
Abstract
Radiation therapy is frequently used to treat non-small cell lung cancers (NSCLCs). We have previously shown that a combination of ionizing radiation (IR) and the staurosporine analog PKC 412, but not Ro 31–8220, increases cell death in NSCLC cells. To identify genes involved in the enhancement of cell death, a total gene profiling in response to co-administration of (i) PKC 412 with IR, or (ii) Ro 31–8220 with IR was implemented. These combined treatments caused upregulation of 140 and 179 genes and downregulation of 253 and 425 genes, respectively. Certain genes were selected and verified by real-time quantitative PCR and, of these genes, robust suppression of Ephrin B3 expression was suggested as a possible cell death-inducing mechanism of combined treatment with IR and PKC 412. Indeed, silencing of Ephrin B3 using siRNA in NSCLC cells resulted in a major alteration of their morphology with an elongated phenotype, decreased proliferation and increased cell death signaling. Moreover, silencing of Ephrin B3 in combination with IR caused a decrease in IR-mediated G2-arrest, induced cellular senescence, inhibited MAPK ERK and p38 phosphorylation, and caused an upregulation of p27kip1 expression. Finally, silencing of Ephrin B3 in combination with IR sensitized U-1810 cells to IR-induced apoptosis. In conclusion, we identify and describe Ephrin B3 as a putative signaling molecule involved in the response of NSCLC cells to combined treatment with PKC 412 and ionizing radiation.
Similar content being viewed by others


Main
Non-small cell lung carcinoma (NSCLC) patients have a poor prognosis with a 5-year survival of about 10%.1 Chemotherapy (CT) and radiotherapy (RT) are standard treatments for inoperable NSCLC cases but often high intrinsic resistance is encountered hampering good clinical management. Mechanisms responsible for intrinsic and acquired resistance to RT include cellular signaling alterations, for example, increased DNA repair and growth factor signaling, or impaired cell death activation. Thus we showed that impaired apoptosis signaling via the Bcl-2 family members Bak and Bax, dysfunction of the mitochondrial release of cytochrome c and subsequent effects on caspase activation, all contribute to pronounced RT resistance of NSCLC cells.2, 3, 4 With the intention of finding treatments that could trigger cell death in ionizing radiation (IR)-resistant NSCLC cells, we showed that staurosporine could circumvent resistance and induce release of cytochrome c and subsequent caspase-3 activation.3 Next, we examined if analogs of staurosporine, PKC 412 and Ro 31–8220, could sensitize NSCLC cells to IR.5, 6 Indeed PKC 412 was demonstrated to sensitize for RT and triggers mitochondria-mediated apoptotic response although Ro 31–8220 did not and instead increased survival signaling.
Increased growth factor receptor signaling through, for example, EGFR has been demonstrated to influence NSCLC's response to IR (reviewed in1). Treatments in which EGFR inhibitors or monoclonal antibodies, such as, cetuximab are used in combination with CT and/or RT. These treatments have unfortunately only increased survival in a small part of the patient cohort, that is, in those whose tumors have an aberrant EGFR signaling network. For the vast majority of NSCLC patients, other pathways are likely driving tumors and should be therapeutically intervened with either alone or in combination with CT/RT. In the current study, we applied a global and non-supervised strategy to explore potential key signaling events conferring RT responsiveness or resistance in NSCLC cells. Consequently, a total gene profiling of the NSCLC cell line U-1810 was carried out after IR alone or IR in combination with either PKC 412 or Ro 31–8220, using the Affymetrix-based gene array. The gene array data together with validation on gene, protein and functional levels suggested Ephrin B3, a ligand of Eph receptors (EphR), as a putative regulator of RT resistance of NSCLC cells.
Results
A combination of IR and PKC 412 increases apoptotic signaling in NSCLC cells
We previously showed that a combination of IR and PKC 412 sensitized NSCLC cells to IR-induced apoptotic cell death,5 which was confirmed here (Figure 1). Thus a combination of IR and PKC 412 triggered increased apoptototic signaling as illustrated by cleavage of PARP into the specific 85-kDa cleavage fragment (Figure 1a), a twofold increase in caspase-mediated cleavage of cytokeratin 187, 8 (Figure 1b) and increased caspase-3 activation and apoptotic nuclear morphology (data not shown). Moreover, IR and PKC 412 combined treatment clearly caused a more prominent inhibition of proliferation than either treatment alone (Figure 1c).

Combined treatment with IR and PKC 412 induces death of U-1810 NSCLC cells. U-1810 cells were exposed to IR (8 Gy) and 24 h post-incubation treated with PKC 412 (1 μ M) for another 24 h before western blot analysis of PARP cleavage (a) or flow cytometric analysis of cytokeratin 18 cleavage (b) was examined. Data presented are the means of three independent experiments. T-test was used for statistical analysis (*=P<0.05). (c) The effect of IR and PKC412 on cell proliferation was analyzed at 72 h post IR in samples treated as in (a) using MTS cell proliferation assay. Data shown are the mean of three independent experiments. T-test was used for statistical analysis (P≤0.05)
Affymetrix-based gene array identifies genes differentially expressed in response to IR alone and to IR in combination with PKC 412 or with Ro 31–8220
To reveal genes uniquely regulated in response to a combination of IR and PKC 412, total gene profiles of U-1810 cells after exposure to IR or to IR combined with either PKC 412 or Ro 31–8220 were analyzed by Affymetrix gene array (Supplementary Figure S1A). Thus at 24 h post IR, U-1810 cells were treated with either PKC inhibitor for 4 h and total RNA was collected. A complete list of regulated genes is found in Supplementary data (http://www.ncbi.nlm.nih.gov/geo/, accession number AAH22499). Genes that showed at least a 1.5-fold upregulation or downregulation after IR alone or in response to a combination of IR and PKC 412 or Ro 31–8220 were selected and a two-step Venn diagram analysis was carried out as outlined (Supplementary Figure S1A). Briefly, the differentially expressed genes in response to IR or to a single or combined treatment with either inhibitor were listed. Subsequently, the genes that showed altered expression in combinatory treatments were evaluated, generating Venn diagrams of ‘unique’ and ‘shared’ genes for the different IR combinations (Supplementary Figure S1A). Co-administration of PKC 412 with IR was found to cause upregulation of 140 genes and downregulation of 253 genes that did not show any change in expression after the combination of IR and Ro 31–8220. Similarly, co-treatment with IR and Ro 31-8820 caused upregulation of 179 genes and downregulation of 425 genes, of which none were altered when PKC 412 was co-administrated with IR.
