
Abstract
Inflammatory responses mediated by activated microglia play a pivotal role in the pathogenesis of human immunodeficiency virus type 1 (HIV-1)-associated neurocognitive disorders. Studies on identification of specific targets to control microglia activation and resultant neurotoxic activity are imperative. Increasing evidence indicate that voltage-gated K+ (Kv) channels are involved in the regulation of microglia functionality. In this study, we investigated Kv1.3 channels in the regulation of neurotoxic activity mediated by HIV-1 glycoprotein 120 (gp120)-stimulated rat microglia. Our results showed treatment of microglia with gp120 increased the expression levels of Kv1.3 mRNA and protein. In parallel, whole-cell patch-clamp studies revealed that gp120 enhanced microglia Kv1.3 current, which was blocked by margatoxin, a Kv1.3 blocker. The association of gp120 enhancement of Kv1.3 current with microglia neurotoxicity was demonstrated by experimental results that blocking microglia Kv1.3 attenuated gp120-associated microglia production of neurotoxins and neurotoxicity. Knockdown of Kv1.3 gene by transfection of microglia with Kv1.3-siRNA abrogated gp120-associated microglia neurotoxic activity. Further investigation unraveled an involvement of p38 MAPK in gp120 enhancement of microglia Kv1.3 expression and resultant neurotoxic activity. These results suggest not only a role Kv1.3 may have in gp120-associated microglia neurotoxic activity, but also a potential target for the development of therapeutic strategies.
Similar content being viewed by others

Main
Human immunodeficiency virus type 1 (HIV-1) invades the central nervous system (CNS) early in the course of infection and provokes progressive cognitive, behavioral, and neurological impairments, which are now collectively referred to as HIV-1-associated neurocognitive disorders (HAND).1 HAND, varying in severity from an asymptomatic to a mild neurocognitive impairment and in its most serious form a devastating dementia (HIV-associated dementia), develops in a subset of individuals infected with HIV-1.1 As HIV-1 infects mononuclear phagocytes (brain perivascular macrophages and microglia), but not neurons, in the CNS, the pathogenesis for HAND is widely believed a consequence of indirect neurotoxicity. Ample evidence suggests that immune and inflammatory responses mediated by the activated microglia, the predominant resident CNS immunocompetent cell type productively infected by HIV-1, play a pivotal role in the HAND pathogenesis.1, 2, 3 As such, studies on how HIV-1 triggers microglia neurotoxic activities and identification of specific target(s) to regulate microglia activation and resultant production of neurotoxins are highly imperative.
Voltage-gated potassium (Kv) channels have recently gained attention as promising targets for therapy of immune- and inflammation-mediated neurological disorders4, 5, 6 and for specific immunomodulation.7 Electrophysiological studies of microglia in cell culture and tissue slices have demonstrated that microglia express several types of Kv channels including inward and delayed rectifier Kv channels and the expression levels of these Kv channels in microglia undergo dramatic changes during the process of activation.8 Exposure to a variety of activating stimuli produce a characteristic pattern of upregulation of Kv1.3 currents8, 9, 10, 11 and elevated Kv1.3 channels appear to be associated with microglia activation and neurotoxic activity.8, 12, 13, 14 Indeed, studies have shown that activation of microglia results in neuronal injury through a process requiring Kv1.3 activity in microglia and blocking microglia Kv1.3 or decrease of Kv1.3 expression inhibits microglia-associated neurotoxicity.15, 16 We hypothesize that HIV-1 infection and/or HIV-1 glycoprotein 120 (gp120) can activate microglia through enhancement of outward K+ current conducted by Kv1.3 channels, resulting in microglia production of neurotoxins and consequent neuronal injury. To test this hypothesis, we studied the effects of HIV-1 gp120 on Kv1.3 current in primary rat microglia cultures. Our previous studies have shown that gp120 increased the outward K+ current in cultured rat microglia, leading to neuronal injury via CXCR4 and cAMP-dependent protein kinase A signaling and enhancement of Kv1.3 expression.17 In this study, we examined the role of Kv1.3 in gp120-stimulated microglia production of neurotoxins and resultant neuronal injury. Our results revealed that incubation of rat microglia with gp120 significantly increased microglia production of neurotoxins and resultant neurotoxic activity. The neurotoxic activity induced by gp120-stimulated microglia was significantly attenuated by a Kv1.3 channel blocker margatoxin (MgTx) or by siRNA knockdown of microglia Kv1.3 gene. Further investigation indicates an involvement of p38 MAP kinase (MAPK) signaling in gp120-associated microglia neurotoxic activity.
