
Abstract
Use of the cre transgene in in vivo mouse models to delete a specific ‘floxed’ allele is a well-accepted method for studying the effects of spatially or temporarily regulated genes. During the course of our investigation into the effect of cyclic adenosine 3′,5′-monophosphate-dependent protein kinase A (PKA) expression on cell death, we found that cre expression either in cultured cell lines or in transgenic mice results in global changes in PKA target phosphorylation. This consequently alters gene expression profile and changes in cytokine secretion such as IL-6. These effects are dependent on its recombinase activity and can be attributed to the upregulation of specific inhibitors of PKA (PKI). These results may explain the cytotoxicity often associated with cre expression in many transgenic animals and may also explain many of the phenotypes observed in the context of Cre-mediated gene deletion. Our results may therefore influence the interpretation of data generated using the conventional cre transgenic system.
Similar content being viewed by others


Main
The ability to manipulate pluripotent embryonic stem cells through homologous recombination has revolutionized mouse genetics in the past two decades. Cre-loxP recombination system has been an integral part of mouse genome manipulation, initially to remove the selectable marker and thus eliminating ‘the selection cassette effects’1, 2 and subsequently to overcome certain deficiencies associated with ubiquitous gene deletions. These deficiencies include embryonic lethality and functional compensation by redundant gene products during ontogeny and can mask the effect of a particular gene deletion.3 Conditional deletion, which results in temporal and spatial ablation of the targeted genes, is achieved through the bacteriophage P1 recombinase, Cre. Cre is a member of the integrase family of site-specific recombinase that catalyzes recombination between ‘lox-P’ DNA elements during normal viral life cycle.4 In mouse gene manipulation, the targeted allele is flanked by two 34-bp lox-P sites (‘floxed’ allele) and the Cre recombinase is expressed by temporarily or spatially regulated promoters.3
Thus, the ‘Cre-loxP’ system has been the mainstay of mouse genetic manipulation for many years, and it has been assumed that this enzyme is inert in mammals and maintains high specificity in vivo. However, there have been reports of nonspecific activity on cryptic or pseudo loxP-like sites similar to the star activity observed with certain restriction enzymes.5 It has also been shown that Cre overexpression in the post-mitotic spermatids results in gross chromosomal abnormalities, leading to male sterility,4 and chromosomal abnormality in mouse embryonic fibroblasts (MEFs), leading to growth arrest.6 Deletion of prkar1a, the ubiquitously expressed regulatory subunit of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated protein kinase A (PKA), should result in increased PKA activity.7 During our investigation of prkar1afl/fl mice, we observed (contrary to expectations) a severely muted PKA response. We subsequently observed that Cre expression led to the induction of protein kinase A inhibitor (PKI) causing a global attenuation of the PKA signal transduction pathway. These observations may explain some of the phenotypes associated with Cre transgene expression, such as cellular growth retardation and cytotoxicity.
Results
Cre expression results in reduction of PKA activity
cAMP regulation of PKA is achieved via a unique three-component signaling system. The catalytic (C) and regulatory (R) subunits of PKA interact to form an inactive holoenzyme complex. Binding of cAMP induces a conformational change in the R1α subunit (the most ubiquitously and predominantly expressed subunit) and leads to the dissociation of the holoenzyme into its constituent subunits, that is, PRKAR1A and PKACα.8 The gene prkar1a is known to be a tumor suppressor in a variety of endocrine and non-endocrine tumors (cardiac and breast myxomas), pituitary hyperplasia, growth hormone secreting adenomas and in osteosarcomas.7, 9, 10 This is attributed to increased PKA activity. In prkar1afl/fl MEFs, adenoviral expression of Cre resulted in the depletion of PRKAR1A with a robust induction of both total and free PKA activity and has been attributed to the immortalization process.11 However, increased PKA activity is associated with induction of apoptosis in many cell types.8, 12, 13 In an effort to understand the regulation of apoptosis by increased PKA activity, we expressed Cre in prkar1afl/fl MEFs by a variety of different methods. We consistently found that there was a reduction in the PKA activity following Cre expression. Expression of Cre by cells infected with a retroviral expression construct resulted in the depletion of PRKAR1A protein and reduction of PKA activity, as observed by decreased phosphorylated CREB protein and the enzyme activity (Figures 1a and b). Similarly, expression of 4-hydroxytamoxifen (4-OHT)- regulated Cre from a lentiviral vector resulted in the depletion of PRKAR1A protein and a reduction in phospho-CREB (pCREB) levels (Figure 1c). To rule out the possibility that this was an artifact of the vector DNA backbone, we transduced prkar1afl/fl MEFs with a recombinant, cell permeable TAT-Cre protein (with a nuclear localization signal or HTNC).14 Addition of the recombinant protein into the culture resulted in the depletion of PRKAR1A protein with a concomitant reduction in the pCREB levels (Figure 1c, lane 7). Further, T cells purified from transgenic mice expressing Cre under the T-cell-specific promoter lck (lck-cre) showed a reduction of pCREB in both the thymic and in the splenic T-cell populations (Figure 1d); particularly in the splenic T cells where the reduction was much more significant.

