
Abstract
The strong interest in cell death, and the shift in emphasis from basic mechanisms to translational aspects fostered the launch last year of the new sister journal of Cell Death and Differentiation, named Cell Death and Disease, to reflect its stronger focus towards clinical applications. Here, we review that first year of activity, which reflects an enthusiastic response by the scientific community. On the basis of this, we now launch two novel initiatives, the start of a new section dedicated to cancer metabolism and the opening of a new editorial office in Shanghai.
Similar content being viewed by others

Main
It is impossible to overstate the relevance of cell death research to human pathology. Compromised cell death pathways clearly contribute to proliferative diseases, and overactive death mechanisms are implicated in degenerative disease. In addition, cell death results from ischaemic episodes, and reducing ischaemic death correspondingly reduces the structural and functional deficits that follow, for example, myocardial and cerebral infarction. It is also likely that manipulation of the form of cell death that follows injuries such as ischaemia may also influence disease outcome. Thus, converting cell loss from a necrotic to an apoptotic phenotype will reduce the associated inflammation, resulting in a smaller final lesion and fewer clinical consequences.
In 1991, a mouse strain was serendipitously discovered for its resemblance to patients with the disease Systemic Lupus Erythematosus, that is, the mice showed a tendency to develop enlarged lymph nodes and spleens along with autoimmune diseases such as immune complex glomerulonephritis.1 The mice, named lpr and gld, had mutations in CD95 (Fas, Apo-1) and CD95 L (FasL, Apo-1L), respectively. The lymphocytes from these mice proliferated normally in response to antigenic challenges, but they failed to be eliminated at the end of the immune response because of the mutation in the death receptor and its ligand (CD95 and CD95L) that impaired the ability to undergo apoptosis, resulting in an abnormal T-cell accumulation in the lymphoid tissues. Some of the lymphocytic clones triggered autoimmune diseases. Furthermore, the normal development of T cells was defective in these mice, with drastically reduced double positive mature T cells, which should normally express either CD4 or CD8 markers along with CD3 receptors for antigen. This was the first clear evidence of defects in the cell death machinery leading to immunological disease, both in mice and human. The molecular mechanisms regulating the activation of death receptors have now been analysed in detail, with their ubiquitylation2, 3, 4 regulating cell fate.5 In addition to elucidating the role of death receptors in inflammation and immunity, the improved understanding of these pathways opens novel therapeutic perspectives, which are only now beginning to be exploited in the clinic.
Cell death is crucial also for the development of malignancies. Here, the list of defective genes linked to the cell death machinery is long and well established, see, for example, the role of p53. This transcription factor regulates cell cycle as well as cell death.6 Humans with defects in p53 (Li–Fraumeni syndrome) are cancer-prone. Loss of p53 function by gene mutation or other mechanisms of inactivation has been shown to be a characteristic of a large majority of human tumours, and many tumours also have lost genes whose products stabilize p53, such as ataxia telangiectasia mutated.7, 8, 9
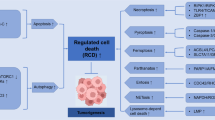
DNA damage is normally detected in the nucleus by specific protein complexes, which then can induce cell cycle arrest and/or classic apoptosis via p53, or via its family members p73 or p63,10, 11 (Figure 1) and deregulation of these pathways leads to cancer progression. The stability of proteins of the p53 family is regulated by specific ubiquitin E3 ligases12 and, correspondingly, inhibition of these E3 enzymes would stabilize the protein with enhancement of its activity. Thus, pharmacological methods for stabilizing the p53 family of transcription factors would seem to offer a potential therapeutic window (Figures 1b and c). Interestingly, in addition to classic apoptosis, the p53 family members are able to interact with components of the kinetochore complex such as BubR1,13, 14 affecting mitosis and leading to mitotic catastrophe. Of course, the p53 family, like other transcription factors, does not act in isolation at enhancer elements, but as a complex with other factors that may increase or reduce their transcriptional activity on specific targets. Unlike the vast literature devoted to p53 and its siblings, however, comparatively little attention has been focused on these cofactors, and a better understanding of how specific cofactors may act to enhance the apoptotic activity of p73 in cancers where p53 is inactive may provide other therapeutic opportunities.