Results
Enhancement of microglia Kv1.3 current by gp120
We have previously showed that gp120 IIIB enhanced whole-cell outward K+ currents in cultured rat microglia.17 To further examine whether gp120 enhancement of microglia outward K+ current is conducted by Kv1.3 channels, we studied the effects of gp120 IIIB on outward K+ currents recorded in microglia in the presence and absence of MgTx, a specific Kv1.3 blocker. Incubation of microglia with gp120 for 2 h significantly enhanced the outward K+ currents with an average instantaneous outward K+ current density of 71.4±4.7 pA/pF (Figure 1a, n=9) when measured at +60 mV. In comparison with the current density of 46.5±1.6 pA/pF (n=12) recorded in control (Ctrl) microglia, the difference was statistically significant (P<0.01), confirming our previous finding that gp120 enhances outward K+ current in microglia. We separated the Kv1.3 current by subtraction of the currents recorded in the presence of MgTx (Figure 1b) from the whole-cell outward K+ current. As shown in Figures 1c and d, gp120 significantly increased the Kv1.3 currents recorded in rat microglia (P<0.01, n=9). The gp120-induced increase of outward K+ currents was blocked by MgTx in a concentration-dependent manner. At concentrations (nM) of 0.1, 1, 10 and 100, MgTx blocked 10.3±4.3, 23.1±6.7, 64.9±3.8 and 93.5±6.7% of gp120-induced increase of outward K+ current, respectively, indicating gp120 increases microglia outward K+ current via Kv1.3 (Figure 1e).

Enhancement of microglia Kv1.3 outward K+ current by gp120. (a) Whole-cell outward K+ current traces recorded from a non-treated (Ctrl) microglia and a different microglial cell treated with gp120 (gp120). Voltage protocol employed to generate outward K+ current is shown above the current traces (voltage step duration is not in proportion to the current traces). (b) Outward K+ currents recorded from the same cells shown in a in the presence of 100 nM MgTx (MgTx) or MgTx plus gp120 (MgTx+gp120). (c) Kv1.3 currents obtained by subtraction of the currents in b from the currents in a. (d) Instantaneous whole-cell current densities (WC) and the instantaneous current densities carried by Kv1.3 channels (Kv1.3) recorded in non-gp120-treated microglia (Ctrl) and gp120-treated microglia (gp120). Note gp120 significantly increased Kv1.3 current. *P<0.01. (e) gp120-induced increase of K+ outward current was blocked by MgTx in a dose-dependent manner
Gp120 upregulation of Kv1.3 expression in microglia
Growing evidence indicate that activated microglia express higher levels of Kv channels, especially the Kv1.3. To assess the effect of gp120 on the levels of Kv1.3 expression in microglia, we monitored mRNA abundance and protein expression levels of gene-related Kv1.3 on rat microglia. First, Kv1.3 mRNA expression was examined using reverse transcription (RT)-PCR. Rat microglia were exposed to gp120 (500 pM) or LPS (0.5 μg/ml, as a positive control) for 24 h. Using gene-specific primers, RT-PCR was performed and standardized to a housekeeping gene (β-actin) in each sample. As shown in Figure 2a, gp120 treatment increased expression levels of Kv1.3 mRNA in microglia by 1.5-fold. Similar results were obtained from microglia treated with LPS as positive controls, with ∼1.9-fold increase of Kv1.3 mRNA expression. As mRNA levels do not always correspond with protein expression, we further examined the expression levels of Kv1.3 channel proteins by western blot using a specific antibody against Kv1.3. In parallel with the results unraveled by RT-PCR, western blot analysis revealed increased levels of Kv1.3 protein expression by 1.5-fold and 2.3-fold in microglia treated with gp120 and LPS, respectively (Figure 2b). These results are in full agreement with our previous observations that gp120 increased Kv1.3 protein expression in cultured rat microglia as detected by western blot and immunocytochemistry.17

gp120 upregulation of microglia Kv1.3 expression. (a) Representative gel showing RT-PCR products for Kv1.3 and internal control β-actin (Left). The density of each band was normalized to its internal control and represented in a bar graph (Right). Results reflect the increase of Kv1.3 mRNA was significantly different in LPS- (increasing 1.9-fold) or gp120- (increasing 1.5-fold) treated microglia when compared with non-treated controls (Ctrl). (b) The left panel shows representative western blot (Kv1.3, and β-actin). Right, each band density was normalized to internal β-actin control. Significant differences were detected in LPS (increasing 2.3-fold) or gp120 (increasing 1.5-fold). *P<0.05 versus Ctrl; ***P<0.001 versus Ctrl
Involvement of Kv1.3 in microglia-induced neurotoxicity
After demonstration of gp120 enhancement of Kv1.3 expression and Kv1.3 current in microglia, we questioned whether the elevated levels of Kv1.3 expression and the enhancement of Kv1.3 current were involved in microglia-induced neurotoxicity. To these ends, we examined neuronal viability and neuronal apoptosis in a microglia-neuronal co-culture system using MTT and TUNEL assays and tested whether the neurotoxicity mediated by gp120-pretreated microglia could be blocked by pre-treatment of microglia with MgTx, 4-aminopyridine (4-AP) or tetraethylammonium (TEA). In our early study, we found gp120-treated microglia induced neuronal injury, which could be attenuated by Kv channel antagonists.17 To further confirm our previous observations, we examined neuronal viability via MTT assay after co-culture of rat cortical neurons with the microglia pretreated with gp120 at different concentrations of 20, 200, 500 or 1000 pM. Being pretreated with gp120 for 24 h, microglia plated on the transwells were co-cultured with rat cortical neurons grown on poly-D-lysine-coated coverslips in 24-well plates for additional 24 h. As shown in Figure 3a, no significant reduction of neuronal viability was induced by microglia pretreated with 20 or 200 pM of gp120 when compared with the cell viability observed in non-gp120-pretreated controls (Ctrl). However, when pretreated with 500 and 1000 pM gp120, microglia caused a significant decrease of neuronal viability, with an average of 72.9±0.8% (P<0.05) and 26.1±1.6% (P<0.001) of survival neurons, respectively. The reduction of neuronal viability induced by gp120-pretreated microglia was blocked by MgTx, 4-AP or TEA (Figure 3a), suggesting Kv1.3 is involved in microglia-associated neurotoxicity. As 500 pM gp120 produced a consistent and significant decrease of neuronal survival, this concentration was chosen in the following TUNEL assay study and other studies reported in this paper.

