BDNF-mediated neuronal morphology and survival through MAPK and Akt pathways.17 RanBP9 is also a ligand for Rho-GEF18 and many studies have shown an essential role for Rho-like GTPases in regulating spine and dendritic morphology.19 Further, like in AD, defects in dendritic spines are common to several forms of cognitive deficits, including mental retardation, and RanBP9 has also been shown to bind to fragile X-linked mental retardation protein (FMRP).20 These evidences taken together imply that RanBP9 might have a primary role in the loss of synapses in AD. Here, we further demonstrate that spinophilin, a specific marker of spines is inversely correlated with RanBP9 protein levels in both mouse and human AD brains. More importantly, mouse synaptosomes showed significant reduction of MTT activity in the APΔE9/RanBP9 mice, suggesting aggravation of functional deficits by RanBP9.
Results
Striking reduction of spinophilin levels in the cortex and hippocampus of APΔE9/RanBP9 triple-transgenic mice
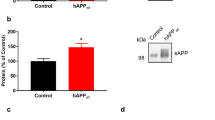
Generation of RanBP9 transgenic mice and APΔE9/RanBP9 triple-transgenic mice have been described previously.21 Loss of synaptic markers is the primary pathological sequelae in AD. Spinophilin is a postsynaptic marker located mainly in the heads of dendritic spines. As a marker of spine density, we first directly quantified the levels of spinophilin by immunoblots. As there can be a great variability in protein concentrations and loading of each sample, we normalized the levels of spinophilin to that of actin. One-way analysis of variance (ANOVA) revealed that normalized levels of spinophilin were reduced by 39% (P<0.001) in APΔE9 brains compared with that in WT controls. The reduction was even more striking in the APΔE9/RanBP9 triple-transgenic mice, which was reduced by 70% (P<0.001) of WT (Figures 1a and b). Also, there was 34% (P<0.01) reduction in spinophilin levels in the triple-transgenic mice compared with that in double-transgenic mice. Exogenous expression of flag-tagged RanBP9 was confirmed by flag antibody (Figure 1a, panel 3) and expression of endogenous RanBP9 was confirmed using a monoclonal anti-RanBP9 antibody that detected both the flag-tagged exogenous RanBP9 and the endogenous RanBP9 (Figure 1a, panel 2). Quantitation revealed that exogenous flag-tagged RanBP9 was expressed more than endogenous RanBP9 in the cortex (Figures 1a and c).

RanBP9 overexpression in APΔE9 mice decreases spinophilin protein levels in the cortex at 12 months of age. (a) Brain homogenates from APΔE9 double-transgenic, APΔE9/RanBP9 triple-transgenic and age-matched WT control mice were subjected to SDS-PAGE electrophoresis and probed with anti-spinophilin antibody to detect spinophilin protein. Immunoblotting using RanBP9 specific monoclonal antibody detected both flag-tagged exogenous RanBP9 and endogenous RanBP9 in the triple-transgenic mice, while WT mice and APΔE9 mice showed expression of endogenous RanBP9 only (panel 2). Flag antibody detected expression of flag-tagged exogenous RanBP9 in APΔE9/RanBP9 triple-transgenic mice only (panel 3). (b) Image J quantification showed decreased levels of spinophilin by 39% in APΔE9 and by 70% in APΔE9/RanBP9 mice compared with WT. Compared between APΔE9 and APΔE9/RanBP9 mice, spinophilin levels further reduced by 34%. (c) Relative expression levels of endogenous and flag-tagged exogenous RanBP9 levels in the cortex of triple transgenic mice. Reduced levels of spinophilin were also confirmed by immunohistochemistry in the APΔE9/RanBP9 triple-transgenic mice compared to APΔE9 and WT mice. ANOVA followed by post hoc Tukey’s test revealed significant differences. ***P<0.001 in both APΔE9 and APΔE9/RanBP9 mice compared to WT mice. **P<0.01 in APΔE9 versus APΔE9/RanBP9 mice. The data are mean±S.E.M., n=5 for WT and APΔE9 and n=4 for APΔE9/RanBP9 genotypes
As there is differential vulnerability of brain regions to synaptic damage in AD and also because exogenous expression of flag-tagged RanBP9 protein levels varied between brain regions, we wanted to quantify and compare spinophilin levels in different brain regions. Thus, we quantified spinophilin levels in the hippocampus as well and found a reduction by 45% (P<0.001) in the triple-transgenic mice compared with that in WT controls, but there was no significant difference between WT and the APΔE9 double-transgenic mice (Figures 2a and b). When compared between double- and triple-transgenic mice, there was ∼34% (P<0.05) further reduction in spinophilin levels in the triple-transgenic mice over the double-transgenic mice, which was also statistically significant (Figures 2a and b). However, quantitation of spinophilin levels in the cerebellum did not reveal significant differences among any genotypes (Figures 3a and b). Interestingly, exogenous expression of RanBP9 was the least in the cerebellum among the three brain regions studied, accounting only to ∼49% of that of endogenous RanBP9 (Figure 1c, 2c and 3c). This might explain the insignificant alterations in the levels of spinophilin in the cerebellum.