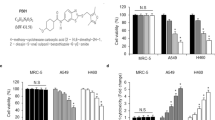
The differentially expressed genes were mapped onto different cellular processes using the GeneSpring GX software (Agilent Technologies, Santa Clara, CA, USA) and the gene array data filtered for genes associated with pathways known to be regulated by IR, that is, signal transduction, cell growth, cell death and cell cycle progression (Figures 2a and b, Supplementary Tables 1–3).

Gene array analysis and gene distribution. (a–b) Gene expression analysis of U-1810 cells treated with IR, PKC 412, or Ro 31–8220 alone, with combinations of IR and PKC 412, or IR and Ro 31–8220 were analyzed after pre-filtering of data using stepwise Venn diagrams. The gene expression analyses were performed at 24 h post IR and with 4h incubation with either PKC 412 or Ro 31–8220. Genes were classified as upregulated relative to control if the expression was above 1.5-fold, or as downregulated if the expression was below 1.5-fold, compared with untreated U-1810 cells. The distribution of regulated genes between the different cellular processes was analyzed using the Gene Spring software. The given values represent the percentage of total altered genes in IR+PKC 412 and IR+Ro 31–8220, respectively. (a) Distribution of upregulated genes in different processes. (b) Distribution of downregulated genes in different processes. (c) Change in Ephrin B3 gene expression in U-1810 cells from Affymetrix gene array analysis was verified using real-time quantitative PCR in response to PKC 412, Ro 31–8220 alone, or in combination with IR (8 Gy). Values are given relative to untreated U-1810 cells. Data are representatives from three independent experiments. Gray bars: Affymetrix gene expression array; Black bars: real-time quantitative PCR. (d) Western blot analysis of Ephrin B3 expression was performed 8, 30 and 48 h post IR and GAPDH was used as a loading control
Validation of gene expression alterations by real-time quantitative PCR
A set of genes that showed at least a 1.5-fold change in expression relative to untreated U-1810 cells was validated using real-time quantitative PCR (RT qPCR) (Figure 2c, Supplementary Figure S1B). These genes were Ephrin B3, the regulatory B subunit of the protein phosphatase 2 gamma isoform(PPP2R2C), the estrogen receptor 1(ESR1) and a member of the RAS oncogene family Rab33A (Rab33A). RT qPCR analyses confirmed the alteration in expression of these genes although the magnitude of expression differed between this assay and gene array analyses (Figure 2c, Supplementary Figure S1B). Importantly, a decreased protein expression of Ephrin B3 in IR and PKC 412 combined treatment was also confirmed (Figure 2d), and Ephrin B3 was selected for validation of its role in the IR-response of NSCLC cells.
Silencing of Ephrin B3 changes cell morphology, delays growth and affects IR responsiveness in human NSCLC cells
Ephrin ligands and their corresponding Ephrin receptors EphR's create an important cellular signaling circuit that has a profound effect on cell-to-cell communication and influences cell adhesion, migration and also invasion potential.9, 10 Binding of Ephrins to EphR's trigger a bidirectional signaling activity in which Eph kinases act as transducers in the EphR-expressing cell and the Src-family kinases are operative downstream the bound Ephrin ligand.9, 10 A number of recent reports demonstrated that the Ephrin/Eph receptor axis is deregulated in tumor cells, resulting in either decreased or increased signaling depending on the Ephrin/Eph receptor subtype and/or tumor origin.11, 12, 13 With respect to Ephrin B3, it has been demonstrated its binding to EphB3 and EphA4. The latter is a receptor that in the absence of its ligand triggers pro-apoptotic signaling.14 As the role of Ephrin B3 in IR-induced signaling has not been evaluated and given the observed decreased expression of Ephrin B3 in response to the combined treatment with IR and PKC 412 (Figure 2c), we decided to evaluate the role of Ephrin B3 in the radiosensitivity of NSCLC cells.
Consequently, Ephrin B3 was silenced using siRNA (hereafter described as Ephrin B3 siRNA) and efficient knockdown was confirmed on RNA and protein level, respectively (Figures 3a and b). Interestingly, silencing of Ephrin B3 greatly altered appearance of U-1810 cells with morphology similar to that of IR and PKC 412 combined (compare Figures 3c and 1c, lower right). Similar results were obtained using another siRNA sequence to silence Ephrin B3 expression, clearly supporting an on-target effect (Supplementary Figure S2). Moreover, in another NSCLC cell line U-1752, siRNA-mediated silencing of Ephrin B3 impaired growth and induced an elongated phenotype, although this was less pronounced than in U-1810 cells (Supplementary Figure S3).

Ephrin B3 siRNA induces elongated cell morphology and decreased cell growth of U-1810 cells. U-1810 cells were treated with Dharmafect only or transfected with non-target small-interfereing RNA (siRNA) or Ephrin B3-targeted siRNA (sequence 1) for 48 h before IR (8 Gy). Silencing of Ephrin B3 was confirmed at the mRNA level (a) using RT qPCR and at protein level using western blot (b). GAPDH was used as a loading control. The experiment presented is one representative out of three independent experiments. (c) The effects of Ephrin B3 siRNA on U-1810 cell morphology were examined using light microscopy (magnification × 400). Non-target siRNA was used as a negative control. (d) The effect of siRNA against Ephrin B3 on cell proliferation was analyzed using CFSE. Quantification of mean CFSE-IFL signal. Data are presented as relative values after normalization against non-target siRNA-treated cells in each experiment. T-tests were performed, P≤0.05
The effect of Ephrin B3 siRNA on cell proliferation was examined using the carboxyfluorescein diacetate N-succinimidyl ester (CFSE) dye, which is diluted upon cell division and hence decreased CFSE signal indicates proliferation.15 Interestingly, Ephrin B3 siRNA decreased cell proliferation at point zero (at time of irradiation), as shown by the increased CFSE fluorescence signal in Ephrin B3 siRNA-expressing cells compared with non-target siRNA-expressing cells (Figure 3d, Supplementary Figure S4). A more prominent inhibition of cell proliferation was observed in Ephrin B3 siRNA-expressing U-1810 cells at 48 h post IR (Figure 3d). As expected, IR caused a reduced proliferation in non-target siRNA-expressing cells and with a further inhibition in cells expressing Ephrin B3 siRNA (Figure 3d). In summary, these results suggest a putative role for Ephrin B3 in regulating cell shape and growth as well as their response to IR.