Involvement of Kv 1.3 in neurotoxic activity induced by gp120-pretreated microglia. Microglia were pre-exposed to Kv channel antagonists, 4-AP (1 mM), TEA (5 mM) or MgTx (1, 10, 100 nM) for 30 min before addition of gp120 (500 pM). After a 24 h treatment, microglia were then co-cultured with neurons for an additional 24 h in a transwell system. (a) Examination of microglia-induced neuronal injury by MTT assay. The reduction of cell viability induced by gp120-pretreated microglia was concentration-dependent, with a significant reduction at 500 and 1000 pM. Such a reduction of neuronal viability by gp120-pretreated microglia was blocked by broad spectrum Kv channel antagonists 4-AP and TEA or by a specific Kv1.3 channel blocker MgTx (10 nM). (c) Apoptotic neurons were evaluated by TUNEL staining and visualized by confocal microscopy at x400 original magnification. Scale bar equals 50 μm. (b) Quantification of apoptotic neurons was obtained by determining the percentage of TUNEL-positive cells. Gp120-pretreated microglia produced a significant increase of neuronal apoptosis, which was blocked either by MgTx in a dose-dependent manner, or by broad spectrum Kv channel blockers 4-AP and TEA. The results are representative of three independent experiments performed in triplicate determinations. *P<0.05 and ***P<0.001 versus Ctrl; #P<0.05 versus gp120 500 pM
The neurotoxicity induced by gp120-pretreated microglia was also evaluated via TUNEL assay. Microglia used for TUNEL studies were exposed to a specific Kv1.3 channel blocker MgTx in different concentrations (1, 10, or 100 nM), 4-AP (1 mM) or TEA (5 mM) for 30 min before addition of gp120 (500 pM) to the culture media. After 24 h, microglia were washed three times and then co-cultured with cortical neurons (0.1 × 106/well in 24-well plates) for another 24 h. Apoptotic neurons were evaluated by TUNEL staining and visualized by confocal microscopy (Figure 3c) Our results showed that neurons co-cultured with gp120-pretreated microglia exhibited 37.2±10.9% of TUNEL positive cells. In comparison with 7.7±4.4% of TUNEL positive neurons co-cultured with non-gp120-pretreated microglia (control), the difference was statistically significant (P<0.05). Exposure of microglia to MgTx at 1, 10 and 100 nM before pretreatment of microglia with gp120 significantly reduced the percentage of apoptotic neurons to 26.8±3.6, 16.0±4.4 and 11.8±3.0%, respectively (Figure 3b). Such a dose-dependent reduction of neuronal apoptosis by MgTx suggests an involvement of Kv1.3 in microglia-induced neuronal toxicity. Moreover, exposure of microglia to 4-AP or TEA also reduced microglia-induced neuronal apoptosis to 11.9±4.5 and 12.5±5.0%, respectively, further supporting the involvement of Kv1.3 in neurotoxic activity induced by gp120-pretreated microglia (Figures 3b and c).