Cre expression downregulates PKA activity. (a) Expression of Cre in MEFs by retroviral vector downregulates PKA activity, as indicated by reduced pCREB levels even under PRKAR1A deletion. (b) PKA activity assay in crude lysates using kemptide as the substrate. (c) Deletion of PRKAR1A either by 4-OHT-regulated, lentivirally-expressed cre (clone nos 1 and 2) or by cell permeable recombinant Cre in culture results in downregulation of pCREB levels. (d) Thymocytes and splenocytes from cre transgenic mice show reduced levels of pCREB. (e) Titratability of the Cre effect. MEFs (prkar1afl/fl) were infected with a lentivirus expressing 4-OHT-inducible Cre. Addition of 4-OHT resulted in the ablation of PRKAR1A protein in cells with concomitant reduction of PKA substrate protein in the cells (right lane). Recovery of the cells from 4-OHT results in a robust induction of PKA activity in the absence of Cre (in these cells, PRKAR1A is still ablated resulting in increased PKA activity). Error bars: ±S.E.M., n=3
We also wanted to test the reversibility of this effect. First, we treated prkar1afl/fl MEFs with 4-OHT (5 nM) to induce Cre expression.8 This resulted in the deletion of prkar1a gene and a dramatic reduction of phosphor-PKA substrate15 levels compared with untreated cells. We then released the cells, from 4-OHT treatment, resulting in a very significant induction of phosphorylated PKA substrates. These results are consistent with the notion that deletion of prkar1a leads to increased PKA activity in the absence of any Cre expression (Figure 1e). In addition to PKA activation, deletion of prkar1a also leads to increased phospho-ERK activation in the absence of PKA, indicating cross-talk between these signal transduction pathways.16
Cre expression induces PKI
To understand the mechanism behind PKA inhibition, we tested three possible scenarios. First: activation of a phosphatase by Cre protein. To address this, we measured the total phosphatase activity in cellular extracts of WT MEFs and MEFs expressing Cre protein using EnzCheck Phosphatase assay kit (Invitrogen, Carlsbad, CA, USA). This kit is capable of continuous measurement of phosphatase activity in a wide range of pH. However, we failed detect any difference in phosphatase activity between WT and Cre-expressing MEFs (Figure 2a). Second: Cre protein acting as a surrogate substrate for PKA. KinasePhos (http://KinasePhos.mbc.nctu.edu.tw) analysis of the Cre protein identified Ser108 as a potential PKA phosphorylation site. Indeed, in vitro kinase assay of bacterially expressed and purified Cre protein was readily phosphorylated by PKA and mutation of Ser108 to Ala abolished most of this phosphorylation (Figure 2b). However, expression of this mutant protein in MEFs still attenuated PKA activity, ruling out the possibility that Cre protein acts as a surrogate substrate for PKA (Figure 2c). Third: Cre protein expression leads to a transcriptional induction of pki genes. PKI is an endogenous thermo-stable peptide that modulates cAMP-dependent PKA function. It selectively inhibits the activity of the free catalytic subunit of cAMP-dependent PKA following cAMP-mediated dissociation of the regulatory and catalytic subunits of the enzyme, and thus regulates the basal level of cAMP/PKA activity17, 18 Quantitative PCR analysis of cDNA from wt and cre transgenic MEFs showed a significant increase in both pki-α and pki-γ transcripts in the cre transgenic MEFs (Figure 2d). Western blot analysis also revealed a significant induction of PKI-α protein in the cre transgenic MEFs, corroborating the finding that cre transgene expression leads to a transcriptional induction of pki genes (Figure 2e).