The p53 family in the DNA damage response and cell death regulation. (a) DNA damage and replicative stress can trigger a DNA damage response that involves the function of p53 to regulate cell cycle arrest or apoptosis. During the absence or functional inactivation of p53, the two p53 family members p63 and p73 can at least partially substitute this role through distinct upstream regulators, but with the same biological effect. (b) The protein stability of p53 is mainly regulated by mdm2, whose interaction with p53 can be inhibited by specific low molecular compounds, currently in clinical trials, leading to restoration of p53 steady state protein levels and function. (c) Similarly, the degradation of p63 and p73 is mainly regulated by the ubiquitin E3 ligase ITCH, which offers also a novel therapeutic target. ITCH, in mediating its activity, undergoes a conformational modification in the C-terminal portion (WWP1/AIP5 versus E6AP) in a specific hinge region
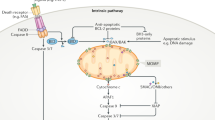
Intrinsic cell death pathways exist in a state of dynamic equilibrium between pro- and anti-apoptotic elements. Death receptor signalling, for example, is inhibited by cFLIP, and activation of procaspase-9 within the apoptosome is restrained by X-IAP until Smac/DIABLO is released from damaged mitochondria. This inherent balance provides an opportunity for influencing death decisions both to enhance and to reduce cell death. In addition to these intrinsic factors, other extrinsic intracellular factors, such as the urocortins, can also powerfully influence the outcome of cell death stimuli.
So far, we have concentrated on the manipulation of apoptosis as a means of influencing pathology – what of necrotic death? It remains unclear how much of the necrosis that occurs, for example, in myocardial infarction, is a catastrophic and irreversible event, and how much occurs as programmed necrosis. The former would probably not be possible to target therapeutically, but the latter mechanism, once its molecular details are better understood, may provide potential therapeutic windows. Finally, in the panoply of cell death modalities, what is the contribution of autophagy to death pathologies – is it protective, and should we therefore promote it, or does it contribute to cell loss? Some evidence suggests that it is the severity and duration of the stimulus that determines this, and short stimuli, such as brief ischaemia, result in an autophagic process, which is protective by providing an intracellular source of nutrients. This, if not resulting in cell salvage, may allow less harmful apoptotic death to proceed instead of necrosis. Since we have hung up our Gilsons much more recently than when we began to help patients, we tend to compartmentalize cell death modes into neat academic entities, but in the real world of patients with cell death pathologies, it is likely these pathways overlap and interact. As such, effective therapies will need to address all cell death subroutines.
Clearly, the study of cell death is pivotal to understand and manipulate disease processes,15, 16 and this was the scientific basis for launching last year a translational sister journal of Cell Death and Differentiation (CDD).
Cell Death and Disease, CDDIS, was launched in January 2010 as the first peer-reviewed open access online journal by Nature Publishing Group that publishes full-length papers, reviews and commentaries describing original research in the field of translational cell death. It seeks to promote diverse and integrated areas of Experimental and Internal Medicine in specialties such as cancer, immunity and neuroscience.
CDD is now in its eighteenth volume and, given the conventional 20-year time lag for the journey from bench to bedside, 2010 seemed an appropriate time to launch CDDIS. This reflects the fact that the academic Cell Death field has now become mature enough to contribute to the physiology and pathology field, a fact reflected in the growing interest in cell death by pharmacists and clinicians. Thus, a significant proportion of papers are focused on translational and therapeutic aspects, and therefore CDDIS seeks to encompass the breadth of translational implications of cell death, and topics of particular concentration will include, but are not limited to, the subspecialities highlighted above.
The original plan was to publish 30 papers in the first year. Instead the response by the scientific community was so positive that CDDIS has published over 100 papers in its first year of life. More important, this success has encouraged NPG to launch more new open access journals, and so now we are witnessing the birth of a number of scientific journals, all based on this novel experimental business model.
Data from Medline show that in 2010, a total of 23 611 papers have been published (compared with 5458 in 1995 or 2219 in 1990) in the area of cell death. By inputting the keywords ‘cell death’ and ‘medicine’, the search yields 5990 (25.4%) papers, while ‘cell death’ with ‘cancer’ yields 9306 (39.4%) papers, with ‘neuron’ 2313 (9.8%) papers, with ‘heart’ 1238 (5.2%) papers and with ‘immunity’ 1091 (4.6%) papers. See Table 1 for more details. In order to compare with a more basic interest, the total number of papers on ‘cell death+caspases’ reduced from 14.9% (2741 out of 18 517) to 12.0% (2838 out of 23 611), in 2005 and 2010, respectively. These numbers indicate both that the number of translational papers and those of a more general interest in cell death remains extremely high, and suggests that we are witnessing a shift in interest from basic molecular mechanisms to the manipulation of cell death in pathology.
Table 2 shows that CDDIS has followed this translational trend, publishing a significant number of papers equally distributed in the different sections.