Silencing of Ephrin B3 sensitizes U-1810 cells to IR-induced apoptosis
Next the effect of Ephrin B3 suppression on apoptotic cell death alone or in combination with IR was examined and PARP cleavage and/or nuclear morphology were analyzed (Figures 4a and b). Silencing of Ephrin B3 alone induced PARP cleavage in U-1810 cells at 48 h as compared to non-target siRNA transfected cells (Figure 4a). Moreover, when non-target siRNA-treated U-1810 cells were irradiated, only a faint band of cleaved PARP was detected at 48 h post IR but with an increase observed at 120 h post IR (Figure 4a). In contrast, silencing of Ephrin B3 in combination with IR initiated a strong PARP cleavage at 48 h post IR and at 120 h post IR this combination induced the most pronounced PARP cleavage of all samples (Figure 4a).

Ephrin B3 siRNA in combination with IR induces an increase in cell-death signaling and alleviates IR-induced G2/M arrest. U-1810 cells were transfected with non-target or Ephrin B3 siRNA for 48 h and then either irradiated (8 Gy) or left untreated before harvesting at the indicated time points. (a) Western blot analysis of PARP cleavage was performed 48 and 120 h post IR and GAPDH was used as a loading control. Results shown are one representative out of three experiments. (b) For analysis of nuclear morphology, cells were fixed in 70% EtOH and stained with Hoechst 33342. Left: cells displaying apoptotic morphology or mitotic catastrophe were quantified in 100 cells in three independent experiments using fluorescence microscopy. Bars represent S.D. Right: pictures showing the nuclear morphology of non-target or Ephrin B3 siRNA-transfected cells, with or without irradiation (8 Gy). (c) Cell cycle analysis was performed using staining of DNA with propidium iodide in which cells with hypodiploid DNA content (subG1 cells) were excluded from the analysis. In quantification only gated cell population was used. The cell cycle distribution after non-target siRNA or Ephrin B3 siRNA either alone or combined with IR (8 Gy) was analyzed at 48 h post IR. Left: flow cytometric histograms showing cell cycle distribution after transfection with Ephrin B3 or non-targeting siRNA in combination with IR. Right: quantification of the number of cells in individual cell cycle phases. Data presented are one representative experiment of three independent experiments
Silencing of Ephrin B3 alone caused a slight increase in the number of U-1810 cells with the characteristics of mitotic catastrophe or apoptotic nuclei morphology (Figure 4b). Importantly, silencing of Ephrin B3 in combination with IR significantly increased the amount of these cells as compared with IR-treated cells transfected with non-target siRNA (Figure 4b). Representative pictures of nuclear morphology in IR-exposed cells with silenced Ephrin B3 are presented in Figure 4b (right panel).
Silencing of Ephrin B3 in combination with IR abrogates IR-induced G2 arrest
The influence of Ephrin B3 ablation on the cell cycle distribution of U-1810 cells at 48 and 24 h post IR was analyzed (Figure 4c and Supplementary Figure S5). As expected, IR caused a G2 cell cycle arrest which was evident 24 h post IR (Supplementary Figure S5), but more pronounced at 48 h post IR (Figure 4c, left). Silencing of Ephrin B3 expression alone only slightly affected cell cycle distribution (compare non-target siRNA with Ephrin B3 siRNA-treated cells; Figure 4c, left). Importantly, silencing of Ephrin B3 in combination with IR alleviated IR-induced G2-arrest and resulted in higher percentage of the cells in the G0/G1 phase (Figure 4c, left and right). These data and the data presented in Figure 3c demonstrate that depletion of Ephrin B3 (i) alone leads to a partial reduction in cell proliferation and (ii) in combination with IR alleviate the normal IR-induced G2-arrest and results in G1 phase accumulation.
Silencing of Ephrin B3 in combination with IR induced senescence of U-1810 cells
Silencing of Ephrin B3 expression caused a phenotype of U-1810 cells, which resembled senescence: a phenotype with few defined molecular alterations, but in which increased β-galactosidase activity is a surrogate marker.16 Histochemical staining for β-galactosidase activity confirmed a senescent phenotype and about 20% of the U-1810 cells with silenced Ephrin B3 or non-target siRNA-treated and irradiated (48 h) cells were senescent (Figure 5a, right). Importantly, silencing Ephrin B3 in combination with IR caused a more pronounced senescence phenotype with flattening and increased cell size. β-Galactosidase positivity was observed in about 40% of the cells (Figure 5a, left and right).