RanBP9 overexpression in APΔE9 mice decreases spinophilin protein levels in the hippocampus at 12 months of age. (a) Brain homogenates from APΔE9 double-transgenic, APΔE9/RanBP9 triple-transgenic and age-matched WT control mice were subjected to SDS-PAGE electrophoresis and probed with anti-spinophilin antibody to detect spinophilin protein. Immunoblotting using RanBP9-specific monoclonal antibody detected both flag-tagged exogenous RanBP9 and endogenous RanBP9 in the triple-transgenic mice, while WT mice and APΔE9 mice showed expression of endogenous RanBP9 only (panel 2). Flag antibody detected expression of flag-tagged exogenous RanBP9 in APΔE9/RanBP9 triple transgenic mice only (panel 3). (b) Image J quantification showed decreased levels of spinophilin by 45% in APΔE9/RanBP9 mice compared with WT, but no change in APΔE9 mice. Compared between APΔE9 and APΔE9/RanBP9 mice, spinophilin levels further reduced by 34%. (c) Relative expression levels of endogenous and flag-tagged exogenous RanBP9 levels in the hippocampus of triple transgenic mice. Reduced levels of spinophilin were also confirmed by immunohistochemistry in the CA1 region of the hippocampus in the APΔE9/RanBP9 triple-transgenic mice compared with APΔE9 and WT mice. ANOVA followed by post hoc Tukey’s test revealed significant differences. ***P<0.001 in APΔE9/RanBP9 mice compared with WT mice. *P<0.05 in APΔE9 versus APΔE9/RanBP9 mice. The data are mean±S.E.M., n=5 for WT and APΔE9 and n=4 for APΔE9/RanBP9 genotypes

RanBP9 overexpression in APΔE9 mice does not alter spinophilin protein levels in the cerebellum at 12 months of age. (a) Brain homogenates from APΔE9 double-transgenic, APΔE9/RanBP9 triple-transgenic and age-matched WT control mice were subjected to SDS-PAGE electrophoresis and probed with anti-spinophilin antibody to detect spinophilin protein. Immunoblotting using RanBP9-specific monoclonal antibody detected both flag-tagged exogenous RanBP9 and endogenous RanBP9 in the triple-transgenic mice, while WT mice and APΔE9 mice showed expression of endogenous RanBP9 only (panel 2). Flag antibody detected expression of flag-tagged exogenous RanBP9 in APΔE9/RanBP9 triple transgenic mice only (panel 3). (b) Image J quantification did not reveal significant changes in the levels of spinophilin in any of the genotypes. (c) Relative expression levels of endogenous and flag-tagged exogenous RanBP9 levels in the cerebellum of triple-transgenic mice. ANOVA did not reveal significant differences. The data are mean±S.E.M., n=5 for WT and APΔE9 and n=4 for APΔE9/RanBP9 genotypes
Immunohistochemical demonstration of reduced spinophilin levels in the APΔE9/RanBP9 triple-transgenic mice
Spinophilin is thought to be involved in spine formation and synaptic transmission. In all brain regions, spinophilin is localized specifically to the heads of dendritic spines and ∼93% of dendritic spines are thought to contain spinophilin.22 Therefore, spinophilin immunoreactivity makes it an excellent marker for quantitative assessment of spine density.22 To assess the role of RanBP9 in the loss of spines, we analyzed spinophilin-immunoreactive puncta in the cortex and CA1 region of the hippocampus within an area of interest (AOI) measuring 30 × 30 μm. As shown in Figure 4, we found 172 spinophilin-immunoreactive puncta in the cortex of WT brains, which was reduced to 134 (P<0.001) in the APΔE9/RanBP9 mice and 151 (P<0.05) in the APΔE9 mice (Figures 4a–c and g). Thus, although immunohistochemical staining showed significant reductions in the spinophilin-immunoreactive puncta, the magnitude of reductions were less than the immunoblots, because the quantitation was done only in an AOI that doesn’t represent the whole brain region. In the CA1 region of the hippocampus, the number of puncta were 209 in the WT, 153 (P<0.001) in the APΔE9/RanBP9 and 180 (P<0.001) in the APΔE9 mice (Figures 4d–f and h). It is important to note that the number of spinophilin puncta were significantly reduced in APΔE9/RanBP9 mice compared with that in APΔE9 mice in both the cortex (P<0.05) and the hippocampus (P<0.01). This suggests that RanBP9, at least partially, is responsible for the loss of spinophilin. Thus, significant reduction in the spine density by RanBP9 is expected to have profound effects on synaptic transmission.

Spinophilin-immunoreactive puncta are significantly reduced in the cortex and hippocampus of APΔE9 and APΔE9/RanBP9 mice. Cortical brain sections from WT (a) APΔE9 (b) and APΔE9/RanBP9 (c) mice were stained with spinophilin antibody and analyzed. Similarly hippocampal brain sections from WT (d) APΔE9 (e) and APΔE9/RanBP9 mice were stained with spinophilin antibody and analyzed. Quantitation of spinophilin by Image-Pro in an AOI in the cortex showed reduced levels in the APΔE9 mice by 12% and in the APΔE9/RanBP9 mice by 22% compared to WT controls. In the hippocampus, the reductions were 14 and 27% in the APΔE9 and APΔE9/RanBP9 mice respectively compared with WT controls. One-way ANOVA followed by post hoc Student–Newman–Keuls multiple comparison test revealed significant differences. The data are mean±S.E.M., n=3 in each group. *P<0.05, **P<0.01 and ***P<0.001 compared with WT controls
Detection of RanBP9 in the synaptosomes of mouse and human brains
To directly assess the role of RanBP9 in the functional loss of synapses, we prepared synaptosomes (nerve terminals) from both mouse and human brains. To determine the specificity of the synaptosome extraction procedure, first, we biochemically characterized the purity of synaptosomes by immunoblotting two proteins for each of the cytosolic, nuclear and synaptic proteins (Figure 5a). As expected, cytosolic proteins, such as TGFβ and stathmin, were completely absent in the synaptosome fractions (S), while considerable amounts of protein were present in both the cytosolic (C) and homogenate fractions (H) (Figure 5a). On the contrary, synaptic proteins, such as synaptophysin and PSD-95, were detected in considerable amounts in the synaptic fractions than in the cytosolic or homogenate fractions (Figure 5a). PCNA, which is exclusively a nuclear protein, could not be detected in the synaptosomal fractions and HDAC2 was not detected in synaptosomal fraction as well, though a little amount of the protein was found in cytosolic fraction (Figure 5a).