How can we make CDDIS evolve in the right direction? Here, we propose two novel initiatives that we hope will be positive for CDDIS.
During the same time frame used above, 1995 versus 2010, matching ‘cell death’ with ‘metabolism’ shows a rise from 3012 to 17 284 (73.5%) papers. See also Table 1. More generally, the number of scientific papers on ‘cancer metabolism’ has increased from 16 924 in 1995 to the respectable number of 44 769 in 2010. For this reason, CDDIS has decided to immediately launch a new section of CDDIS dedicated to Cancer Metabolism. Christina Munoz-Pinedo will help us to achieve this aim.
An interesting new trend in recent years is the rise of high quality science in China and, indeed, several leading scientists have moved back from the United States and Europe to mainland China encouraged by the huge financial investment that has been put in place in China. Accordingly, in May of this year CDDIS organized a scientific meeting in Shanghai in the presence of several members of the Editorial board, including Andreas Strasser, Tak W Mak, Martin Bushell, Xin Lu, Doug R Green, Yufang Shi and both of us. To reflect the increase in high quality scientific output from China, and the global influence this will have in the future, we are opening a new Editorial Office in Shanghai, headed by Yufang Shi.
We hope that these novel initiatives will help us to build on a successful first 18 months and to reach a wider international audience. Our goal, as always, is to provide a satisfying and stimulating forum of knowledge exchange, particularly for those seeking to exploit basic cell death research in the clinic as well as for those more interested in how cell death modalities relate to disease rather than in the molecular complexities of the pathways involved.
Abbreviations
- SLE:
-
disease Systemic Lupus Erythematosus
- ATM:
-
ataxia telangiectasia mutated
References
Cohen PL, Eisenberg RA . Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Ann Rev Immunol 1991; 9: 243–269.
Wertz IE, Dixit VM . Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ 2010; 17: 14–24.
Silke J, Brink R . Regulation of TNFRSF and innate immune signalling complexes by TRAFs and cIAPs. Cell Death Differ 2010; 17: 35–45.
Wicovsky A, Salzmann S, Roos C, Ehrenschwender M, Rosenthal T, Siegmund D et al. TNF-like weak inducer of apoptosis inhibits proinflammatory TNF receptor-1 signaling. Cell Death Differ 2009; 16: 1445–1459.
Berghe TV, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 2010; 17: 922–930.
Fan Y, Lee TV, Xu D, Chen Z, Lamblin AF, Steller H et al. Dual roles of Drosophila p53 in cell death and cell differentiation. Cell Death Differ 2010; 17: 912–921.
Marine JC . p53 stabilization: the importance of nuclear import. Cell Death Differ 2010; 17: 191–192.
Marchenko ND, Hanel W, Li D, Becker K, Reich N, Moll UM . Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ 2010; 17: 255–267.
Bernassola F, Ciechanover A, Melino G . The ubiquitin proteasome system and its involvement in cell death pathways. Cell Death Differ 2010; 17: 1–3.
Tomasini R, Mak TW, Melino G . The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol 2008; 18: 244–252.
Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G . The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 2011; 12: 259–265.
Bernassola F, Karin M, Ciechanover A, Melino G . The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 2008; 14: 10–21.
Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Develpment 2008; 22: 2677–2691.
Tomasini R, Tsuchihara K, Tsuda C, Lau SK, Wilhelm M, Rufini A et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc Natl Acad Sci USA 2009; 106: 797–802.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. Classification of cell death: recommendations of the Nomenclature Committee on cell death 2009. Cell Death Differ 2009; 16: 3–11.
Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16: 1093–1107.
Alajez NM, Shi W, Hui AB, Bruce J, Lenarduzzi M, Ito E et al. Enhancer of Zeste homolog 2 (EZH2) is overexpressed in recurrent nasopharyngeal carcinoma and is regulated by miR-26a, miR-101, and miR-98. Cell Death Dis 2010; 1: e85.
Tenedini E, Roncaglia E, Ferrari F, Orlandi C, Bianchi E, Bicciato S et al. Integrated analysis of microRNA and mRNA expression profiles in physiological myelopoiesis: role of hsa-mir-299-5p in CD34+ progenitor cells commitment. Cell Death Dis 2010; 1: e28.
Yelamanchili SV, Chaudhuri AD, Chen LN, Xiong H, Fox HS . MicroRNA-21 dysregulates the expression of MEF2C in neurons in monkey and human SIV/HIV neurological disease. Cell Death Dis 2010; 1: e77.