Ephrin B3 suppression in combination with IR causes senescence and concomitant alteration of Akt, MAPKs ERK, and p38 phosphorylation, and p27kip1 expression. U-1810 cells were treated as indicated in Figure 4. At 48 h post IR, the cells were stained for β-galactosidase activity as a marker of senescence. (a) Examples of senescent cells are given in the left panel. The percentage of cells out of 100 cells counted in three different fields is presented in the right panel. Data shown are the mean ±S.D. of three separate siRNA transfections. (b) Western blot analysis of Akt, p42/p44 ERK, and p38MAPK (phospho and total) (left) and p21WAF1/Cip1, p16INK4A and p27Kip1, cyclin B1 and cdc2 (Tyr15) (right) at 0 or 48h post IR. Ku80 was used as a loading control. Western blot presented is one representative experiment of three performed experiments. (c) The expression of EphA4 and Ephrin A1 in U-1810 cells treated as in (b) was examined. Ku80 was used as a loading control
Silencing of Ephrin B3 decreases MAPK ERK and p38 phosphorylation
Silencing of Ephrin B3 expression had pronounced effects on basal as well as IR-induced cell growth and cell cycle progression in NSCLC cells. We therefore analyzed how Ephrin B3 siRNA, alone or in combination with IR, influenced the phosphorylation status of growth-promoting signaling cascades, that is, p42/p44 MAPK ERK, p38MAPK, and Akt (Figure 5b, left). Interestingly, in Ephrin B3 siRNA-transfected cells, a decreased ERK and p38MAPK phosphorylation relative to non-target siRNA-transfected cells was evident at 48 h post-transfection, that is, at the time of IR (labeled 0 h; Figure 5b, left). Also at 48 h, post IR Ephrin B3 siRNA-treated cells showed less ERK and p38MAPK phosphorylation relative to non-target siRNA-transfected cells, an effect which was evident upon single treatment and in combination with IR (Figure 5b, left). Thus IR resulted in a significant increase of p38 phosphorylation, which was blocked in Ephrin B3-silenced cells (Figure 5b, left). Similarly, silencing of Ephrin B3 was found to decrease phosphorylation of Akt at Ser473 already at time of irradiation (0 h) with an effect still evident at 48 h post IR (Figure 5b, left). Our results thereby demonstrate that both Akt- and MAPK-mediated survival signaling is clearly impaired upon Ephrin B3 silencing in these NSCLC cells and may in part explain the observed RT-sensitizing effect of Ephrin B3 ablation in NSCLC cells.
Silencing of Ephrin B3 increases p27 Kip1 expression
Silencing of Ephrin B3 expression inhibited IR-induced G2/M accumulation and caused accumulation of U-1810 cells in G0/G1. We, therefore, examined molecular components involved in the regulation of these cell cycle phases, that is, p21WAF1/Cip1, p16INK4A, and p27Kip1, as well as cyclin B1 and phospho-cdc2 (Tyr15), the latter known to be of importance in the G2/M-phase progression (Figure 5b, right). A decreased expression of p21WAF1/Cip1 was evident in Ephrin B3-silenced cells compared with both non-irradiated and irradiated non-target siRNA-transfected cells (Figure 5b, right). A weak expression of p16INK4A was observed in non-target siRNA-expressing U-1810 cells, whereas Ephrin B3 siRNA-transfected cells did not express p16INK4A at the time of irradiation although expression was restored at 48 h post IR (Figure 5b, right). Interestingly, in line with an increased proportion of Ephrin B3-repressed cells in G0/G1 phase, analysis of p27Kip1 expression showed that Ephrin B3 siRNA-transfected U-1810 cells had a higher p27kip1 expression than corresponding cells with non-target siRNA. Moreover, whereas irradiated non-target siRNA-transfected cells showed downregulation of p27kip1, the opposite was observed in Ephrin B3-suppressed cells in which an increase in p27kip1 expression was evident after IR (Figure 5b, left). Altogether these data suggest that Ephrin B3 in NSCLC cells may influence IR-induced cell cycle progression by altering p27kip1 but without influencing p21WAF1/Cip1or p16INK4A.
We also analyzed how siRNA targeting Ephrin B3 influenced expression of cyclin B1 (Figure 5b, right). In line with the decreased number of cells in the G2/M phase caused by Ephrin B3 silencing, a decrease in cyclin B1 expression was observed in both untreated and irradiated cells (compare non-targeting siRNA and Ephrin B3 siRNA at 0 h and 48 h post IR, Figure 5b, right). Similarly, a reduced phosphorylation of cdc2 was evident in Ephrin B3-silenced cells upon irradiation as compared with their non-target siRNA-transfected irradiated counterparts (Figure 5b, right). Thus, these results support that Ephrin B3 silencing alters IR-induced G2 accumulation.
Ephrin B3 silencing-mediated effects do not likely involve EphA4 receptor
Next, we examined putative Ephs involved in the effects of Ephrin B3 silencing in U-1810 NSCLC cells. Ephrin B3 is reported to be a ligand for EphA4, a dependence receptor that in absence of ligand binding activates caspase-mediated apoptotic signaling.14 However, when the EphA4 expression was examined in these cells, only trace amounts of EphA4 was observed (Figure 5c), making this receptor less likely to be responsible for the pronounced effects observed upon Ephrin B3 silencing. We also examined EphA4 expression after Ephrin B3 silencing but no alteration in EphA4 level was observed.
Ephrin B3 is also reported to bind EphB3 (for review see:10). However, western blot analyses of the EphB3 expression in U-1810 cells showed only trace amount of this receptor (data not shown), which allows us to conclude that the primary choices for EphR binding to Ephrin B3, that is, EphA4 and EphB3, do not appear to be of major importance in NSCLC U-1810 cells. In order to understand more about the important signaling effects responsible for obtained results, we took an unbiased approach with mass spectrometry and profiled U-1810 cells with or without Ephrin B3 expression for alteration in their phosphoproteomes.17 In these experiments the EphA2 turned out to be differentially phosphorylated on Ser897 in the presence or absence of Ephrin B3. Thus, silencing of Ephrin B3 expression caused impaired Ser897 EphA2 phosphorylation and in fact resulted in degradation of the EphA2 protein likely as a cause of inactivation of a pro-survival signaling hub controlled by Akt1, CK2, and HSP90AA1. This study strongly suggests that EphA2 is one of the corresponding receptors of Ephrin B3 in lung cancer cells. Indeed, EphA2 is highly expressed in U-181017 and in other NSCLC cell lines (Efazat et al., manuscript in preparation). Altering Ephrin B3 expression levels do not influence Ephrin A1 expression, another ligand for the EphA2 receptor (Figure 5c). Yet it remains to be proven that Ephrin B3 indeed directly binds to EphA2.