Possible mechanism of PKA inhibition in cre-expressing cells. (a) Phosphatase assay in cell lysate from MEFs. (b) Cre is a PKA substrate in in vitro kinase assay and the phosphorylation can be abolished by S180A mutation. The middle lane is the assay performed in presence of PKA-specific inhibitor (Upstate). The sequence shown below is the potential phosphorylation site. (c) Phosphorylation-deficient mutant of Cre still downregulates pCREB and pERK when expressed in MEFs. (d) Quantitative PCR analysis of PKIα and PKIγ in MEFs. (e) Western blot analysis of MEFs expressing Cre showing PKIα induction. Error bars: ±S.E.M., n=3
Global attenuation of signal transduction pathways in the cre transgenics
PKA is one of the main downstream effectors of increased cAMP levels in the cell (the other being exchange protein activated by cAMP or EPAC).19 The cAMP-dependent PKA pathway regulates a variety of cellular process including cellular proliferation, differentiation and apoptosis.20 Furthermore, the PKA pathway forms part of complex network of signal transduction pathways including MAPK and mTOR.21, 22 We wanted to test whether downregulation of PKA activity in the Cre transgenic cells had any effect on the level of phosphorylation of other cellular proteins. To this end, we conducted a total phosphoprotein analysis using a Kinexus antibody microarray (Kinexus Bioinformatics, Vancouver, BC, Canada). Comparison of phosphoprotein levels between WT and Cre-expressing MEFs revealed a substantial difference between these cells (Figure 3a). These results were further validated for a set of 13 proteins by western blot analysis (Figure 3b). In agreement with the western blot data, where Cre-expressing cells had reduced levels of ERK activation (Figure 1e), Kinexus analysis also showed a substantial reduction in the phosphorylation of both forms of ERK (pERK40/42).

Cre transgene alters global phosphorylation pattern in MEFs. (a) Phosphoprotein profiling by Kinex antibody microarray (806 pan-specific and phospho-specific antibodies) of WT and cre transgenic MEFs. Antibodies with very low signals (Z score less than −0.5) were removed from further consideration. (b) Antibodies whose signals differed substantially (where the difference in Z scores was greater than 0.5) were selected for secondary validation through immunoblotting
Transcriptional profiling of Cre transgenic MEFs
In addition to the direct transcriptional effect mediated through the transcription factor CREB, PKA can exert its effect by altering calcium mobilization in the cell.23 In eukaryotic cells, calcium ions are also involved in adaptive responses that involve global regulation of a wide variety of transcription factors.24 In this work, we limited our studies to qPCR analysis of nur77, areg, nr4a2, il-6, rgs2 and dusp1, which are known targets of PKA.25 As evident from Figure 4a, in Cre-expressing MEFs, all these transcripts (except for dusp1) were substantially downregulated. Of these genes, il-6 transcripts in particular were substantially downregulated. IL-6 is one of the preeminent cytokines involved in the inflammatory response. To validate this finding, we measured IL-6 secretion both in MEFs and in in vitro con A-induced T-cell blasts. Both in MEFs and in Con-A-activated T-cell blasts, Cre expression resulted in three- to fivefold reduction in secreted IL-6 (Figures 4b and c).

Cre expression results in downregulation of PKA transcriptional targets. (a) qPCR analysis of transcripts from wt and cre transgenic MEFs. (b) IL-6 measurement in the culture supernatant of MEFs. (c) IL-6 measurement in the culture supernatant of concanavalin A-activated splenic T cells. Error bars: ±S.E.M., n=3
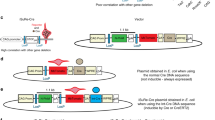
Although Cre recombinase binds to its cognate DNA sequence with very high affinity, it can also bind to non-target DNA with 20-fold lower affinity.26 Therefore, we were interested to know whether the nuclease activity associated with its promiscuous DNA binding could explain Cre’s pleotropic effects. To this end, we created an active site mutant of Cre Y325A.27 Retroviral infection of MEFs (c-mycfl/fl) with the wild-type cre resulted in site-specific recombination and the removal of the target allele (Figure 5a, lower panel) whereas the mutant Y324A failed to do so. However, the wild-type Cre recombinase expression with the nuclease activity resulted in the downregulation of pCREB whereas the mutant had no effect, suggesting that the pleotropic effect of Cre is associated with binding to its cognate DNA sequence (Figure 5a, lower panel).