Bhatnagar N, Li X, Padi SK, Zhang Q, Tang MS, Guo B . Downregulation of miR-205 and miR-31 confers resistance to chemotherapy-induced apoptosis in prostate cancer cells. Cell Death Dis 2010; 1: e105.
Lian J, Tian H, Liu L, Zhang XS, Li WQ, Deng YM et al. Downregulation of microRNA-383 is associated with male infertility and promotes testicular embryonal carcinoma cell proliferation by targeting IRF1. Cell Death Dis 2010; 1: e94.
Cummins NW, Badley AD . Mechanisms of HIV-associated lymphocyte apoptosis. Cell Death Dis 2010; 1: e99.
Wu PC, Wang Q, Dong ZM, Chu E, Roberson RS, Ivanova IC et al. Expression of coxsackie and adenovirus receptor distinguishes transitional cancer states in therapy-induced cellular senescence. Cell Death Dis 2010; 1: e70.
El-Fadili AK, Zangger H, Desponds C, Gonzalez IJ, Zalila H, Schaff C et al. Cathepsin B-like and cell death in the unicellular human pathogen Leishmania. Cell Death Dis 2010; 1: e71.
Ch’ng JH, Kotturi SR, Chong AG, Lear MJ, Tan KS . A programmed cell death pathway in the malaria parasite plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis 2010; 1: e26.
Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP . Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis 2010; 1: e65.
Calandrella N, De Seta C, Scarsella G, Risuleo G . Carnitine reduces the lipoperoxidative damage of the membrane and apoptosis after induction of cell stress in experimental glaucoma. Cell Death Dis 2010; 1: e62.
Pasupuleti N, Matsuyama S, Voss O, Doseff AI, Song K, Danielpour D et al. The anti-apoptotic function of human αA-crystallin is directly related to its chaperone activity. Cell Death Dis 2010; 1: e31.
Allen-Petersen BL, Miller MR, Neville MC, Anderson SM, Nakayama KI, Reyland ME . Loss of protein kinase C delta alters mammary gland development and apoptosis. Cell Death Dis 2010; 1: e17.
Díaz-Manera J, Touvier T, Dellavalle A, Tonlorenzi R, Tedesco FS, Messina G et al. Partial dysferlin reconstitution by adult murine mesoangioblasts is sufficient for full functional recovery in a murine model of dysferlinopathy. Cell Death Dis 2010; 1: e61.
Karaoz E, Genç ZS, Demircan PÇ, Aksoy A, Duruksu G . Protection of rat pancreatic islet function and viability by coculture with rat bone marrow-derived mesenchymal stem cells. Cell Death Dis 2010; 1: e36.
Francis KR, Wei L . Human embryonic stem cell neural differentiation and enhanced cell survival promoted by hypoxic preconditioning. Cell Death Dis 2010; 1: e22.
Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis 2010; 1: e87.
Flourakis M, Lehen’kyi V, Beck B, Raphaël M, Vandenberghe M, Abeele FV . Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis 2010; 1: e75.
Chan KS, Wong CH, Huang YF, Li HY . Survivin withdrawal by nuclear export failure as a physiological switch to commit cells to apoptosis. Cell Death Dis 2010; 1: e57.
Li Q, Li H, Roughton K, Wang X, Kroemer G, Blomgren K et al. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis 2010; 1: e56.
Rello-Varona S, Kepp O, Vitale I, Michaud M, Senovilla L, Jemaà M et al. An automated fluorescence videomicroscopy assay for the detection of mitotic catastrophe. Cell Death Dis 2010; 1: e25.
McKeller MR, Herrera-Rodriguez S, Ma W, Ortiz-Quintero B, Rangel R, Candé C et al. Vital function of PRELI and essential requirement of its LEA motif. Cell Death Dis 2010; 1: e21.
Deng L, Feng J, Broaddus RR . The novel estrogen-induced gene EIG121 regulates autophagy and promotes cell survival under stress. Cell Death Dis 2010; 1: e32.
Silver N, Proctor GB, Arno M, Carpenter GH . Activation of mTOR coincides with autophagy during ligation-induced atrophy in the rat submandibular gland. Cell Death Dis 2010; 1: e14.
McCoy F, Hurwitz J, McTavish N, Paul I, Barnes C, O’Hagan B et al. Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis 2010; 1: e108.
Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I et al. Caspase-mediated cleavage of beclin-1 inactivates beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 2010; 1: e18.
Engel T, Tanaka K, Jimenez-Mateos EM, Caballero-Caballero A, Prehn JH, Henshall DC . Loss of p53 results in protracted electrographic seizures and development of an aggravated epileptic phenotype following status epilepticus. Cell Death Dis 2010; 1: e79.