Blockade of Kv1.3 reduces gp120-induced microglia production of neurotoxins
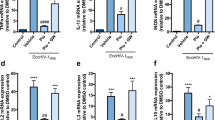
It is widely accepted that activated microglia produce neurotoxins such as cytokines, chemokines, nitric oxide (NO) and reactive oxygen species (ROS) resulting in neuronal injury. To understand the mechanisms underlying the neurotoxic activity mediated by gp120-treated microglia, we examined cytokine, chemokine, NO and ROS production in gp120-treated microglia and the blockade of microglia production of neurotoxins by Kv channel antagonists. In these experiments, purified microglia were exposed to gp120 (500 pM) in the presence or absence of Kv channel blockers 4-AP (1 mM), TEA (5 mM) or MgTx (10 nM) in the culture media (added 30 min before the addition of gp120). The culture supernatants were then harvested 24 h later and used subsequently for the detection of bioactive molecules. As illustrated in Figures 4a and b, the production levels of chemokines (cytokine-induced neutrophil chemoattractant-2(CINC-2) and CINC-3) and cytokines (IL-1α, IL-6, and TNF-α) were significantly increased in the supernatants recovered from gp120-treated microglia in comparison with the results obtained from the supernatants recovered from non-gp120-treated microglia (control). The gp120-induced production of bioactive molecules was markedly (P<0.01) reduced by Kv1.3 blocker MgTx, or by broad spectrum Kv channel blockers 4-AP and TEA, suggesting the involvement of Kv1.3 in gp120-induced microglia production of bioactive molecules. However, gp120 did not enhance microglia production of interferon-γ (IFN-γ), a cytokine was reported to be produced by microglia18, 19 (Figures 4a and b). In addition to detection of cytokine and chemokine production, we also measured NO levels in microglia-conditioned media. Treatment of microglia with gp120 significantly increased NO production with an average of 767±64 nM when compared with 195±31 nM detected in matched controls. Addition of Kv channel blockers significantly reduced gp120-induced NO production. The average concentration was 438±48, 300±34, or 312±35 nM when 4-AP, TEA or MgTx was added to the cultures, respectively (Figure 4c). In comparison with the NO concentration detected from gp120-treated microglia, the difference was statistically significant (P<0.01). Similar results were obtained in microglia ROS production. As shown in Figure 4d, the amount of ROS produced by gp120-treated microglia was 179.6±15.6% of the control level (untreated microglia). Addition of 4-AP, TEA or MgTx to the cultures significantly decreased the levels of ROS to 120.9±11.8, 115.6±8.3 or 121.1±11.7% of the control level. These results indicate involvement of Kv1.3 channels in gp120-induced microglia production of neurotoxins.

Blockade of gp120-activated microglia production of neurotoxins by Kv channel blockers. Microglia were pre-exposed to 4-AP (1 mM), TEA (5 mM) or MgTx (10 nM) before addition of gp120. After a 24 h incubation, the supernatants were harvested for cytokines and NO analysis and the cells were subjected to measure ROS production as a function of H2O2 levels. (a) Collected supernatants were subjected to cytokine array blots (RayBiotech). Ovals encompass replicated blots indicating the coordinates of cytokines, CINC 2 (oval 1), CINC 3 (oval 2), IFN-γ (oval 3), IL-1α (oval 4), IL-6 (oval 5), and TNF-α (oval 6). (b) Densitometric analysis of cytokine array blots were achieved by digital image analysis with NIH Image J software, and mean densities±S.D. were determined for replicate determinations. Kv channel antagonists significantly ameliorated cytokine (CINC 2, CINC 3, IL-1α, IL-6 and TNF-α) production in gp120-activated microglia. (c) NO production was determined by using Griess reagent kit (Molecular Probes). Results, represented in bar graphs, showed that gp120 significantly increased the levels of NO production, and such an increase was diminished by either 4-AP, TEA or MgTx. (d) ROS production was measured using an Amplex Red assay. An elevated level of ROS production was reflected in Gp120-infected microglia group. This increased ROS production was ameliorated by 4-AP, TEA or MgTx, respectively. ***P<0.001 versus Ctrl; #P<0.05, ##P<0.01, ###P<0.001 versus gp120-treated group
Knockdown Kv1.3 gene abrogated neurotoxic activity of gp120-stimulated microglia
To further confirm the role Kv1.3 had in neurotoxic activity mediated by gp120-stimulated microglia, we exploited gene silencing using siRNA knockdown Kv1.3 gene (KCNA3) in microglia. Microglia were either transfected with Kv1.3-siRNA or nonspecific GAPD control siRNA (control siRNA). After 48 h or 72 h of transfection, microglia were incubated with or without gp120 (500 pM) for additional 24 h. Expression of Kv1.3 mRNA or protein was examined by RT-PCR or western blot. As shown in Figure 5a, Kv1.3 mRNA expression in gp120-stimulated microglia was efficiently inhibited after 48 h transfection with Kv1.3 siRNA, when compared with Kv1.3 mRNA expression in gp120-stimulated microglia transfected with control siRNA. In parallel with the results on Kv1.3 mRNA expression, Kv1.3 protein expression was also diminished after 72 h transfection. These results demonstrated that transfection of microglia with Kv1.3 siRNA inhibited gp120-stimulated increase of Kv1.3 expression. As gp120-activated microglia induced neuronal apoptosis via Kv1.3 (Figure 3), we next examined whether knockdown of Kv1.3 gene suppresses the neurotoxic activity produced by gp120-activated microglia. Supernatants, collected from microglia pre-transfected with Kv1.3-siRNA for 72 h and then incubated with or without gp120 for additional 24 h, were added to rat cortical neuronal cultures at 1 : 10 ratio and incubated for 24 h before being analyzed by MTT assay and TUNEL staining. Knockdown of Kv1.3 abolished neurotoxic activity mediated by gp120-activated microglia as illustrated by its significant improvement of neuronal survival in MTT assay study (Figure 5b). TUNEL staining revealed similar results to the knockdown of Kv1.3 gene, which decreased apoptotic neurons (Figure 5c). These results indicated that knockdown of the Kv1.3 gene significantly abrogated neurotoxic activity of gp120-stimulated microglia.