PKA inhibitory activity of Cre is dependant on its nuclease activity. (a) MEFs (prkar1afl/fl) were infected with retroviruses expressing either the wild-type- or recombinase-deficient mutant cre (Y324A). Recombinase activity was measured by the deletion of the floxed allele by PCR using specific primers (bottom two panels) and pCREB level was measured by western blot analysis (top panels). (b) ROSA-Cre expression does not result in PKA downregulation. CMRC cells (c-mycfl/fl; rosa26cre) were treated with 1.0 μM 4-OHT, and cells were analyzed after 48 h and after another 48 h in the absence 4-OHT (recovery) for pCREB levels (upper panels) and for allele-specific deletion (bottom panels). Primer pair 24/25: wt allele (400 bp), floxed allele (500 bp); 26/27: c-myc-deleted allele or Δ/Δ allele (600 bp)
Next, we tested the new-generation Cre recombinase fused to a mutant estrogen hormone-binding domain (ERT).28 In this system, ERT-Cre expression is regulated by the ROSA26 (R26) locus and is therefore ubiquitous. ERT-Cre protein remains inactive in the cytoplasm until addition of the non-steroidal estrogen analog 4-OHT. To test whether the ERT-Cre cre could lessen its effect on PKA activity, we used CMRC MEFs (c-mycfl/fl; r26-cre) that were treated with 4-OHT. This led to the deletion of the c-myc allele without any reduction of pCREB levels in these MEFs (Figure 5b). These results demonstrate that although the nuclease activity is closely associated with its PKA inhibitory property, this problem could be mitigated by using the new-generation ERT-Cre, which is fused to the estrogen receptor domain, and appear to have no PKA inhibitory activity.
Discussion
The problems associated with cre transgene expression are well documented. These include proliferation defects, cytotoxicity and chromosomal abnormalities29 and epigenetic effects.30 The present study highlights yet another problem associated with the use of the cre transgene expression to achieve conditional deletion of desired genes, that is, attenuation of the PKA signaling pathway with downregulation of its transcriptional targets and the global changes in the phophoprotein levels in the cell. In the present study, we expressed Cre using a variety of systems including 4-OHT regulatable lentiviral expression system, retroviral expression system, tissue-specific transgene expression (lck-cre) and even bacterially expressed recombinant Cre (i.e., HTNC) with consistent results (except for LysM-Cre transgenics, data not shown). These findings obviate the possibility that this effect was mediated by a specific vector backbone or due to the random integration event. Further, we also showed that the effect on the PKA pathway could be due to the upregulation of the PKA inhibitor, PKI, although the molecular basis of this effect is yet to be elucidated. However, it is clear that this effect is dependent on its nuclease activity. This is consistent with the finding that cytotoxicity observed with cre expression is intrinsic to its nuclease activity.29 As the PKA pathway has an important role in the phosphorylation of a variety of substrates including transcription factors involved in cell activation and proliferation,31 it is tempting to speculate that Cre-mediated cytotoxicity may be a result of shutting down the PKA pathway.
Given the importance of the PKA signaling pathway in the regulation of transcription factors involved in cell cycle and proliferation, it is likely that its downregulation will have significant implications for cell survival and proliferation. The effect on IL-6 secretion is notable; IL-6 is an interleukin with both a pro-inflammatory and anti-inflammatory effects, and is involved in the acute phase of the innate immune response.32 Its anti-inflammatory effects are mediated (at least in part) by downregulating expression of TNF-α and MIP-2α.33 The deregulation of IL-6 impacts numerous disease states, including many types of cancer.34
Given the present observation described here, it is worth noting that PubMed has more than 6000 entries on cre transgenic mice dating back to the past 25 years. The generally accepted controls for cre transgenic mice are non-floxed or non-cre transgenic floxed mice, so the observed phenotype of a particular cre transgenic animal might easily be a combination of gene deletion and Cre-mediated effects on PKA signaling (such as dampening of IL-6 secretion). Therefore, it may be worthwhile revisiting some of these experiments (especially those involve cAMP pathway or cytokine signaling) using the new-generation cre transgenics such as the ‘self-excising cre’29 or using the ROSA-Cre system where CRE-ERT2 is integrated at the rosa26 locus, which eliminates random integration effects and the effect on the PKA pathway by regulated administration of 4-OHT.35 Even better alternative would be what Matthaei30 suggested, that is, LacO-LacIR-based repression of desired gene and regulating the gene activity by IPTG in the drinking water.