Characterization of synaptosomes and the demonstration of RanBP9 protein in the mouse and human brain synaptosomes. (a) The purity of the synaptosome preparations was verified by immunoblotting the cytosolic (C), homogenate (H) and synaptosomal (S) fractions for two cytosolic proteins, TGFβ and stathmin, two nuclear proteins PCNA and HDAC2 and two synaptic markers, PSD-95 and synaptophysin. Please note enrichment of synaptic proteins and the absence of nuclear protein PCNA and the cytosolic protein TGFβ in the synaptosome fractions attesting to the purity of synaptosomes. (b) Shows amount of RanBP9 in the mouse brain synaptosomes relative to cytosolic and homogenate fractions. (c) Human brain synaptosomes contain much more RanBP9 protein relative to the cytosolic and homogenate fractions. One-way ANOVA followed by posthoc Student–Newman–Keuls multiple comparison test revealed significant differences. The data presented in b and c are mean±S.E.M., n=3, in each group. ***P<0.001 versus cytosolic fractions
If RanBP9 has an essential role in synaptic damage, first of all, RanBP9 must be present at the synapses. So, we were curious to know whether RanBP9 protein was present in the synaptosomal fractions. Normalized levels of RanBP9 were comparable in the homogenate and synaptosomal fractions, but was slightly higher in the homogenate fractions of mouse brains (Figure 5b). Surprisingly, in the human brains, synaptosomal fractions contained nearly three-fold more RanBP9 protein than the homogenate fractions (Figure 5c), suggesting that RanBP9 must have an essential function at the synapses in the human brain.
Marked reduction of synaptosome viability in the APΔE9/RanBP9 triple-transgenic mice
We assessed mitochondrial activity as an index of synaptosome viability by using MTT reduction assay. This assay is also an index of bioenergetic behavior of synaptosomes. Initially, we compared the basal synaptosomal mitochondrial activity between the three genotypes. Under basal conditions, mitochondrial activity was significantly reduced in both the APΔE9 synaptosomes by 23.3% (P<0.001) compared with that in WT, and the APΔE9/RanBP9 triple-transgenic synaptosomes by 39% (P<0.001) when compared with that in WT (Figure 6a), suggesting that RanBP9 overexpression has adverse effects on the viability of synapses. We also noted 32% (P<0.001) decreased synaptosomal viability in the APΔE9/RanBP9 mice compared with that in APΔE9 mice (Figure 6a), suggesting that RanBP9 overexpression exacerbates the synaptic functional impairment.

RanBP9 exacerbates bioenergetic defects in the synaptosomes of APΔE9/RanBP9 mice. (a) Mitochondrial activity was measured as an index of synaptosomal functional integrity using MTT reduction assay. Basal mitochondrial activity in the synaptosomes was reduced by 23.3% in APΔE9 and by 39% in the APΔE9/RanBP9 mice compared with WT controls. Importantly, we noted 32% reduced activity in the APΔE9/RanBP9 mice compared to APΔE9 mice. (b), Under ATP-stimulated conditions used at 10.0 μM, MTT formazan production was increased by 22% in the WT, 18% in the APΔE9 and only 7% in the APΔE9/RanBP9 synaptosomes. (c) Under KCl- stimulated conditions, used at 25, 50 or 100 mM concentrations mitochondrial activity was increased more in the WT and APΔE9 synaptosomes compared with APΔE9/RanBP9 mice. At 100 mM KCl, the increase in the activity was 48% in the WT, 53% in the APΔE9 and only 37% in the APΔE9/RanBP9 synaptosomes. ANOVA followed by post hoc Tukey’s test revealed significant differences. *P<0.05, **P<0.01 ***P<0.001 in APΔE9/RanBP9 mice compared with WT mice or APΔE9 mice. The data are mean±S.E.M., n=4 in each group
Next, to determine whether synaptosomal preparations are compromised functionally, we measured MTT activity under ATP- and KCl- stimulated conditions. ATP stimulation of synaptosomes at 10.0 μM induced an increase in MTT formazan production by 22% (P<0.01) in WT, 18% (P<0.05) in the APΔE9 mice and only ∼7% in the APΔE9/RanBP9 triple-transgenic mice (Figure 6b). Stimulation of synaptosomes at 25 mM KCl increased formazan production by 12.1%, at 50 mM KCl by 29.9% and at 100 mM KCl by 47.7% in WT mice (Figure 6c). Comparable values were obtained for APΔE9 mice, that is, 3.75% at 25 mM, 49.59% at 50 mM and 53.3% at 100 mM (Figure 6c). However, in the APΔE9/RanBP9 triple-transgenic mice, the formazan production was 13.5% at 25 mM KCl, 32.5% at 50 mM KCl and 37% at 100 mM KCl (Figure 6c). Thus, these results clearly suggest that synaptosomes from the triple-transgenic mice are functionally impaired.
Increased activated microglia reflects exacerbated inflammatory response in APΔE9/RanBP9 triple-transgenic mice
Microglia is very sensitive to their microenvironment and serves the role of immune surveillance and host defense. In response to neuronal injury, microglia is transformed into activated states such as ameboid microglia.23 Therefore, we wanted to use activated microglia as a marker of inflammation in the brains of transgenic mice. Expression of the lysosomal protein, CD68, is upregulated in the activated microglia24 and, therefore, we used CD68 immunoreactivity as a marker of activated microglia. Although we didn’t quantify the numbers, we consistently observed more activated microglia in the APΔE9/RanBP9 brains compared with that in both APΔE9 and WT mice (Figure 7). We could hardly see any CD68-immunoreactive microglia in the WT brains. However in the APΔE9 brains, we observed few CD68-immunoreactive cells, whereas in the APΔE9/RanBP9 triple-transgenic mice, we found many positively immunoreactive microglia (Figure 7).