Knockdown Kv1.3 by siRNA abrogated the neurotoxic activity of gp120-activated microglia. Microglia were transfected with siRNA targeting Kv1.3 (KCNA3) or nonspecific GAPD control siRNA for 48 or 72 h and stimulated with gp120 (500 pM) for an additional 24 h. Cells were harvested for preparation of mRNA (48 h post-transfection/24 h gp120 stimulation) and proteins (72 h post-transfection/24 h gp120 stimulation). Supernatants were subjected to neuronal culture. Neuronal apoptosis and viability assay were determined using TUNEL staining and MTT assay. (a) Representative gels show RT-PCR products for Kv1.3 mRNA and internal control β-actin (up 2 rows), and western blots show Kv1.3 channel protein and internal control β-actin protein expression of microglia (bottom 2 rows). (b) Knockdown of Kv1.3 gene abolished the neurotoxic activity induced by the culture supernatants recovered from gp120-activated microglia as evaluated by MTT assay. The collected supernatants (1 : 10 dilution) were subjected to primary neuronal culture for 24 h and percentage of survival neurons were evaluated by MTT assay. An increased viability was observed in neurons that treated with supernatants collected 72 h post-Kv1.3 siRNA transfection/24 h post-gp120 stimulation when compared with neurons treated with supernatants collected 72 h post-control siRNA transfection/24 h post-gp120 stimulation (P<0.05). Supernatant collected from post-transfection of control siRNA had no significant effect on altering neuronal viability in comparison with the neuronal viability induced by supernatant recovered from gp120-stimulated microglia without siRNA transfection. (c) Transfection of microglia with siRNA targeting Kv1.3 gene inhibited the neurotoxic activity induced by the culture supernatants collected from gp120-activated microglia. In contrast, the supernatants collected from microglia transfected with control siRNA (ctrl-siRNA) and then stimulated with gp120 (500 pM) produced a notable increase of neuronal death. Each experiment was done in triplicate and data were obtained from three experiments. *P<0.05 versus Ctrl, #P<0.05 versus ctrl siRNA
gp120 enhanced Kv1.3 expression and resultant microglia neurotoxic activity via p38 MAPK signaling
The above results showed that gp120 induces microglia neurotoxic activity by enhancing microglia Kv1.3 current and resultant microglia production of neurotoxins. We further analyzed potential intracellular signaling on gp120-mediated upregulation of microglia Kv1.3 expression and resultant microglia neurotoxic activity. Microglia were exposed to gp120 (500pM) in the absence or presence of 4-AP (1 mM), TEA (5 mM), MgTx (10 nM) or SB203580 (SB, 10 μM) for 24 h. Immunocytochemistry, western blot and TUNEL assay were performed to evaluate involvement of p38 MAPK in microglia Kv1.3 expression and microglia-induced neurotoxicity in microglia–neuronal co-cultures. Our results showed that gp120-mediated upregulation of Kv1.3 channel expression was associated with activation of microglia p38 MAPK and blocking p38 MAPK by SB significantly reduced the gp120-associated upregulation of Kv1.3 expression (Figures 6a and b). Pretreatment of microglia with SB also attenuated the neurotoxic activity mediated by gp120-treated microglia as demonstrated in a microglia-cortical neuronal transwell co-culture system (Figure 6e). These results indicate an involvement of p38 MAPK in intracellular signaling of gp120-associated microglia neurotoxic activity. Another piece of evidence supporting the involvement of p38 MAPK in gp120-associated microglia neurotoxicity came from western blot studies demonstrating gp120 upregulation of p38 MAPK phosphorylation and the blockade of gp120-mediaed upregulation of p38 MAPK phosphorylation by 4-AP, TEA and MgTx (Figures 6c–e).

p38 MAPK is involved in gp120-mediated microglia upregulation of Kv1.3 expression and resultant neurotoxic activity. Microglia were exposed to gp120 (500 pM), with or without 4-AP (1 mM), TEA (5 mM) MgTx (10 nM) or SB203580 (10 μM). Immunocytochemistry, western blot and TUNEL assay were performed after 24 h incubation for evaluation of microglia Kv1.3 expression and resultant neurotoxic activity. (a) gp120 enhanced the expression levels of Kv1.3 in microglia and this enhancement was blocked by a p38 MAPK inhibitor SB203580 (SB). The microglia were stained with rat monoclonal Mac-1 (CD11b; 1 : 500 dilution, red), whereas Kv1.3 was stained with rabbit polyclonal antibody (1 : 200 dilution, green). Images were visualized by fluorescent microscopy. Representative images showed that gp120 enhanced Kv1.3 expression, which was blocked by SB. (b) Western blot analysis illustrated that the gp120 enhancement of Kv1.3 expression in microglia was blocked by SB, demonstrating involvement of p38 MAPK in gp120-induced increase of Kv1.3 expression. (c) Further analysis by western blot revealed that gp120 upregulated p38 MAPK phosphorylation, with remarkable effects during the first 2 h. (d) The blockade of gp120 upregulation of p38 MAPK phosphorylation by 4-AP, TEA or MgTx, suggests a link between Kv channel activation and p38 MAPK phosphorylation. (e) A bar graph illustrating p-p-38/p-38 relative density. Note gp120 significantly increased the relative density and the gp120-induced increase was attenuated by 4-AP, TEA or MgTx. ***versus Ctrl, P<0.001; ##versus gp120, P<0.01; ###versus gp120, P<0.001). (f) Neuronal apoptosis induced by gp120-stimulated microglia was significantly attenuated (gp120-microglia versus SB: 33.8% versus 12.8%) by the treatment of gp120-stimulated microglia with SB, indicating that activation of p38 MAPK is required for neurotoxic activity mediated by gp120-stimulated microglia. Data were presented as Mean±S.D. obtained from three independent experiments. Compared with Control: *P<0.05 versus non-gp120-stimulated microglia (Ctrl); #P<0.05 versus gp120-stimulated microglia