Materials and Methods
Cell culture and lentiviral infection
MEFs were cultured in DMEM supplemented with 10% fetal calf serum (Invitrogen) at 37 °C in a humidified 10% CO2 incubator. Immortalized MEFs were generated from E15 embryos in accordance with standard procedures and were infected with SV40 large T antigen expressing lentiviruses. To generate lentiviral particles, 293T cells were transfected with packaging constructs pCMV ðR8.2 and VSVg and the relevant lentiviral plasmid at a ratio of 1 : 0.4 : 0.6 using Fugene 6.0 transfection reagent (Roche Diagnostics, Castle Hill, NSW, Australia) following the manufacturer’s instructions. The virus containing supernatants were harvested, filtered (0.8 μM) and supplemented with polybrene (4 μg/ml). Target cells were infected with virus supernatant as described.8
The generation of prkar1afl/fl mouse is already described 14 and was a kind gift from Dr. Hua Gu (Columbia University, NY, USA). c-mycfl/fl MEFs were a kind gift from Dr. Ignacio de Alboran (Madrid).36
Antibodies
Phospho-(Ser/Thr) PKA substrate (#9621), anti-ERK (#4695) and phospho-ERK (#9102) antibodies were purchased from Cell Signaling (Boston, MA, USA). pCREB antibodies were from Abcam (ab5803, Cambridge, MA, USA), Prkar1a antibodies were from BD Biosciences (610609, North Ryde, NSW, Australia) and PKIα antibodies were from Santa Cruz Biotechnology (sc-50349, Santa Cruz, CA, USA). Anti mouse, anti rabbit and anti rat HRP-conjugated secondary antibodies were purchased from GE Healthcare (Rydalmere, NSW, Australia).
Kinase assay
In vitro PKA assays were carried out using the PKA assay kit (Upstate, Lake Placid, NY, USA). Each assay contained 5–10 ng of the recombinant hexa-his-tagged cre protein in a total volume of 60 μl reaction buffer that contained the following: 10 μl of 1 × assay dilution buffer, 5 μl of 20 μM cAMP, 10 μl of nonspecific kinase inhibitor cocktail, 1 μl of PKA catalytic subunit (2.5–10 U) and 10 μl of Mg-ATP cocktail. Ten microliter of PKA inhibitor was added to the control reactions. The reactions were incubated at 30 °C for 30 min before adding 2 × Laemmli loading buffer and separating on SDS-polyacrylamide gel electrophoresis gel (Novex, Invitrogen, Carlsbad, CA, USA), followed by transfer onto PVDF membrane. The radioactive proteins were detected in a Typhoon 9410 phosphorimager (GE Healthcare). For measuring PKA activity in crude lysates, 5 μl of 1 mM Kemptide was used as the substrate in a reaction similar to the one used for the recombinant Cre protein. At the end of the reaction, 25 μl of the reaction was spotted on P81 paper squares, air dried and washed three times in 0.75% phosphoric acid and once in acetone before reading in a scintillation counter.
Phosphatase assay
Total phosphatase in the cell lysate was measured using EnzCheck Phosphatase Assay Kit (E12020, Molecular Probes, Eugene, OR, USA) with 10 μg of cell lysate following the manufacturer’s instructions.
Cell permeable cre purification
HTNC (His-tagged, TAT-fusion Cre with nuclear localization signal) was originally gifted by K. Rajewsky obtained from Addgene (Cambridge, MA, USA). Recombinant protein from E. coli was purified by nickel affinity chromatography. Endotoxin was removed using Endotoxin removal beads (Miltenyl Biotech, North Ryde, NSW, Australia) and filtered through 0.8-μ filters before using in cell culture at 100 μg/ml.