RanBP9 overexpression in APΔE9 mice increases CD68 positive activated microglia at 12 months of age. Immunohistochemical staining of the brain sections from the cortex and the CA1 region of the hippocampus from WT, APΔE9 and APΔE9/RanBP9 mice with CD68 antibody. A representative image for each genotype, all at the same magnification showed relatively more activated microglia in the APΔE9/RanBP9 mice compared with APΔE9 mice and almost complete absence in the WT brains
RanBP9 levels are inversely correlated to spinophilin levels in Alzheimer’s brain synaptosomes
To confirm the relationship between RanBP9 and spinophilin, we quantified their protein levels in the synaptosomes of brains from patients with Alzheimer’s disease and compared with age-matched controls. Results revealed a decreasing trend for spinophilin from Braak stage 2 to Braak stages 3, 5 and 6, though statistical significance was reached only for Braak stage 6 that showed 48% reduction (P<0.05) (Figure 8a and b). The Braak stages 2–6 were classified based on the appearance of the neurofibrillary tangles that increase from stage 2–6. This suggests that spinophilin levels are inversely correlated to the severity of Alzheimer’s disease in terms of the accumulation of neurofibrillary tangles. Levels of RanBP9-FL in the synaptosomes were also increased only in the brains at Braak stage 6 by 64% (P<0.05) (Figure 8a and c). The increase was even more for RanBP9-N60, nearly three-fold (284%), making it the most significant (P<0.001) (Figures 8a and d). We also demonstrated nearly four-fold increased levels of RanbP9-FL in the whole-brain lysates of J20 APP transgenic mice.7 The present results in the synaptosomes suggest that RanBP9-FL is also increased in addition to RanBP9-N60. Thus, there is a clear inverse relationship between RanBP9 and spinophilin levels in the synaptosomes.

RanBP9 protein levels are inversely correlated to spinophilin levels in the synaptosomes of AD brains. (a) Synatosomes from hippocampus of AD with varying severity of the disease starting from Braak stage 2 (Br-2) to Braak stage 6 (Br-6) classified based on the appearance of neurofibrillary tangles and age-matched control brains were prepared as detailed in the legend to figure 5. Equal amounts of total proteins were subjected to western blots to detect spinophilin, RanBP9-FL, RanBP9 N60 and actin as a loading control using specific antibodies. (b) Quantitation of spinophilin levels by imageJ revealed reduction in AD brain synaptosomes only in Braak stage 6 (Br-6) by 48% compared to age-matched controls. (c) Quantitation of levels of RanBP9-FL also showed alteration only in Br-6 which increased by 64%. (d) Quantitation of RanBP9-N60 also showed increase to 287% compared to age-matched controls. ANOVA followed by post hoc Tukey’s test revealed significant differences. *P<0.05, **P<0.01 ***P<0.001 in Br-6 stage of AD synaptosomes compared to age-matched controls. The data are mean±S.E.M. and n numbers for each are indicated on the blots
Impaired learning in APΔE9/RanBP9 triple-transgenic mice
We tested six mice for each of the genotypes such as APΔE9, APΔE9/RanBP9 and non-transgenic littermate WT controls in the T maze performance at 12 months of age. The test included six trials per session, for a total of six sessions, one session per day. In general, the time taken to reach an arm was more during the initial sessions, which gradually reduced to ∼20 s per trial (Figure 9a). Similarly the number of correct responses in terms of reaching the goal arm was less in the initial sessions, which gradually increased to >80% correct responses by session 6 (Figure 9b). When data were analyzed by repeated measures analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparison tests, significant differences among different genotypes were revealed. At 12 months, APΔE9 mice reached the goal arm only 50% of the trials compared with 70% for the WT controls, reaching statistical significance (P<0.01), while the APΔE9/RanBP9 triple-transgenic mice made correct choices only 34% of the times, which was also highly significant (P<0.001). Interestingly, the 16% difference among the APΔE9 and APΔE9/RanBP9 mice was also significant (P<0.05) (Figure 9c). Calculated in another way in terms of percent errors, Errors occurrence in APΔE9 mice committed errors was 50% (P<0.01) of the times compared to only 30% errors for the WT controls, while the triple-transgenic mice showed errors at 63% (P<0.001) of the trials (Figure 9c). These data indicate that APΔE9 mice display significant deficits in learning skills at 12 months of age and that RanBP9 further exacerbate this deficit.

Impaired spatial learning in a T maze paradigm in APΔE9 and APΔE9/RanBP9 transgenic mice at 12 months of age. (a) Analysis of latency in terms of time taken in seconds to reach the T maze arm was plotted against the sessions, one session per day, six trials per session. (b) Percentage of correct responses to reach the goal arm was plotted against the sessions as in a. (c) Analysis of the percentage of correct responses averaged among all the sessions were found to be significantly decreased in APΔE9 mice by 20% and in APΔE9/RanBP9 mice by 36% compared with WT controls. Percentage of errors significantly increased in APΔE9 mice by 21% and in APΔE9/RanBP9 mice by 34%. Repeated measures ANOVA followed by post hoc Tukey–Kramer multiple comparison test revealed significant differences. *P<0.05, **P<0.01 ***P<0.001. The data are mean±S.E.M. and n=6 per genotype
Discussion
The primary objective of the present study is to test whether RanBP9 overexpression in a mouse model of AD has any synaptic toxicity. Our previous observations that RanBP9 overexpression led to robust increase in the levels of Aβ and amyloid plaque burden, which was accompanied by significant decrease in the levels of synaptic proteins,21 formed a strong basis for the present study. Here, we have further demonstrated that RanBP9 overexpression in APΔE9 mice led to striking reduction in the levels of spinophilin, a marker of dendritic spines. We also found that RanBP9 overexpression led to remarkable loss of synaptic functional integrity as revealed by MTT formazan reduction experiments using synaptosomes isolated from APΔE9/RanBP9 triple-transgenic mice. Increased number of activated microglia by RanBP9, which is well known to release several inflammatory cytokines, might partially account for synaptic toxicity. Finally, we directly confirmed in Alzheimer’s brain synaptosomes that RanBP9 protein levels are inversely correlated to spinophilin levels, further strengthening our conclusion that RanBP9, in fact, may have a major role in the synaptic toxicity and loss of spines in AD.