Discussion
HIV-1gp120 is a viral envelope glycoprotein with neurotoxic activity in HIV-1-infected brains. It is shed off from virions and/or secreted from HIV-1-infected macrophages and microglia. This soluble viral protein has the potential to diffuse and interact directly with surrounding and distant neural cells possibly via chemokine receptor CXCR4,20, 21 or act indirectly on local and distant neural cells by stimulating uninfected microglia to produce neurotoxins. Whereas the indirect mechanism of HIV-1-associated neuronal injury has widely been studied, few have investigated the role of microglia Kv channels, in particular the Kv1.3, in neurotoxic activity mediated by gp120-activated microglia. We have previously demonstrated that gp120 enhanced outward K+ current in cultured rat microglia via chemokine receptor CXCR4 and cAMP-dependent PKA pathways.17 Biological significance of gp120-induced enhancement of microglia outward K+ current was demonstrated by experimental results showing the neurotoxic activity induced by gp120-pretreated microglia, evaluated by TUNEL staining and MTT assays, was significantly attenuated by Kv channel blockers. These results suggest that gp120 triggers microglia neurotoxic activity by enhancing microglia outward K+ current.17 In the present study, we further investigated the involvement of Kv channels in neurotoxic activity induced by gp120-treated microglia and its intracellular signaling pathways. Our results showed that gp120 enhanced microglia Kv1.3 current and production of cytokines, chemokines, NO and ROS resulting in microglia neurotoxic activity. These findings were supported by experimental results that cytokine production and neurotoxic activity induced by gp120-treated microglia were blocked either by Kv1.3 antagonists or by knockdown of Kv1.3 gene in microglia. The gp120-induced increase of microglia production of neurotoxins and resultant neurotoxic activity was mediated via p38 MAPK signaling.
In addition to being the target for HIV-1, soluble viral protein gp120 and cytokines, microglia also function as a source of neurotoxins in many infectious, inflammatory and degenerative brain diseases as well as a source of neurotrophins in normal brain.13, 22, 23, 24 Upon challenging, microglia undergo dramatic phenotypic, immunochemical, and functional changes, collectively referred to as ‘activation’ and the activated microglia produce a variety of bioactive molecules with potential toxicity to neurons. Ample evidence indicates that immune and inflammatory responses mediated by activated microglia have a pivotal role in the pathogenesis of HAND.2, 3 Studies have shown that HIV-1-infected and immune-activated microglia (and brain macrophages) release a number of soluble substances including, but not limited to, pro-inflammatory cytokines, chemokines, excitatory amino acids, NO and ROS, as well as viral proteins, which can diffuse and injure surrounding and distant neurons, contributing to the HAND pathogenesis.23, 25 Therefore, it is important to identify potential target(s) to regulate microglia activation and their resultant production of neurotoxins in order to control microglia-associated neurotoxicity.
In this study, we examined the involvement of Kv1.3 on microglia production of chemokines, cytokines (the representative chemokines and cytokines analyzed are shown in Figure 4b), NO and ROS. Our results showed gp120 stimulated microglia production of chemokines (CINC-2, CINC-3) and cytokines (IL-1α, IL-6 and TNF-α, but not IFN-γ), as well as NO and ROS. The gp120-induced microglia production of chemokines, cytokines, NO and ROS were significantly reduced by a specific Kv1.3 blocker MgTx in addition to its blockade by broad spectrum Kv channel blockers 4-AP and TEA, suggesting an involvement of Kv1.3 in microglia production of neurotoxic substances and resultant neurotoxicty. Interestingly, gp120 did not increase microglia production of IFN-γ, a cytokine expressed and produced by microglia.18, 19, 26 The failure of gp120 in increasing microglia production of IFN-γ may indicate microglia production of a small amount of IFN-γ or microglia may require other mechanisms for full production of IFN-γ and secrete it to the culture supernatant.18, 19, 26
Microglia expresses several types of Kv channels and the levels of their expression undergo dramatic changes during the process of activation.8, 13 The activated microglia express Kv1.3 channels and Kv1.3 current was predominantly detected in activated microglia,27, 28 suggesting a role for Kv1.3 in regulating microglia function. It has been reported that activated microglia kill neurons and blocking microglia Kv1.3 channels inhibits microglia neurotoxicity,15 indicating the involvement of Kv1.3 in microglia-mediated neurotoxicity. In an agreement with the aforementioned findings, our results showed that gp120 elevated the levels of microglia Kv1.3 expression, enhanced Kv1.3 current, and increased microglia production of cytokines and resultant neurotoxic activity, which were blocked by a specific Kv1.3 blocker MgTx or by siRNA knockdown of Kv1.3 gene. These results clearly indicate the involvement of microglia Kv1.3 channels in the neurotoxic activity mediated by gp120-pretreated microglia.