Meley D, Spiller DG, White MR, McDowell H, Pizer B, Sée V . p53-mediated delayed NF-κB activity enhances etoposide-induced cell death in medulloblastoma. Cell Death Dis 2010; 1: e41.
Barton CE, Johnson KN, Mays DM, Boehnke K, Shyr Y, Boukamp P et al. Novel p63 target genes involved in paracrine signaling and keratinocyte differentiation. Cell Death Dis 2010; 1: e74.
Mitchell G, Fillinger J, Sittadjody S, Avila J, Burd R, Limesand K . IGF1 activates cell cycle arrest following irradiation by reducing binding of ΔNp63 to the p21 promoter. Cell Death Dis 2010; 1: e50.
Gonzalez-Cano L, Herreros-Villanueva M, Fernandez-Alonso R, Ayuso-Sacido A, Meyer G, Garcia-Verdugo JM et al. p73 deficiency results in impaired self renewal and premature neuronal differentiation of mouse neural progenitors independently of p53. Cell Death Dis 2010; 1: e109.
Straub WE, Weber TA, Schäfer B, Candi E, Durst F, Ou HD et al. The C-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis 2010; 1: e5.
Yuan M, Luong P, Hudson C, Gudmundsdottir K, Basu S . c-Abl phosphorylation of ΔNp63α is critical for cell viability. Cell Death Dis 2010; 1: e16.
Fricker M, O’Prey J, Tolkovsky AM, Ryan KM . Phosphorylation of Puma modulates its apoptotic function by regulating protein stability. Cell Death Dis 2010; 1: e59.
Tischner D, Woess C, Ottina E, Villunger A . Bcl-2-regulated cell death signalling in the prevention of autoimmunity. Cell Death Dis 2010; 1: e48.
Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M . A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis 2010; 1: e40.
Esposti MD . Bcl-2 antagonists and cancer: from the clinic, back to the bench. Cell Death Dis 2010; 1: e37.
Jiang CC, Lai F, Tay KH, Croft A, Rizos H, Becker TM et al. Apoptosis of human melanoma cells induced by inhibition of B-RAFV600E involves preferential splicing of bimS. Cell Death Dis 2010; 1: e69.
Rigaud O, Fortunel NO, Vaigot P, Cadio E, Martin MT, Lundh O et al. Exploring ultrashort high-energy electron-induced damage in human carcinoma cells. Cell Death Dis 2010; 1: e73.
Janson V, Johansson A, Grankvist K . Resistance to caspase-8 and -9 fragments in a malignant pleural mesothelioma cell line with acquired cisplatin-resistance. Cell Death Dis 2010; 1: e78.
Heidari N, Hicks MA, Harada H . GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis 2010; 1: e76.
Gonzalez-Mejia ME, Voss OH, Murnan EJ, Doseff AI . Apigenin-induced apoptosis of leukemia cells is mediated by a bimodal and differentially regulated residue-specific phosphorylation of heat-shock protein-27. Cell Death Dis 2010; 1: e64.
Wemeau M, Kepp O, Tesnière A, Panaretakis T, Flament C, De Botton S et al. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis 2010; 1: e104.
Sears D, Luong P, Yuan M, Nteliopoulos G, Man YK, Melo JV et al. Functional phosphoproteomic analysis reveals cold-shock domain protein A to be a Bcr-Abl effector-regulating proliferation and transformation in chronic myeloid leukemia. Cell Death Dis 2010; 1: e93.
Bennett HL, Fleming JT, O’Prey J, Ryan KM, Leung HY . Androgens modulate autophagy and cell death via regulation of the endoplasmic reticulum chaperone glucose-regulated protein 78/BiP in prostate cancer cells. Cell Death Dis 1: e72.
Tomiyama A, Tachibana K, Suzuki K, Seino S, Sunayama J, Matsuda KI et al. MEK-ERK-dependent multiple caspase activation by mitochondrial proapoptotic Bcl-2 family proteins is essential for heavy ion irradiation-induced glioma cell death. Cell Death Dis 2010; 1: e60.
Hervouet E, Vallette FM, Cartron PF . Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death Dis 2010; 1: e8.
Romano S, D’Angelillo A, Staibano S, Ilardi G, Romano MF . FK506-binding protein 51 is a possible novel tumoral marker. Cell Death Dis 2010; 1: e55.
Karasawa T, Wang Q, David LL, Steyger PS . CLIMP-63 is a gentamicin-binding protein that is involved in drug-induced cytotoxicity. Cell Death Dis 2010; 1: e102.