In this study, data from both immunohistochemistry and biochemical experiments have provided compelling evidence that overexpression of RanBP9 in APΔE9 mice leads to robust loss of spinophilin protein levels. Interestingly, the extent of spinophilin reduction was dependent on the overexpression levels of exogenous RanBP9. Thus, an inverse correlation between RanBP9 and spinophilin, which is a marker of spines in different regions of mouse brain, suggests loss of spines may be at least partially due to RanBP9. As both the mouse models of AD25, 26 and actual AD27, 28, 29 have deficits in dendritic spine density, the current results on the effect of RanBP9 on a marker of dendritic spines strongly implicate a crucial role of RanBP9 in the loss of dendritic spines and therefore in the pathogenesis of AD. As dendritic spines are the primary targets of excitatory synaptic inputs to pyramidal neurons, RanBP9-induced significant reduction in spine density is likely the reason for the observed hippocampal-dependent spatial learning and memory deficits in the APΔE9/RanBP9 triple-transgenic mice in a T maze learning paradigm.
The molecular mechanism by which RanBP9 reduces spine density may be through its binding to APP, LRP and BACE1,4 thereby increasing Aβ generation and plaque burden,21 which are known to reduce spine density especially in the vicinity of the plaques. It is also possible that RanBP9 might decrease spine density independent of its effect on Aβ and amyloid plaques. Substantial amounts of RanBP9 protein are present in the neurites of primary neurons as well as throughout the dendritic branches in the adult mouse brains with a punctate appearance.21 We also demonstrated that RanBP9 activates/dephosphorylates cofilin,8 a key regulator of actin dynamics. Interestingly, the neuronal inclusions from AD contain cofilin, actin and hyperphosphorylated tau. As abnormalities in the actin cytoskeleton are detected early during the course of AD, RanBP9-mediated altered cofilin signaling can lead to the formation of cofilin–actin rods, which may attract hyperphosphorylated tau and promote tau pathology followed by synaptic dysfunction. The present study also demonstrated that activated form of RanBP9-N60 is increased in the synaptosomes of AD patients, which is inversely correlated to spinophilin levels. We also recently demonstrated that RanBP9 overexpression is known to cause cell death through Cofilin pathway by increasing neuroinflammation.7 These evidences taken together with the increased levels of activated form of RanBP9 in AD brains seen in the present study clearly suggest a pivotal role for RanBP9 in the loss of synapses. Multiple evidences from other laboratories also support a critical regulatory role for RanBP9 at the synapses. RanBP9 has been shown to drastically reduce the growth and branching of neurites from dorsal root ganglion (DRG) neurons16 and cerebellar primary neurons.30 RanBP9 is a ligand for Rho-GEF,31 and as Rho-GEF’s are known to regulate dendritic and spine morphology,19 RanBP9 is also naturally expected to have a critical role in the maintenance of spine and dendritic morphology.
Another important novel finding in the present study is that the bioenergetic defects in the synaptosomes of APΔE9/RanBP9 triple-transgenic mice are much more severe than that observed in the APΔE9 double-transgenic mice, suggesting that RanBP9 positively contributes to the synaptosomal abnormality. Synaptic terminals have high energy demands and they are very sensitive to bioenergetic defects. As AD is characterized by extensive loss of synapses, which is now believed to be responsible for the cognitive impairment, synaptosomal mitochondrial abnormalities are predicted to be prevalent in AD and AD mouse models. In line with these expectations, the basal MTT activity, a measure of mitochondrial membrane integrity was significantly reduced by ∼23% in the APΔE9 brains and by 39% in the triple-transgenic mice. Several studies in the literature also have reported about ∼20% decline of respiration in mitochondria isolated from whole brains, including the 3xTg-AD mice,32, 33 thy-1 APP mice34 and Tg2576 mice.35 Another recent study found mitochondrial abnormalities isolated from synaptosomes, but not whole brains, as early as 4 months of age in J20 mice.36 Even more important is our finding that both ATP- and KCl- stimulated MTT formazan reduction was highly significant in APΔE9/RanBP9 synaptosomes compared with that in both the WT controls and the APΔE9 mice. The observed deficit in synaptosomal mitochondrial activity confirms our conclusion that RanBP9 exacerbates synaptic damage in the APΔE9 mouse model of AD, as synaptic mitochondria are vital for the maintenance of synaptic function and transmission.37, 38 Abnormalities of mitochondrial function are known to occur in the AD brains as well.39, 40 The worsening of MTT reduction in response to ATP- and KCl- stimulations in the triple-transgenic mice and inverse correlation of RanBP9 levels with that of spinophilin in AD brain synaptosomes, as well as mouse model of AD, is an explicit evidence that RanBP9, in fact, contributes to functional loss of synapses through loss of spines and mitochondrial deficits.
We previously demonstrated robust microgliosis and astrocytosis within the degenerating hippocampus of RanBP9 transgenic mice using Iba1 and GFAP immunohistochemistry, respectively.8 In order to determine whether loss of spines in the cortex and hippocampus were accompanied by changes in the number of activated microglia, in the present study, we used CD68, an excellent marker of activated microglia24 unlike Iba1, which is immunoreactive to both active and resting microglia.41 Here, we showed that microglia are not activated in either the APΔE9 or WT mice and that RanBP9 overexpression in the APΔE9 mice is required for the accumulation of activated microglia. It is widely known that microglia are the brain-resident macrophages to provide the initial response to injury within the CNS. However, activation of microglia leads to the synthesis of a variety of proinflammatory mediators, especially TNF-α and IL-1β, which if produced chronically may perpetrate bystander neural insult. Thus activated microglia observed in the present study might at least in part account for the loss of spines and synapses.
In summary, we have demonstrated by both immunostainings and immunoblots the inverse correlation between RanBP9 and spinophilin in the APΔE9 mouse model as well as AD brains and that such loss of spines is associated with perturbation of synaptic mitochondrial functional integrity. These findings provided concrete evidence that RanBP9 has a major role in the mitochondrial deficits through loss of spines and synapses. Intervention of RanBP9 protein levels in AD brains might prevent loss of spines and synapses and, therefore, RanBP9 may be an excellent therapeutic target for AD.