qPCR analysis
Total RNA was extracted using Trizol reagent (Invitrogen). cDNA was synthesized using Superscript III RT system (Invitrogen). Quantitative PCR was carried out using SYBRGreen reagent (Invitrogen) following the manufacturer’s instructions. The gene-specific primers were as follows: areg (s): 5′-AGAACCTGGAGGTGGTGACATG-3′, areg (as) 5′-TCGTTTCCAAAGGTGCACTGTG-3′; dusp1 (s): 5′-GTACCCCTCTCTACGATCAGGGG-3′, dusp1 (as): 5′-CCAGGAGCTGATGTCTGCCTT-3′; il6 (s): 5′-ACGGCCTTCCCTACTTCACA-3′, il6 (as): 5′-ATGGTACTCCAGAAGACCAGAGGA-3′; nr4a2 (s): 5′-GATCCGGGCTCCCTTCACAA-3′, nr4a2 (as): 5′-CGAAGGTCTGCCCATCCACT-3′; nur77 (s): 5′-CCTGTTGCTAGAGTCTGCCTTC-3′, nur77 (as): 5′-CCTGTTGCTAGAGTCTGCCTTC-3′; rgs2 (s): 5′-GGCCAGTAAATATGGGCTGG-3′; rgs2 (as): 5′-GTGGTGAAGCAGCCACTTGTA-3′; cre (s): 5′-GGGATTGCTTATAACACCCTGTTACG-3′, cre (as): 5′-TATTCGGATCATCAGCTACACCAGAG-3′; pki-α (s): 5′-TGCAGATTTCATTGCTTCAGGA-3′, pki-α (as): 5′-CAGACTTGGCTGCTTCTCCC-3′; pki-γ (s): 5′-GACTTCATCTCCTGCGACCG-3′, pki-γ (as): 5′-CAGGATGAGGTGTTCGCATCA-3′.
Abbreviations
- cAMP:
-
cyclic adenosine 3′,5′-monophosphate
- PKA:
-
protein kinase A
- MEF:
-
mouse embryonic fibroblasts
- 4-OHT:
-
4-hydroxytamoxifen
- IL-6:
-
interleukin-6
References
Schwenk F, Baron U, Rajewsky K . A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucl Acids Res 1995; 23: 5080–5081.
Fiering S, Epner E, Robinson K, Zhuang Y, Telling A, Hu M et al. Targeted deletion of 5'HS2 of the murine beta-globin LCR reveals that it is not essential for proper regulation of the beta-globin locus. Genes Dev 1995; 9: 2203–2213.
Kuhn R, Schwenk F, Aguet M, Rajewsky K . Inducible gene targeting in mice. Science 1995; 269: 1427–1429.
Schmidt EE, Taylor DS, Prigge JR, Barnett S, Capecchi MR . Illegitimate Cre-dependent chromosome rearrangements in transgenic mouse spermatids. Proc Natl Acad Sci USA 2000; 97: 13702–13707.
Adams DJ, van der Weyden L . Are we creating problems? Negative effects of Cre recombinase. Genesis 2001; 29: 115.
Loonstra A, Vooijs M, Beverloo HB, Al Allak B, van Drunen E, Kanaar R et al. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci USA 2001; 98: 9209–9214.
Molyneux SD, Di Grappa MA, Beristain AG, McKee TD, Wai DH, Paderova J et al. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J Clin Invest 2010; 120: 3310–3325.
Moujalled D, Weston R, Anderton H, Ninnis R, Goel P, Coley A et al. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep 2011; 12: 77–83.
Sandrini FKL, Bei T, Farmakidis C, Yasufuku-Takano J, Takano K, Prezant TR et al. PRKAR1A, one of the Carney complex genes, and its locus (17q22-24) are rarely altered in pituitary tumours outside the Carney complex. J Med Genet 2002; 39: e78.
Bossis I, Stratakis CA . PRKAR1A: normal and abnormal functions. Endocrinology 2004; 145: 5452–5458.
Nadella KS, Kirschner LS . Disruption of protein kinase a regulation causes immortalization and dysregulation of D-type cyclins. Cancer Res 2005; 65: 10307–10315.
Myklebust JHJD, Blomhoff HK, Levy FO, Naderi S, Reed JC, Smeland EB . Activation of the cAMP signaling pathway increases apoptosis in human B-precursor cells and is associated with downregulation of Mcl-1 expression. J Cell Physiol 1999; 180: 71–80.
Zhang L, Insel PA . The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem 2004; 279: 20858–20865.
Pasqualucci LKY, Gu H, Dalla-Favera R . PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci USA 2006; 103: 395–400.
Moir RD, Lee J, Haeusler RA, Desai N, Engelke DR, Willis IM . Protein kinase A regulates RNA polymerase III transcription through the nuclear localization of Maf1. Proc Natl Acad Sci USA 2006; 103: 15044–15049.