Wang Y, Nangia-Makker P, Balan V, Hogan V, Raz A . Calpain activation through galectin-3 inhibition sensitizes prostate cancer cells to cisplatin treatment. Cell Death Dis 2010; 1: e101.
Lacombe S, Sabatier L, Wien F, Gauduel YA . Spatio-temporal radiation biology: new insights and biomedical perspectives. Cell Death Dis 2010; 1: e4.
Ruela-de-Sousa RR, Fuhler GM, Blom N, Ferreira CV, Aoyama H, Peppelenbosch MP . Cytotoxicity of apigenin on leukemia cell lines: implications for prevention and therapy. Cell Death Dis 2010; 1: e19.
Sayeed A, Meng Z, Luciani G, Chen LC, Bennington JL, Dairkee SH . Negative regulation of UCP2 by TGFβ signaling characterizes low and intermediate-grade primary breast cancer. Cell Death Dis 2010; 1: e53.
Meltser V, Ben-Yehoyada M, Reuven N, Shaul Y . c-Abl downregulates the slow phase of double-strand break repair. Cell Death Dis 2010; 1: e20.
Lei WW, Zhang KH, Pan XC, Wang DM, Hu Y, Yang YN et al. Histone deacetylase 1 and 2 differentially regulate apoptosis by opposing effects on extracellular signal-regulated kinase 1/2. Cell Death Dis 2010; 1: e44.
Guardiola-Serrano F, Rossin A, Cahuzac N, Lückerath K, Melzer I, Mailfert S et al. Palmitoylation of human FasL modulates its cell death-inducing function. Cell Death Dis 2010; 1: e88.
Schneider-Jakob S, Corazza N, Badmann A, Sidler D, Stuber-Roos R, Keogh A et al. Synergistic induction of cell death in liver tumor cells by TRAIL and chemotherapeutic drugs via the BH3-only proteins Bim and Bid. Cell Death Dis 2010; 1: e86.
Reis CR, van der Sloot AM, Natoni A, Szegezdi E, Setroikromo R, Meijer M et al. Rapid and efficient cancer cell killing mediated by high-affinity death receptor homotrimerizing TRAIL variants. Cell Death Dis 2010; 1: e83.
Cheng JP, Betin VM, Weir H, Shelmani GM, Moss DK, Lane JD . Caspase cleavage of the Golgi stacking factor GRASP65 is required for Fas/CD95-mediated apoptosis. Cell Death Dis 2010; 1: e82.
Schneider B, Münkel S, Krippner-Heidenreich A, Grunwald I, Wels WS, Wajant H et al. Potent antitumoral activity of TRAIL through generation of tumor-targeted single-chain fusion proteins. Cell Death Dis 2010; 1: e68.
Tadokoro D, Takahama S, Shimizu K, Hayashi S, Endo Y, Sawasaki T . Characterization of a caspase-3-substrate kinome using an N- and C-terminally tagged protein kinase library produced by a cell-free system. Cell Death Dis 2010; 1: e89.
Jalmar O, García-Sáez AJ, Berland L, Gonzalvez F, Petit PX . Giant unilamellar vesicles (GUVs) as a new tool for analysis of caspase-8/Bid-FL complex binding to cardiolipin and its functional activity. Cell Death Dis 2010; 1: e103.
Matteucci C, Minutolo A, Balestrieri E, Marino-Merlo F, Bramanti P, Garaci E et al. Inhibition of NF-κB activation sensitizes U937 cells to 3′-azido-3′-deoxythymidine induced apoptosis. Cell Death Dis 2010; 1: e81.
Lam D, Shah S, de Castro IP, Loh SH, Martins LM . Drosophila happyhour modulates JNK-dependent apoptosis. Cell Death Dis 2010; 1: e66.
Tomlinson V, Gudmundsdottir K, Luong P, Leung KY, Knebel A, Basu S . JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis 2010; 1: e29.
Brandt B, Abou-Eladab EF, Tiedge M, Walzel H . Role of the JNK/c-Jun/AP-1 signaling pathway in galectin-1-induced T-cell death. Cell Death Dis 2010; 1: e23.
Chu KM, Minogue S, Hsuan JJ, Waugh MG . Differential effects of the phosphatidylinositol 4-kinases, PI4KIIα and PI4KIIIβ, on Akt activation and apoptosis. Cell Death Dis 2010; 1: e106.
Lanzillotta A, Sarnico I, Ingrassia R, Boroni F, Branca C, Benarese M et al. The acetylation of RelA in Lys310 dictates the NF-κB-dependent response in post-ischemic injury. Cell Death Dis 2010; 1: e96.