Material and methods
Chemicals and antibodies
MTT substrate, (3-(4,-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (cat# M5655), dimethyl sulfoxide (DMSO) (cat# 472301), adenosine 5′-triphosphate disodium salt (ATP) (cat# A2383), potassium chloride (cat# P9541) and glutaraldehyde (cat# G-7776) were all purchased from Sigma-Aldrich (St. Louis, MO, USA). Monoclonal antibody against RanBP9 was produced by immunizing mice with a peptide corresponding to 146–729 amino acids of RanBP9 as described previously.6 Anti-flag-tag antibody (M2, cat# F3165) was purchased from Sigma (St. Louis, MO, USA). Monoclonal anti-HDAC2, clone 3F3 (cat# 05-814) was purchased from Millipore (Temecula, CA, USA). Polyclonal Stathmin antibody (cat# 3352) and polyclonal spinophilin antibody (cat# 9061S) were purchased from Cell Signaling (Danvers, MA, USA). Proliferating cell nuclear antigen (PCNA) (cat# 3350R-100) was purchased from Biovision (Milpitas, CA, USA). Monoclonal ant-Drebrin antibody (cat# D029-3) was purchased from MBL international corporation (Woburn, MA, USA). PSD-95 polyclonal antibody (cat# EP2652Y) was obtained from Epitomics (Burlingame, CA, USA). Anti-synaptophysin mouse monoclonal antibody (cat# 573822) was purchased from EMD Chemicals Inc. (Gibbstown, NJ, USA). Polyclonal anti-TGF beta 1 antibody (cat# NBP1-67698) and monoclonal CD68 antibody, clone SPM281 (cat# NBP1-74570) were purchased from Novus biologicals (Littleton, CO, USA). Mouse monoclonal antibody against beta-actin (cat# A00702) was purchased from Genscript USA Inc. (Piscataway, NJ, USA). Secondary antibodies such as peroxidase-conjugated AffiniPure goat anti-mouse (Code# 115-035-146) and anti-rabbit (code# 111-035-144) IgGs were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Tissue extraction and immunoblotting
Mouse brain tissues from three different genotypes, viz., wild-type (WT), APP/PS1 double-transgenic mice expressing APP with Swedish mutation and PS1 with ΔE9 mutation (APΔE9) and APΔE9/RanBP9 triple-transgenic mice were dissected out in to cortex, hippocampus and cerebellum on ice. Brain lysates were prepared from 12-months-old mice from all three genotypes. In brief, we anesthetized the mice with isoflurane, decapitated immediately and rapidly removed the brain tissue in to 1% NP40 buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.02% sodium azide, 400 nM microcystine-LR, 0.5 mM sodium vanadate and 1% sodium Nonidet P-40) containing complete protease inhibitor cocktail for use with mammalian cell and tissue extracts (Sigma). Tissue was homogenized using Power Gen 125 (Fisher Scientific, Pittsburgh, PA, USA) and centrifuged at 100 000 g for 1 h. Protein concentrations from each sample were measured by BCA method (Pierce Biotechnology Inc., Rockford, IL, USA). Equal amounts of proteins were loaded into each well and subjected to SDS-PAGE electrophoresis. The proteins were then transferred onto PVDF membranes, blocked with 5% milk and incubated overnight with primary antibodies, followed by 1 hour incubation with HRP-conjugated secondary antibodies. The protein signals were detected using Super Signal West Pico Chemiluminescent substrate (Pierce Biotechnology Inc.).
Spinophilin and CD68 Immunohistochemistry
Brain sections (16 μm) from 12-month-old WT, APΔE9 double-transgenic (Dbl) and APΔE9/RanBP9 triple-transgenic (Tpl) mice were washed with PBS 1 × , three times each for 5 min. Antigen retrieval was carried out by immersing slides in 10 mM citric acid (pH 6.0) for 10 min at 90–95 °C. Sections were washed with PBS 1 × for 5 min, three times, and incubated in blocking solution (10% normal goat serum, 1% BSA, 0.1% Triton X-100 in PBS 1X) for 1 h at room temperature. The sections were incubated overnight with anti-CD68 mouse monoclonal antibody (Novus Biologicals, Littleton, CO, USA) or polyclonal anti-spinophilin antibody (Cell Signaling, Danvers, MA, USA) in blocking solution (1 : 200) at 4 °C. After washing in PBS 1 × for 5 min, three times, the sections were incubated with Alexa Fluor 568 goat anti-mouse IgG (Invitrogen, Carlsbad, CA, USA) in blocking solution (1 : 500) at room temperature for 2 h in the dark. Finally, slides were washed with PBS 1x for 5 min, three times, covered with mounting medium for fluorescence with DAPI (Vector Laboratories) and sealed with nail clear. Sections were visualized in a fluorescent microscope (Axio Examiner D1) and a confocal microscope (Nikon C1Si laser scanning multispectral confocal microscope; Nikon, Melville, NY, USA). Activated microglia reacting to CD68 was counted in a defined area of cortex and hippocampus (CA1) regions using Image-Pro Plus (Media Cybernetics, Rockville, MD, USA) software package. Positive activated microglia was defined as those whose nuclei and processes were evidently stained for CD68 and whose nuclei were colocalized with DAPI. Similarly spinophilin-immunoreactive puncta in the CA1 region of the hippocampus and cortex were assessed and compared between different genotypes. To count the spinophilin-immunoreactive puncta, an AOI measuring 30 × 30 μm were selected within the stratum pyramidale at the CA1 region of the hippocampus and at lateral parietal association cortex (layers V and VI). Image histograms were flattened and normalized by adjusting the threshold to the histogram mean using the standard deviation and a common factor. Spines were automatically counted using Image-Pro software.