Discussion
Targeted therapy directed against EGFR has had a major impact on treatment outcome for patients with an EGFR-driven LC, both as monotherapy and in combination with CT/RT. Unfortunately, ablation of EGFR signaling is only a personalized cancer therapy for a minor fraction of NSCLC patients as the rest of the NSCLC tumors are driven by other proliferation-regulating cascades. Hence, further characterization of signaling alterations in NSCLC and how they influence the CT/RT response is needed to enable a personalized cancer therapy approach for more of the NSCLC patients. In this study, gene expression analysis of NSCLC cells refractory to RT identified Ephrin B3, a ligand of EphR, as a putative driver of RT resistance. Moreover, siRNA-mediated Ephrin B3 suppression demonstrated a role of this protein in NSCLC proliferation and cell death propensity. Importantly, ablation of Ephrin B3 altered MAPKs and Akt signaling, interfered with the IR-induced G2/M cell cycle arrest, increased apoptosis, mitotic catastrophe and senescence signaling and thereby increased RT sensitivity.
Binding of Ephrin to the EphR may cause activation and deactivation of the Eph and Ephrin downstream signaling cascades resulting in altered cell-to-cell interactions and cell migration.9, 10 A critical role of the Ephrin/EphR signaling hub in tumor cell signaling is evident9, 10, 18, 19, 20, 21 and recently certain mutations in the kinase domain or in the Ephrin ligand-binding domain of EphR's of NSCLC were described,11 suggesting a role for EphR's signaling in this tumor type.
Here we found a novel function of Ephrin B3 in regulating tumor cell response to IR. Thus, silencing Ephrin B3 in combination with IR resulted in prominent inhibition of cell proliferation and increased apoptotic cell death. In searching for EphR involved in the effects mediated by Ephrin B3 silencing in NSCLC cells, we first focused on EphA4, a receptor previously shown to be dependent on Ephrin B3 for maintenance of survival signaling,14 and EphB3, an Ephrin B3 binding partner. Only trace amounts of EphA4 and EphB3 were observed in NSCLC cells before and after Ephrin B3 silencing, making it unlikely that EphA4 or EphB3 has a major function in these cells. In a phosphoproteome analysis, we described that impaired Ephrin B3 expression inhibited Ser897 EphA2 phosphorylation, compromised Akt phosphorylation and caused degradation of the EphA2 receptor.17 Therefore, we propose that Ephrin B3 mediates its RT-sensitizing effects, as well as its growth inhibitory propensity, in part, by altering EphA2 signaling, although the molecular pathways require further characterization. However, in this study, we rule out that Ephrin B3 silencing would exert its effect by altering Ephrin A1 expression as it remained highly expressed when Ephrin B3 was silenced. Further work is required to pinpoint the Eph receptor binding-partners of Ephrin B3. Nevertheless, given the discovery that EphA2 is overexpressed in NSCLC and the recent data demonstrating a link between certain EphA2 mutations and NSCLC invasiveness, understanding the role of Ephrin B3 in this signaling context may provide novel therapeutic approaches for NSCLC.
Mitotic catastrophe has previously been linked to the cellular response to radiation (reviewed in22, 23). We found that silencing of Ephrin B3 in combination with IR increased mitotic catastrophe in about 15% of the cells. This finding is in line with our previous publication demonstrating that a combination of PKC 412 and IR increases the number of cells undergoing mitotic catastrophe.6 Moreover, we previously showed that mitotic catastrophe triggered by DNA-damage can, dependent on tumor cells signaling status, follow by apoptosis or necrosis.23 With respect to Ephrin silencing in combination with IR in NSCLC U-1810 cells, the induction of mitotic catastrophe occurred independent of functional p53, a molecule previously shown to be required for efficient mitotic catastrophe.
Ephrins/EphR's are known to influence MAPKs and PI3K/Akt signaling.9, 10 Indeed we found that Ephrin B3 siRNA-transfected U-1810 cells showed a prominent inhibition of Akt and p38MAPK, as well as p42/p44 ERK phosphorylation (Figure 5b). Importantly, in IR-treated non-target siRNA-expressing U-1810 cells, both Akt, ERK and p38MAPK phosphorylation was evident, whereas in irradiated Ephrin B3 siRNA-expressing cells, significantly less phosphorylation was observed. Thus, our results suggest that Ephrin B3 in part regulates Akt, ERK and p38MAPK phosphorylation and it may be speculated that Ephrin B3 influences IR sensitivity in this way.
After Ephrin B3 siRNA and IR combined treatment, an elongated, flattened, and increased cell size was noted (Figure 3c). These features are hallmarks of senescence, a form of cell death previously described as a potent barrier to tumorigenesis (reviewed in Kuilman and Peeper24). By staining the cells with β-galactosidase, a senescence effect was evident after silencing of Ephrin B3 in combination with IR (Figure 5a). To our knowledge, this is the first observation demonstrating a role for Ephrin B3 signaling in senescence. Nevertheless, senescence factors are often used for cell-to-cell communication,24 which fits well with the bi-directional properties of Ephrin/EphR signaling. Albeit a role for p53 and Rb in senescence signaling is evident,24 it is clear that senescence may also work in tumor cells expressing mutant p53.25 Although the NSCLC cells used in the current experiments do not express the p53 protein,26 they still become senescent thus further supporting the existence of a non-p53-dependent route to this cell state. It has been suggested that senescent cells have an increased p21CIP1/WAF1 and/or p16INK4A expression relative to ‘normal’ tumor cells. Although we did indeed detect a G0/G1 arrest upon Ephrin B3 silencing, surprisingly, an increase in p21WAF1/Cip1 or p16INK4A expression was not observed, but instead cells lacking Ephrin B3 expression at basal level, as well as after IR, showed less of both these markers (Figure 5b). Thus, neither p21WAF1/Cip1 nor p16INK4A are likely involved in the Ephrin B3 silencing-mediated effects on cell cycle progression. In fact, a slight upregulation of p27Kip1 in U-1810 cells upon silencing of Ephrin B3 alone as compared with non-targeting siRNA-expressing cells at the 48 h time point was detected (Figure 5b). However after IR, latter cells showed G2/M accumulation and low p27Kip1 expression, which was in marked contrast to Ephrin B3-silenced cells, where an increased level of p27Kip1 over basal level was detected (Figure 5b). As p27kip1 expression has been shown to be highest in the G0 cell cycle phase,27, 28 this increase in p27Kip1 fits well with the observed G0/G1 accumulation of cells with silenced Ephrin B3 expression. One common component of several EphR is Src. v-Src transformation in which cells acquire a rounded phenotype and increased migration potential is reported to require downregulation of p27Kip1. In the context of Ephrin B3 suppression, it may be speculated that this suppression may decrease Src activity, increase p27Kip1 stability and consequently cause G0/G1 arrest. Further work is, however, required to clarify whether p27Kip1 is a prerequisite or a consequence of induction of senescence after Ephrin B3 suppression.