The intracellular signaling pathways underlying gp120-Kv1.3-associated microglia neurotoxic activity are largely unknown. Studies have shown that phosphorylation (activation) of p38 MAPK is involved in the processes of microglia/macrophage activation, leading to pro-inflammatory cytokine production and neuronal death.3, 15, 29, 30 Thus, inactivation of p38 MAPK pharmacologically or genetically would diminish or abolish microglia/macrophage-associated neurotoxic activity.31 Indeed, inhibition of p38 MAPK phosphorylation by a specific blocker SB203580 prevents gp120-associated neuronal death in mixed neuronal-glial cultures.31 In an attempt to explore whether p38 MAPK is involved in gp120-associated elevation of microglia Kv1.3 expression and resultant neurotoxic activity, we examined effects of SB203580, a specific p38 MAPK inhibitor, on gp120 enhancement of microglia Kv1.3 expression and effects of MgTx on p38 MAPK phosphorylation. Our results revealed gp120 upregulation of Kv1.3 expression resulting in neuronal apoptosis via p38 MAPK as inhibition of p38 MAPK by SB203580 blocked gp120 upregulation of Kv1.3 expression and resultant neurotoxic activity. We also observed gp120 enhancement of microglia p38 phosphorylation and pretreatment of microglia with MgTx attenuated gp120-induced p38 phosphorylation. These findings are in agreement with those reports showing the activation of p38 MAPK as a requirement for microglia/macrophage in gp120-induced neurotoxicity.31, 32, 33
It has been shown that Kv channel blockers have promising effects in the treatments of multiple sclerosis, spinal cord injury cognitive decline.34, 35, 36 The US Food and Drug Administration has recently approved Dalfampridine (Ampyra), a Kv channel blocker, as a treatment to improve walking in patients with multiple sclerosis (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm198463.htm). These shed light on the future to use Kv1.3 blockers to combat against HAND and other neurodegenerative disorders in which microglia-associated immune and inflammatory responses are involved in their pathophysiological processes. Although the pharmacological blockade of microglia Kv1.3 channels might generate therapeutic benefit, it might also have the risk of causing immune suppression as Kv1.3 is expressed in T cells.7, 28 However, as Kv1.3 is mainly expressed in human effector memory T cells (CD4+CCR7−CD45RA−), which mediate Th1 cell inflammatory response,7, 37 blocking Kv1.3 pharmacologically may have, if any, limited effects on immunity because innate immune response, Th2 cell response, B-cell immunity and immunological synapse formation are basically not affected.37, 38 Indeed, specific blockers of Kv1.3 are being investigated as therapeutics for diverse autoimmune disorders involving effector memory T cells. Several lines of evidence indicate that suppression of T effector memory cells by Kv1.3 does not increase susceptibility to infection nor compromise immune responses to vaccination,39, 40 although this will have to be thoroughly investigated in the future.
In the scenario of HIV-1 brain infection, the pathogenesis of HAND involves immune and inflammatory responses, which are orchestrated by cells of the innate immune system, namely microglia and macrophages. Activated microglia and macrophages release neurotoxins resulting in neuronal damage and cognitive impairment as seen in patients with HAND. Our results clearly demonstrated that gp120 stimulates microglia production of neurotoxins leading to neuronal injury via microglia Kv1.3. Thus, to minimize and/or prevent microglia/macrophage production of neurotoxins and resultant neuronal injury, which may occur in HIV-1-infected brain, it is most likely achievable to minimize and/or prevent microglia/macrophage production of neurotoxins and resultant neuronal injury by blocking microglia Kv1.3 with specific blockers. As there is very limited information available on how the Kv1.3 can be ‘best’ utilized for optimal therapeutic benefit, our findings might have potential significance in the development of therapeutic strategies for HAND, and perhaps other neurodegenerative disorders. Although much work remains to be done to bring specific Kv1.3 blockers to the clinic, the future is encouraging.
Materials and Methods
Materials
HIV-1gp120 IIIB was purchased from Immunodiagnostics, Inc. (Woburn, MA, USA). Aliquots of gp120 were kept as 100 nM stock solution at −80°C. The stock solution was diluted to desired concentrations with artificial cerebrospinal fluid (ACSF) 2–5 min before test. All chemicals, unless otherwise specified, were from Sigma (St. Louis, MO, USA).
Isolation and purification of rat microglia and neurons
Highly purified microglia were prepared from cerebral cortices of 1–2 days old neonatal rats and neuronal cultures were prepared from day 18 embryonic Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA). The cortical tissues were dissected in cold Hanks’ Balanced Salt solution (Gibco, Grand Island, NY, USA) and digested with 0.25% trypsin (Invitrogen, Carlsbad, CA, USA). Isolated cells were suspended under different culture conditions to differentiate into neurons or microglia. For neuronal differentiation, isolates were seeded at a density of 0.10 × 106 cells per well in 24-well plates or 1.0 × 106 cells per well in poly-D-lysine-coated 6-well-plates and maintained for 10 days in neurobasal medium supplemented with 2% B27, 1% penicillin/streptomycin and 0.25 mM L-glutamine (Invitrogen). The purity of neuronal cells (>90%) was determined by staining with microtubule-associated protein-2 (1 : 1000, Chemicon International, Temecula, CA, USA) antibody.