Wabnitz GH, Goursot C, Jahraus B, Kirchgessner H, Hellwig A, Klemke M et al. Mitochondrial translocation of oxidized cofilin induces caspase-independent necrotic-like programmed cell death of T cells. Cell Death Dis 2010; 1: e58.
Zhao C, Tan YC, Wong WC, Sem X, Zhang H, Han H et al. The CD14 (+/low) CD16(+) monocyte subset is more susceptible to spontaneous and oxidant-induced apoptosis than the CD14(+)CD16(−) subset. Cell Death Dis 2010; 1: e95.
Karlberg M, Ekoff M, Labi V, Strasser A, Huang D, Nilsson G . Pro-apoptotic Bax is the major and Bak an auxiliary effector in cytokine deprivation-induced mast cell apoptosis. Cell Death Dis 2010; 1: e43.
Sikkink LA, Ramirez-Alvarado M . Cytotoxicity of amyloidogenic immunoglobulin light chains in cell culture. Cell Death Dis 2010; 1: e98.
Gorvel L, Al Moussawi K, Ghigo E, Capo C, Mege JL, Desnues B . Tropheryma whipplei, the Whipple's disease bacillus, induces macrophage apoptosis through the extrinsic pathway. Cell Death Dis 2010; 1: e34.
Ben Mosbah I, Alfany-Fernández I, Martel C, Zaouali MA, Bintanel-Morcillo M, Rimola A et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis 2010; 1: e52.
Knauer SK, Heinrich UR, Bier C, Habtemichael N, Docter D, Helling K et al. An otoprotective role for the apoptosis inhibitor protein survivin. Cell Death Dis 2010; 1: e51.
Fujita E, Dai H, Tanabe Y, Zhiling Y, Yamagata T, Miyakawa T et al. Autism spectrum disorder is related to endoplasmic reticulum stress induced by mutations in the synaptic cell adhesion molecule, CADM1. Cell Death Dis 2010; 1: e47.
Paoletti R, Maffei A, Madaro L, Notte A, Stanganello E, Cifelli G et al. Protein kinase Cθ is required for cardiomyocyte survival and cardiac remodeling. Cell Death Dis 2010; 1: e45.
Giampietri C, Petrungaro S, Coluccia P, Antonangeli F, Giannakakis K, Faraggiana T et al. c-Flip overexpression affects satellite cell proliferation and promotes skeletal muscle aging. Cell Death Dis 2010; 1: e38.
Manning JA, Kumar S . A potential role for NEDD1 and the centrosome in senescence of mouse embryonic fibroblasts. Cell Death Dis 2010; 1: e35.
Upreti M, Koonce NA, Hennings L, Chambers TC, Griffin RJ . Pegylated IFN-α sensitizes melanoma cells to chemotherapy and causes premature senescence in endothelial cells by IRF-1 mediated signaling. Cell Death Dis 2010; 1: e67.
Schmid R, Schiffner S, Opolka A, Grässel S, Schubert T, Moser M et al. Enhanced cartilage regeneration in MIA/CD-RAP deficient mice. Cell Death Dis 2010; 1: e97.
Bose R, Moors M, Tofighi R, Cascante A, Hermanson O, Ceccatelli S . Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis 2010; 1: e92.
Blagosklonny MV . Linking calorie restriction to longevity through sirtuins and autophagy: any role for TOR. Cell Death Dis 2010; 1: e12.
Kroemer G, Nicotera P, Melino G . Cell death and disease: a new journal for a central area of pathophysiology. Cell Death Dis 2010; 1: e11.
Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 2010; 1: e10.
Ciavardelli D, Silvestri E, Viscovo AD, Bomba M, Gregorio DD, Moreno M et al. Alterations of brain and cerebellar proteomes linked to Aβ and tau pathology in a female triple-transgenic murine model of Alzheimer's disease. Cell Death Dis 2010; 1: e90.
Osato K, Sato Y, Ochiishi T, Osato A, Zhu C, Sato M et al. Apoptosis-inducing factor deficiency decreases the proliferation rate and protects the subventricular zone against ionizing radiation. Cell Death Dis 2010; 1: e84.
Dribben WH, Eisenman LN, Mennerick S . Magnesium induces neuronal apoptosis by suppressing excitability. Cell Death Dis 201; 1: e63.
Ruiz A, Matute C, Alberdi E . Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis 2010; 1: e54.
Wang G, Bieberich E . Prenatal alcohol exposure triggers ceramide-induced apoptosis in neural crest-derived tissues concurrent with defective cranial development. Cell Death Dis 2010; 1: e46.