In conclusion, in this study, we identified Ephrin B3 as a putative molecule involved in NSCLC proliferation as well as of a driver of radioresistance. Recently Eph receptor mutations in lung cancer have been identified, suggesting that the Ephrin/Eph pathways might be worth to further explore for therapeutic purposes in this tumor type. Our results suggest that interfering with Ephrin B3 binding to Eph might be a therapeutic avenue for at least certain NSCLC cases. Further work regarding Ephrin B3 and downstream signaling in NSCLC is, therefore, required to establish a therapeutic strategy with clinical potential.
Material and Methods
Cell culture, cell counting, and treatments
A human lung carcinoma cell line U-1810 derived from an undifferentiated large cell NSCLC case was used.29 For certain experiments U-1752, a NSCLC cell line with squamous cell histology was used.30 Both cell lines are refractory to RT with a surviving fraction 2 Gy value of about 0.9.29, 30 All cells were maintained in RPMI 1640 medium (Sigma Aldrich, Stockholm, Sweden) supplemented with 10% heat-inactivated fetal calf serum, 1 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified incubator with 5% CO2. For all experiments, cells were seeded 24 h prior to the treatment at 0.5 × 106 cells per 10-cm dish and were sub-confluent at time of irradiation. Irradiation was carried out at room temperature using γ photons (γ-IR; adsorbed dose 8 Gy, 0.9 Gy/min from a Co60 source). At 24 h post IR, cells were pulsed with either the staurosporine analog PKC 412 (1 μM), Ro 31–8220 (10 μM) or vehicle (DMSO) for the indicated times. The PKC 412 was a gift from Novartis Pharma AG (Basel, Switzerland) and Ro 31–8220 was purchased from Sigma (Sigma Aldrich). For studies of cell morphology, photographs of cells were taken at indicated time points using an optical light microscope (Nikon Eclipse TS100, Stockholm, Sweden) at × 400 magnification.
MTS cell proliferation assay
U-1810 cells were plated in 96-well plates at final density 4 × 103 cells in growth media. 24 h after the seeding, cells were irradiated (8 Gy) and 24 h after IR treated with PKC 412 (1 μ M). At 72 h post IR CellTiter 96 AQueous non-radioactive cell proliferation Assay (Promega, SDS, Falkenberg, Sweden) was performed according to the manufacture’s protocol. Results presented are the means±S.E.M. of the three independent experiments, P>0.05.
Affymetrix gene expression analysis
RNA was isolated from 10 million U-1810 cells that were untreated or treated with PKC-412, Ro 31–8220, irradiation or a combination of IR and PKC 412 or Ro 31–8220 at the indicated time points post-treatment. The RNA was extracted using the RNeasy Mini kit, according to the manufacturer’s instructions (RNeasy Midi Handbook; Qiagen, Germantown, MD, USA). The RNA purity and concentration were assessed using a NanoDrop ND-1000 UV–vis Spectrophotometer (NanoDrop Technologies Wilmington, DE, USA).
Gene expression analyses were performed using the Affymetrix platform (Affymetrix Inc., Santa Clara, CA, USA). The synthesis of cRNA, labeling and hybridization were performed according to the protocols provided by the manufacturer. The data were analyzed by the GeneSpring GX software (Agilent Technologies). Normalization of gene expression data was accomplished in two ways: per chip normalization and per gene normalization. For per chip normalization, all expression data on a chip were normalized to the 50th percentile of all values on that chip. For per gene normalization, the data for a given gene was normalized to the median expression level of that gene across all samples. The data sets were then assigned to three groups for the experiments (control, PKC 412, and Ro 31–8220 either alone or in combination with IR). The expression profiles from the three independent experiments were compared using ANOVA (parametric test, variances assumed equal) to identify genes that were differentially expressed between the different treatments. All further analyses were carried out on the remaining 863 probe sets.
Cytokeratin 18 cleavage
Caspase-mediated cytokeratin 18 cleavage is accompanied by the appearance of a specific neo-epitope in the cytokeratin 18 molecule.7, 8, 31 An antibody that recognizes this epitope, M30 CytoDEATH-FITC antibody (Roche Diagnostics Scandinavia AB, Stockholm, Sweden), was used. Ice-cold-methanol-fixed cells (−20 °C, 30 min) were stained with the M30 CytoDEATH-FITC antibody and the percentage of cells showing positive staining was analyzed with flow cytometry by measuring the FITC fluorescence in the FL1 of fluorescence-associated cell sorting calibur (BD Biosciences, Stockholm, Sweden). Results presented are the means±S.E.M. of the three independent experiments, P>0.05.
Real-time quantitative PCR validation
Reverse-transcribed cDNAs from the indicated samples were used as templates. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was applied as a positive control and also used for normalization of the different template values. To amplify the cDNA, 100 ng of the reverse-transcribed cDNA from each sample was subjected to 40 cycles of real-time quantitative PCR in a 20-μl total reaction volume. The Ephrin B3, protein phosphatase 2 gamma isoform, estrogen receptor 1, Rab33A and GAPDH primers and probes were purchased from Applied Biosystems (Applied Biosystems, Stockholm, Sweden) and were designed to flank the target DNA sequence. The mRNA gene expressions of each gene in the treated U-1810 cells were given as fold increase or reduction compared with untreated U-1810 cells after normalization against GAPDH.