Isolation of synaptosomes
To isolate synaptosomes, mice were euthanized under isoflurane anesthesia and frontal cortices from WT, APΔE9 and APΔE9/RanBP9 mice were weighed and dounced in a grinder using Syn-PER synaptic protein extraction reagent (cat# 87793) purchased from Thermo Scientific (Rockford, IL, USA). Alzheimer’s and control brain tissues were provided by the ‘Harvard Brain Tissue Resource Center’, which is supported in part by PHS grant number R24MH068855′′ at the McLean hospital. Immediately before use, protease inhibitor mixture for mammalian cells from Sigma (cat# P8340, St. Louis, MO, USA) was added to the Syn-PER reagent. Human brain tissue was similarly dounced using Syn-PER synaptic protein extraction reagent at volume ratio of ten using a 7-ml Dounce tissue grinder with 15 up and down even strokes. The homogenate was centrifuged at 2000 g for 10 min to remove cell debris. The resulting supernatant was centrifuged at 15 000 g for 20 min. The supernatant formed the cytosolic fraction and the synaptosome pellet was gently resuspended in Syn-PER synaptic protein extraction reagent. The amount of total proteins in the homogenate, cytosolic fraction and synaptosomes were measured by BCA method and compared. The quality of synaptosome preparation was verified by immunoblotting for two cytosolic proteins (TGFβ and stathmin), two nuclear proteins (PCNA and HDAC2) and two synaptic proteins (synaptophysin and PSD-95).
Synaptosomal functional assay
We made use of MTT reduction assay to measure mitochondrial activity as an index of the functional status of the synaptosomes. To determine the enzyme activity, we used the calorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) metabolic activity assay using cell growth determination kit (cat # CGD-1, Sigma-Aldrich) carried out according to the manufacturer’s instructions. Briefly, 50 μl of the synaptosome preparation equivalent to 2.0 mg of total protein was added to 500 μl of MTT solution (stock, 5 mg/ml in PBS and working concentration was prepared by 10-fold dilution). Following 3 h incubation at 37 °C, the resulting formazan crystals were dissolved in 100 μl of DMSO and the absorbance was read at 570 nm within an hour using the Smart Spec Plus spectrophotometer (Bio-Rad) after subtracting the background absorbance at 690 nm.
To assess synaptosomal functional integrity, ATP stimulations were carried out by the addition of stock solution of ATP to a final concentration of 10 μM to the synaptosomes prepared from WT, APΔE9 and APΔE9/RanBP9 mice. Next, depolarization of synaptosomes was carried out by incubating synaptosomes at the final concentrations of 0, 25, 50 and 100 mM KCl.
Behavioral testing
APΔE9, APΔE9/RanBP9 and littermate WT controls were tested in T maze learning paradigm to assess spatial learning skills. We tested six mice per genotype six sessions per mice, six trials per session in a total of six sessions. The methods used are as described in our previous publication.42
Statistical analysis
Immunoblot signal for spinophilin was quantified using Image J software. Spinophilin levels in WT, APΔE9 and APΔE9/RanBP9 mice were analyzed by one-way ANOVA. The values for MTT activity were expressed in optical density units as mean±S.E.M. ANOVA followed by the Newman–Keul’s post hoc test was used for comparisons among WT, APΔE9 and APΔE9/RanBP9 mice using Instat3 software (GraphPad Software, San Diego, CA, USA). We used two-tailed P-value assuming populations may have different standard errors. The data presented are mean±S.E.M. The data were considered significant only if the P <0.05, * indicates P<0.05, **P<0.01 and ***P<0.001.
Abbreviations
- Aβ:
-
amyloid β peptide
- AD:
-
Alzheimer’s disease
- APP:
-
amyloid precursor protein
- ATP:
-
adenosine triphosphate
- BACE-1:
-
β site app cleaving enzyme
- CNS:
-
central nervous system
- GFAP:
-
glial fibrillary acidic protein
- HDAC2:
-
histone deacetylase 2
- KCl:
-
potassium chloride
- LRP:
-
low-density lipoprotein-related protein
- MTT:
-
3-(4,-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NGS:
-
normal goat serum
- NP40:
-
Nonidet-P40
- PFA:
-
paraformaldehyde
- PSD-95:
-
postsynaptic density protein 95
- PVDF:
-
polyvinylidene fluoride
- RanBP9:
-
Ran-binding protein 9
References
Murrin LC, Talbot JN . RanBPM, a scaffolding protein in the immune and nervous systems. J Neuroimmune Pharmacol 2007; 2: 290–295.
Suresh B, Ramakrishna S, Baek KH . Diverse roles of the scaffolding protein RanBPM. Drug Discov Today 2012; 17: 379–387.
Arefin AS, Mathieson L, Johnstone D, Berretta R, Moscato P . Unveiling clusters of RNA transcript pairs associated with markers of Alzheimer’s disease progression. PLoS One 2012; 7: e45535.
Lakshmana MK, Yoon IS, Chen E, Bianchi E, Koo EH, Kang DE . Novel role of RanBP9 in BACE1 processing of amyloid precursor protein and amyloid beta peptide generation. J Biol Chem 2009; 284: 11863–11872.
Lakshmana MK, Chen E, Yoon IS, Kang DE . C-terminal 37 residues of LRP promote the amyloidogenic processing of APP independent of FE65. J Cell Mol Med 2008; 12: 2665–2674.
Woo JA, Roh SE, Lakshmana MK, Kang DE . Pivotal role of the RanBP9 in integrin-dependent focal adhesion signaling and assembly. FASEB J 2012; 26: 1672–1681.
Lakshmana MK, Chung JY, Wickramarachchi S, Tak E, Bianchi E, Koo EH et al. A fragment of the scaffolding protein RanBP9 is increased in Alzheimer’s disease brains and strongly potentiates amyloid-beta peptide generation. FASEB J 2010; 24: 119–127.
Woo JA, Jung AR, Lakshmana MK, Bedrossian A, Lim Y, Bu JH et al. Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell Death Differ 2012; 19: 1413–1423.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991; 30: 572–580.
Samuel W, Masliah E, Hill L.R, Butters N, Terry R . Hippocampal connectivity and Alzheimer’s dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology 1994; 44: 2081–2088.
DeKosky ST, Scheff SW . Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 1990; 27: 457–464.