Smith FD, Langeberg LK, Scott JD . Plugging PKA into ERK scaffolds. Cell Cycle 2011; 10: 731–732.
Ashby CD, Walsh DA . Characterization of the interaction of a protein inhibitor with adenosine 3',5'-monophosphate-dependent protein kinases. I. Interaction with the catalytic subunit of the protein kinase. J Biol Chem 1972; 247: 6637–6642.
Dalton GD, Dewey WL . Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides 2006; 40: 23–34.
Ster J, De Bock F, Guerineau NC, Janossy A, Barrere-Lemaire S, Bos JL et al. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc Natl Acad Sci USA 2007; 104: 2519–2524.
Liu J, Li XD, Ora A, Heikkila P, Vaheri A, Voutilainen R . cAMP-dependent protein kinase activation inhibits proliferation and enhances apoptotic effect of tumor necrosis factor-alpha in NCI-H295R adrenocortical cells. J Mol Endocrinol 2004; 33: 511–522.
Waltereit R, Weller M . Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol 2003; 27: 99–106.
Miller AL, Garza AS, Johnson BH, Thompson EB . Pathway interactions between MAPKs, mTOR, PKA, and the glucocorticoid receptor in lymphoid cells. Cancer Cell Int 2007; 7: 3.
Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Biol Chem 2003; 160: 919–928.
Cruzalegui FH, Bading H . Calcium-regulated protein kinase cascades and their transcription factor targets. Cell Mol Life Sci 2000; 57: 402–410.
Li X, Liu H, Qin L, Tamasi J, Bergenstock M, Shapses S et al. Determination of dual effects of parathyroid hormone on skeletal gene expression in vivo by microarray and network analysis. J Biol Chem 2007; 282: 33086–33097.
Abremski K, Hoess R . Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. J Biol Chem 1984; 259: 1509–1514.
Lee L, Sadowski PD . Strand selection by the tyrosine recombinases. Prog Nucleic Acid Res Mol Biol 2005; 80: 1–42.
Vooijs M, Jonkers J, Berns A . A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep 2001; 2: 292–297.
Silver DP, Livingston DM . Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol Cell 2001; 8: 233–243.
Matthaei KI . Genetically manipulated mice: a powerful tool with unsuspected caveats. J Physiol 2007; 582: 481–488.
Montminy M . Transcriptional regulation by cyclic AMP. Ann Rev Biochem 1997; 66: 807–822.
van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF . Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis 1997; 176: 439–444.
Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest 1998; 101: 311–320.
Hong DS, Angelo LS, Kurzrock R . Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer 2007; 110: 1911–1928.
Hameyer D, Loonstra A, Eshkind L, Schmitt S, Antunes C, Groen A et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol Genomics 2007; 31: 32–41.
de Alboran IM, O'Hagan RC, Gärtner F, Malynn B, Davidson L, Rickert R et al. Analysis of C-MYC function in normal cells via conditional gene targeted mutation. Immunity 2001; 14: 45–55.
Acknowledgements
We thank Dr. Andrew Coley for reviewing the manuscript, and Dr. Ignacio de Alboran and Dr. Hua Gu for reagents. We also thank the staff at the central animal house, La Trobe University. LG and YYL are supported by Co-operative Research Centre for Biomarker Translation and HP is supported by Australian Research Council Future Fellowship (FT0990683) and by Australian Research Council project grant (DP110100417).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Ciliberto
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Gangoda, L., Doerflinger, M., Lee, Y. et al. Cre transgene results in global attenuation of the cAMP/PKA pathway. Cell Death Dis 3, e365 (2012). https://doi.org/10.1038/cddis.2012.110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.110
Keywords
This article is cited by
-
Cre toxicity in mouse models of cardiovascular physiology and disease
Nature Cardiovascular Research (2022)
-
Elevated intracellular cAMP exacerbates vulnerability to oxidative stress in optic nerve head astrocytes
Cell Death & Disease (2018)
-
Transgenic Mouse Technology in Skin Biology: Generation of Complete or Tissue-Specific Knockout Mice
Journal of Investigative Dermatology (2014)
-
Loss of Prkar1a leads to Bcl-2 family protein induction and cachexia in mice
Cell Death & Differentiation (2014)
-
CREB-binding protein (CBP) regulates β-adrenoceptor (β-AR)−mediated apoptosis
Cell Death & Differentiation (2013)