Western blot analysis
The total protein content was extracted using a lysis buffer (6 M urea and 2% SDS in 200 mM ammonium bicarbonate and protease inhibitor cocktail (Roche, Mannheim, Germany)). The lysates were sonicated, cleared from the insoluble cell membranes and debris by centrifugation (10 000 g/10 min), and the protein content was measured using the bicinchoninic acid (BCA) assay (Interchim, Montiuçon Cedex, France). Western blot analyses were carried out as previously described4 and the following primary antibodies were used: Ephrin B3, Ku80 (Abcam, Cambridge, UK), Akt (phospho ser473- and total), Cyclin B1, ERK, p38 (phospho- and total), phospho-cdc2 (Tyr15) (Cell Signaling, Danvers, MA, USA), PARP, p21WAF1/Cip1, and p27Kip1 (BD Pharmingen, Franklin Lakes, NJ, USA), EphA4 (S-20) and p16INK4A (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and GAPDH (Trevigen, Gaithersburg, MD, USA).
Silencing of Ephrin B3 expression with siRNA
Ephrin B3 siRNA was purchased from Qiagen. The 5′-CCAGGAGTATAGCCCTAAT-3′ target sequence (sequence 1) corresponding to 748–766 bp, which is described as unique relative to other Ephrin members32 and Dharmafect (Dharmacon, Lafayette, CO, USA) were used according to the manufacturers’ instructions. To rule out the off-target effect of the Ephrin B3 siRNA, another sequence 5′-GCTCGCACCACGATTACTA-3′ corresponding to 786–804 bp (sequence 2), also previously noted as specific for Ephrin B3 ligand,32 was applied in corresponding experiments (see Supplementary Figure S2). For each experiment 500 000 cells were seeded onto 10-cm dishes and transfected after 6 h using 100 nM siRNA. Non-target siRNA (Dharmacon) was used as control. In all these experiments, cells were not confluent even at late time points as they started to be arrested in G1 and grow very slowly shortly after transfection. Hence, a risk that other Ephrins may signal onto Ephs as a consequence of confluency could be considered small. Irradiation was performed 48 h after transfection when silencing of Ephrin B3 expression was achieved and the cells were then harvested at indicated time points post IR.
Cell division analysis using carboxyfluorescein diacetate N-succinimidyl ester (CFSE)
The CFSE is a cell-permeable dye, that is, non-fluorescent until it is cleaved by intracellular esterases to yield a fluorescent carboxyfluorescein succinimidyl ester, that is, retained within the cells. During each cell division, the fluorescent label will be progressively diluted, allowing cell proliferation to be monitored by fluorescence-associated cell sorting.15 Briefly, the same volume of U-1810 cells from the same stock suspension was seeded onto each plate. The cells were incubated at room temperature for 5 min in complete medium containing 2.5 μmol/l CFSE. Thereafter, the cells were washed and incubated in fresh medium followed by siRNA transfection. At 48 h post-transfection (=0 h) or at 48 h post IR (=48 h), the cells were collected for fluorescence-associated cell sorting analysis without prior fixation. CFSE fluorescence was detected in the FL1 channel and the data is given as CFSE-associated IFL signal.
Cell cycle distribution
Three million cells were pelleted at 500 × g and resuspended in 100 μl of ice-cold PBS. Next, the cells were fixed by a drop-wise addition of 1 ml of ice-cold 70% ethanol while vortexing. Upon analysis, the cells were centrifuged at 200 × g, washed in PBS and resuspended in 1 ml of propidium iodide staining solution (40 μg/ml propidium iodide and 10 μg/ml RNase-A in PBS). Analysis of the cell cycle distribution was carried out on a FACScan Instrument (BD Biosciences, San Jose, CA, USA) in the FL2 channel and analyzed using ModFit LT Verity Software (Topsham, ME, USA). In these experimental settings, cell debris, as well as the subG1 population, was excluded from the analysis by gating of the cells.
Analysis of nuclear morphology
Nuclear morphology was evaluated as the number of cells with abnormal nuclei as defined in Figure 4b (right). The cells were harvested after the indicated treatments using cell dissociation solution (CDS, Sigma Aldrich). After washing in PBS, the cells were fixed for 10 min in 4% PFA solution. The nuclei of fixed cells were stained with mounting media containing Hoechst 33342 (Vector Laboratory, Burlingame, CA, USA). The number of cells with nuclear morphology indicative of an apoptotic or mitotic catastrophe phenotype was quantified using a fluorescent microscope (ZEISS Axioplan 2 imaging microscope, ZEISS, Thornwood, NY, USA). At least 100 cells were assessed in each sample and apoptosis is presented as the percentage of cells with fragmented nuclei.
Analysis of senescent cells
To reveal a senescent phenotype of the cells, the Senescence Cells Histochemical Staining Kit (Sigma Aldrich) was applied according to the manufacturer’s instructions. Briefly, cells were seeded onto 60-mm plates and fixed for 7 min at room temperature. The cells were then washed with PBS and stained with X-gal-based staining mixture overnight at 37 °C without CO2. Triplicates samples were analyzed in which the percentage of blue-stained cells expressing β-galactosidase out of 100 total cells examined in three representative fields was evaluated.
Accession codes
Abbreviations
- CK18:
-
cytokeratin 18
- CFSE:
-
carboxyfluorescein diacetate N-succinimidyl ester
- CT:
-
chemotherapy
- IR:
-
ionizing radiation
- NSCLC:
-
non-small cell lung carcinoma
- RT:
-
radiotherapy
- RT qPCR:
-
real-time quantitative PCR
- STS:
-
staurosporine
References
Viktorsson K, Lewensohn R . Apoptotic signaling pathways in lung cancer. J Thorac Oncol 2007; 2: 175–179.
Viktorsson K, Ekedahl J, Lindebro MC, Lewensohn R, Zhivotovsky B, Linder S et al. Defective stress kinase and Bak activation in response to ionizing radiation but not cisplatin in a non-small cell lung carcinoma cell line. Exp Cell Res 2003; 289: 256–264.
Joseph B, Marchetti P, Formstecher P, Kroemer G, Lewensohn R, Zhivotovsky B . Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene 2002; 21: 65–77.
Stahl S, Fung E, Adams C, Lengqvist J, Mork B, Stenerlow B et al. Proteomics and pathway analysis identifies JNK signaling as critical for high linear energy transfer radiation-induced apoptosis in non-small lung cancer cells. Mol Cell Proteomics 2009; 8: 1117–1129.