Lanz TA, Carter DB, Merchant KM . Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis 2003; 13: 246–253.
Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML . Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol 2004; 33: 377–387.
Tsai J, Grutzendler J, Duff K, Gan WB . Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci 2004; 7: 1181–1183.
Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci 2005; 25: 7278–7287.
Togashi H, Schmidt EF, Strittmatter SM . RanBPM contributes to Semaphorin3A signaling through plexin-A receptors. J Neurosci 2006; 26: 4961–4969.
Yin YX, Sun ZP, Huang SH, Zhao L, Geng Z, Chen ZY . RanBPM contributes to TrkB signaling and regulates brain-derived neurotrophic factor-induced neuronal morphogenesis and survival. J Neurochem 2010; 114: 110–121.
Valiyaveettil M, Bentley AA, Gursahaney P, Hussien R, Chakravarti R, Kureishy N et al. Novel role of the muskelin-RanBP9 complex as anucleocytoplasmic mediator of cell morphology regulation. J Cell Biol 2008; 182: 727–739.
Nakayama AY, Harms MB, Luo L . Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci 2000; 20: 5329–5338.
Menon RP, Gibson TJ, Pastore A . The C terminus of fragile X mental retardation protein interacts with the multi-domain Ran-binding protein in the microtubule-organising centre. J Mol Biol 2004; 343: 43–53.
Lakshmana MK, Hayes CD, Bennet SP, Bianchi E, Reddy KM, Koo EH et al. Role of RanBP9 on amyloidogenic processing of APP and synaptic protein levels in the mouse brain. FASEB J 2012; 26: 2072–2083.
Hao J, Janssen WG, Tang Y, Roberts JA, McKay H, Lasley B et al. Estrogen increases the number of spinophilin immunoreactive spines in the hippocampus of young and old rehesus monkeys. J CompNeurol 2003; 465: 540–550.
Liberski PP, Sikorska B, Bratosiewicz-Wasik J, Gajdusek DC, Brown P . Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: from apoptosis to autophagy. Int J Biochem Cell Biol 2004; 36: 2473–2490.
Lee YB, Nagai A, Kim SU . Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res 2002; 69: 94–103.
Alpar A, Ueberham U, Bruckner MK, Seeger G, Arendt T, Gartner U . Different dendrite and dendritic spine alterations in basal and apical arbors in mutant human amyloid precursor protein transgenic mice. Brain Res 2008; 1099: 189–198.
Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 2006; 103: 5161–5166.
Catala I, Ferrer I, Galofre E, Fabregues I . Decreased numbers of dendritic spines on cortical pyramidal neurons in dementia. A quantitative Golgi study on biopsy samples. Hum Neurobiol 1988; 6: 255–259.
Davies CA, Mann DM, Sumpter PQ, Yates PO . A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci 1987; 78: 151–164.
de Ruiter JP, Uylings HB . Morphometric and dendritic analysis of fascia dentata granule cells in human aging and senile dementia. Brain Res 1987; 402: 217–229.
Cheng L, Lemmo S, Lemmon V . RanBPM is an L1-interacting protein that regulates L1- mediated mitogen-activated protein kinase activation. J Neurochem 2005; 94: 1102–1110.
Bowman AL, Catino D, Strong JC, Randall WR, Kontrogianni-Konstantopoulos A, Bloch RJ 2008 The rho-guanine nucleotide exchange factor domain of obscurin regulates assembly of titin at the Z-disk through interactions with Ran binding protein 9. Mol Biol Cell 2008; 19: 3782–3792.
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD . Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 2009; 106: 14670–14675.
Cassano T, Serviddio G, Gaetani S, Romano A, Dipasquale P, Cianci S et al. Glutamatergic alterations and mitochondrial impairment in a murine model of Alzheimer disease. Neurobiol Aging 2012; 33: e1121–1112.
Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M et al. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 2009; 30: 1574–1586.
Gillardon F, Rist W, Kussmaul L, Vogel J, Berg M, Danzer K et al. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 2007; 7: 605–616.
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS . Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci USA 2010; 107: 18670–18675.
Li Z, Okamoto K, Hayashi Y, Sheng M . The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004; 119: 873–887.
Chang DT, Honick AS, Reynolds IJ . Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci 2006; 26: 7035–7045.
Lin MT, Beal MF, Alzheimer's APP . mangles mitochondria. Nat Med 2006; 12: 1241–1243.
Maurer I, Zierz S, Moller HJ . A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging 2000; 21: 455–462.
Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J . Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007; 27: 2596–2605.
Hayes CD, Dey D, Palavicini JB, Wang H, Araki W, Lakshmana MK . Chronic cladribine administration increases amyloid beta peptide generation and plaque burden in mice. PLoS One 2012; 7: e45841.
Acknowledgements
This work was supported by National Institute of Aging (NIA)/NIH grant numbers (1R03AG032064-01, M.K. Lakshmana), 1R01AG036859-01, M.K. Lakshmana). We are grateful to receive Alzheimer’s and control brain tissues provided by the ‘Harvard Brain Tissue Resource Center, which is supported in part by PHS grant number R24MH068855′ at the McLean Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Palavicini, J., Wang, H., Bianchi, E. et al. RanBP9 aggravates synaptic damage in the mouse brain and is inversely correlated to spinophilin levels in Alzheimer’s brain synaptosomes. Cell Death Dis 4, e667 (2013). https://doi.org/10.1038/cddis.2013.183
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.183
Keywords
This article is cited by
-
Differential splicing of neuronal genes in a Trem2*R47H mouse model mimics alterations associated with Alzheimer’s disease
BMC Genomics (2023)
-
Autophagy-Dependent Increased ADAM10 Mature Protein Induced by TFEB Overexpression Is Mediated Through PPARα
Molecular Neurobiology (2021)
-
Tau Protein Squired by Molecular Chaperones During Alzheimer’s Disease
Journal of Molecular Neuroscience (2018)
-
Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits
Acta Neuropathologica (2017)
-
How Does p73 Cause Neuronal Defects?
Molecular Neurobiology (2016)