Almasieh M, Zhou Y, Kelly ME, Casanova C, Di Polo A . Structural and functional neuroprotection in glaucoma: role of galantamine-mediated activation of muscarinic acetylcholine receptors. Cell Death Dis 2010; 1: e27.
Sancho-Pelluz J, Alavi MV, Sahaboglu A, Kustermann S, Farinelli P, Azadi S et al. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis 2010; 1: e24.
Zhu S, Zhang Y, Bai G, Li H . Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington's disease. Cell Death Dis 2011; 2: e115.
Yacoubian TA, Slone SR, Harrington AJ, Hamamichi S, Schieltz JM, Caldwell KA et al. Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson's disease. Cell Death Dis 2010; 1: e2.
Lee MH, Lin SR, Chang JY, Schultz L, Heath J, Hsu LJ et al. TGF-β induces TIAF1 self-aggregation via type II receptor-independent signaling that leads to generation of amyloid β plaques in Alzheimer's disease. Cell Death Dis 2010; 1: e110.
Sivananthan SN, Lee AW, Goodyer CG, LeBlanc AC . Familial amyloid precursor protein mutants cause caspase-6-dependent but amyloid β-peptide-independent neuronal degeneration in primary human neuron cultures. Cell Death Dis 2010; 1: e100.
Sassone J, Colciago C, Marchi P, Ascardi C, Alberti L, Di Pardo A et al. Mutant Huntingtin induces activation of the Bcl-2/adenovirus E1B 19-kDa interacting protein (BNip3). Cell Death Dis 2010; 1: e7.
Burnstock G, Verkhratsky A . Long-term (trophic) purinergic signalling: purinoceptors control cell proliferation, differentiation and death. Cell Death Dis 2010; 1: e9.
Di Guardo G, Marruchella G . Prions and neuronal death. Cell Death Dis 2010; 1: e6. No abstract available.
Cordeiro MF, Guo L, Coxon KM, Duggan J, Nizari S, Normando EM et al. Imaging multiple phases of neurodegeneration: a novel approach to assessing cell death in vivo. Cell Death Dis 2010; 1: e3.
Bunk EC, König HG, Bernas T, Engel T, Henshall DC, Kirby BP et al. BH3-only proteins BIM and PUMA in the regulation of survival and neuronal differentiation of newly generated cells in the adult mouse hippocampus. Cell Death Dis 2010; 1: e15.
Chi S, Cai W, Liu P, Zhang Z, Chen X, Gao L et al. Baifuzi reduces transient ischemic brain damage through an interaction with the STREX domain of BKCa channels. Cell Death Dis 2010; 1: e13.
Rodríguez JJ, Witton J, Olabarria M, Noristani HN, Verkhratsky A . Increase in the density of resting microglia precedes neuritic plaque formation and microglial activation in a transgenic model of Alzheimer's disease. Cell Death Dis 2010; 1: e1.
Corona C, Masciopinto F, Silvestri E, Viscovo AD, Lattanzio R, Sorda RL et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis 2010; 1: e91.
Flanagan L, Sebastià J, Tuffy LP, Spring A, Lichawska A, Devocelle M et al. XIAP impairs Smac release from the mitochondria during apoptosis. Cell Death Dis 2010; 1: e49.
Tejedo JR, Tapia-Limonchi R, Mora-Castilla S, Cahuana GM, Hmadcha A, Martin F et al. Low concentrations of nitric oxide delay the differentiation of embryonic stem cells and promote their survival. Cell Death Dis 2010; 1: e80.
Nicolai J, Burbassi S, Rubin J, Meucci O . CXCL12 inhibits expression of the NMDA receptor's NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Dis 2010; 1: e33.
Scheiman J, Jamieson KV, Ziello J, Tseng JC, Meruelo D . Extraribosomal functions associated with the C terminus of the 37/67 kDa laminin receptor are required for maintaining cell viability. Cell Death Dis 2010; 1: e42.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Knight, R., Melino, G. Cell death in disease: from 2010 onwards. Cell Death Dis 2, e202 (2011). https://doi.org/10.1038/cddis.2011.89
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.89
Keywords
This article is cited by
-
Die for the community: an overview of programmed cell death in bacteria
Cell Death & Disease (2015)
-
1000 successes as CDDIS reaches 1000 published papers!
Cell Death & Disease (2014)
-
Interaktion von Anästhetika und Analgetika mit Tumorzellen
Der Anaesthesist (2014)
-
Cancer metabolism: current perspectives and future directions
Cell Death & Disease (2012)
-
Novel transglutaminase 1 mutations in patients affected by lamellar ichthyosis
Cell Death & Disease (2012)