Hemstrom TH, Joseph B, Schulte G, Lewensohn R, Zhivotovsky B . PKC 412 sensitizes U1810 non-small cell lung cancer cells to DNA damage. Exp Cell Res 2005; 305: 200–213.
Hemstrom TH, Sandstrom M, Zhivotovsky B . Inhibitors of the PI3-kinase/Akt pathway induce mitotic catastrophe in non-small cell lung cancer cells. Int J Cancer 2006; 119: 1028–1038.
Rupa JD, de Bruine AP, Gerbers AJ, Leers MP, Nap M, Kessels AG et al. Simultaneous detection of apoptosis and proliferation in colorectal carcinoma by multiparameter flow cytometry allows separation of high and low-turnover tumors with distinct clinical outcome. Cancer 2003; 97: 2404–2411.
Hagg M, Biven K, Ueno T, Rydlander L, Bjorklund P, Wiman KG et al. A novel high-through-put assay for screening of pro-apoptotic drugs. Invest New Drugs 2002; 20: 253–259.
McCarron JK, Stringer BW, Day BW, Boyd AW . Ephrin expression and function in cancer. Future Oncol 2010; 6: 165–176.
Pasquale EB . Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer 2010; 10: 165–180.
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455: 1069–1075.
Alam SM, Fujimoto J, Jahan I, Sato E, Tamaya T . Coexpression of EphB4 and ephrinB2 in tumour advancement of ovarian cancers. Br J Cancer 2008; 98: 845–851.
Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest 2009; 119: 1727–1740.
Furne C, Ricard J, Cabrera JR, Pays L, Bethea JR, Mehlen P et al. EphrinB3 is an anti-apoptotic ligand that inhibits the dependence receptor functions of EphA4 receptors during adult neurogenesis. Biochim Biophys Acta 2009; 1793: 231–238.
Quah BJ, Warren HS, Parish CR . Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc 2007; 2: 2049–2056.
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92: 9363–9367.
Stahl S, Branca RM, Efazat G, Ruzzene M, Zhivotovsky B, Lewensohn R et al. Phosphoproteomic profiling of NSCLC cells reveals that ephrin B3 regulates pro-survival signaling through Akt1-mediated phosphorylation of the EphA2 receptor. J Proteome Res 2011; 10: 2566–2578.
Brannan JM, Dong W, Prudkin L, Behrens C, Lotan R, Bekele BN et al. Expression of the receptor tyrosine kinase EphA2 is increased in smokers and predicts poor survival in non-small cell lung cancer. Clin Cancer Res 2009; 15: 4423–4430.
Brannan JM, Sen B, Saigal B, Prudkin L, Behrens C, Solis L et al. EphA2 in the early pathogenesis and progression of non-small cell lung cancer. Cancer Prev Res 2009; 2: 1039–1049.
Germain S, Eichmann A . VEGF and ephrin-B2: a bloody duo. Nat Med 2010; 16: 752–754.
Faoro L, Singleton PA, Cervantes GM, Lennon FE, Choong NW, Kanteti R et al. EphA2 mutation in lung squamous cell carcinoma promotes increased cell survival, cell invasion, focal adhesions, and mammalian target of rapamycin activation. J Biol Chem 2010; 285: 18575–18585.
Jonathan EC, Bernhard EJ, McKenna WG . How does radiation kill cells? Curr Opin Chem Biol 1999; 3: 77–83.
Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B . DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ 2008; 15: 555–566.
Kuilman T, Peeper DS . Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 2009; 9: 81–94.
Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010; 464: 374–379.
Sirzen F, Heiden T, Nilsson A, Bergh J, Skog S, Lewensohn R . Characterisation of the G1/S cell cycle checkpoint defect in lung carcinoma cells with different intrinsic radiosensitivities. Anticancer Res 1997; 17: 3381–3386.
Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D et al. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci USA 2009; 106: 9268–9273.
Larrea MD, Wander SA, Slingerland JM . p27 as Jekyll and Hyde: regulation of cell cycle and cell motility. Cell Cycle 2009; 8: 3455–3461.
Bergh J, Nilsson K, Ekman R, Giovanella B . Establishment and characterization of cell lines from human small cell and large cell carcinomas of the lung. Acta Pathol Microbiol Immunol Scand A 1985; 93: 133–147.
Brodin O, Lennartsson L, Nilsson S . Single-dose and fractionated irradiation of four human lung cancer cell lines in vitro. Acta Oncol 1991; 30: 967–974.
Caulin C, Salvesen GS, Oshima RG . Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J Cell Biol 1997; 138: 1379–1394.
Nakada M, Drake KL, Nakada S, Niska JA, Berens ME . Ephrin-B3 ligand promotes glioma invasion through activation of Rac1. Cancer Res 2006; 66: 8492–8500.
Acknowledgements
This research was supported by grants to RL, BZ and KV from the Swedish Cancer Society, the Stockholm Cancer Society, the Swedish Research Foundation, the Swedish Childhood Cancer Foundation, the Swedish National Board of Health and Welfare, the Stockholm County Council, the Karolinska Institutet Research Fund and the European Union FP6 (Chemores), and FP7 (Apo-Sys). VOK was supported by a fellowship from the Swedish Institute and Karolinska Institutet and AHV from the Wenner-Gren Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ståhl, S., Kaminskyy, V., Efazat, G. et al. Inhibition of Ephrin B3-mediated survival signaling contributes to increased cell death response of non-small cell lung carcinoma cells after combined treatment with ionizing radiation and PKC 412. Cell Death Dis 4, e454 (2013). https://doi.org/10.1038/cddis.2012.188
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.188
Keywords
This article is cited by
-
Eph receptors and ephrins in cancer progression
Nature Reviews Cancer (2024)
-
Dying to communicate: apoptotic functions of Eph/Ephrin proteins
Apoptosis (2018)
-
Enhancing radiosensitization in EphB4 receptor-expressing Head and Neck Squamous Cell Carcinomas
Scientific Reports (2016)
-
DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53
Cell Death & Differentiation (2016)
-
Systems biology approaches to develop innovative strategies for lung cancer therapy
Cell Death & Disease (2014)