For microglia differentiation, isolated cells (30 × 106) were plated into T75 cm2 flasks in high-glucose Dulbecco's modified Eagle's medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1% penicillin/streptomycin, and 1 μg ml–1 macrophage colony-stimulating factor. After 10 days in culture, flasks were gently shaken to detach microglial cells to culture media. The culture media were then plated in a fresh T75 flask for 30 min at 37°C. The suspensory glial cells were then removed via changing culture media. The resulting culture was 98–100% microglia as determined by staining with OX-42 antibody (Serotec, Oxford, UK), a marker for microglia CR3/CD11b receptors. All animal-use procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Nebraska Medical Center (# 00-062-07).
Electrophysiology
Whole-cell outward K+ currents were recorded from primary rat microglia cultures at room temperature (RT, ∼22 °C). The microglia cells were perfused with oxygenated (bubbled with 95% O2, 5% CO2) ACSF contained (in mM): 140 NaCl, 5 KCl, 2.0 CoCl2, 1 MgCl2, 10 D-glucose, 10 HEPES (pH 7.4 adjusted with NaOH, osmolarity: 310 mOsm). Patch-clamp electrodes, made from borosilicate glass capillaries, had tip resistance of 4–8 MΩ when filled with pipette solution contained (in mM): 135 K-gluconate, 10 KCl, 1 CaCl2, 1 MgCl2, 10 EGTA, 0.5 Tris-GTP, 2 Mg-ATP, 10 HEPES (Adjusted pH to 7.3 with KOH, osmolarity: 300 mOsm). After establishment of the whole-cell configuration, the cells were allowed to stabilize for 3–5 min before recording. The recorded cells were voltage-clamped at −60 mV and whole-cell outward K+ current was induced by voltage steps from the holding potential of −60 to −40 mV in the first step and then stepped to +60 mV in increments of 10 mV. The seal resistance was 1–10 GΩ. Junction potentials were corrected and the cell capacitance was compensated (∼70%) in most cells. Current signals were amplified with an Axopatch 200B amplifier, filtered at 1 kHz and digitized at 5 kHz using a Digidata 1440A digitizer. The current and voltage traces were displayed and recorded in a Dell computer using pClamp 10.1 data acquisition/analysis system. The outward K+ current density (pA/pF) was calculated by dividing the cell capacitance from the peak current generated by a voltage step.
TUNEL and MTT assays
Neuronal apoptosis were detected using the in situ cell death detection kit, AP (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer's instructions. Briefly, pretreated microglia, at a density of 0.5 × 106 cells per transwell inserts (pore diameter, 1 μm, BD Biosciences, Franklin Lakes, NJ, USA), were co-cultured with cortical neurons growing on poly-D-lysine-coated coverslips at a density of 1.0 × 105cells per well in 24-well plates. After a 24 h incubation, microglia were removed, neurons were washed and fixed with 4% paraformaldehyde, followed by permeabilizing with 0.1% Triton X-100. Neurons were then processed for TUNEL staining (green). ProLong Gold antifade reagent (Molecular Probes, Eugene, OR, USA) with 4′,6′-diamidino-2-phenylindol (DAPI) was added to stain neuronal nuclei and mount coverslips. Cells were visualized by Zeiss LSM 510 META NLO microscope (Zeiss MicroImaging, Inc., Thornwood, NY, USA) in a 40 × oil-immersion objective. Apoptotic neurons (green fluorescent cells) were counted and normalized to the total number of cells as determined by DAPI nuclear stain. For MTT assays, neurons after co-cultured with pretreated microglia were washed in PBS and then incubated with fresh neuronal culture media containing MTT (500 μg/ml) for 3 h. At the end of the incubation period, the MTT solution was replaced with 500 μl of dimethyl sphingosine for cell lyses, and the optical density (OD) of absorption was then measured at 570 nm.
Detection of oxidative stress
To assess hydrogen peroxide (H2O2) production from activated microglia, ROS and NO were monitored as an indication of H2O2 accumulation using an Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit or a Griess reagent kit (Molecular Probes), respectively. Pre-treated microglia seeded at 1 × 105 cells per well in a 96-well fluorometer plate were incubated with Krebs-Ringer buffer containing 50 μM Amplex Red reagent, 1 U ml–1 horseradish peroxidase (HRP) for 30 min at 37°C. Fluorescence was measured using a fluorescence microplate reader (BioTek Instruments, Winooski, VT, USA) with excitation at 530 nm and detection at 580 nm. Background fluorescence determined at 0 min incubation for a non-H2O2 control reaction was subtracted. For NO production, supernatants (50 μl) collected from pre-treated microglia were mixed with 50 μl of the sulfanilamide solution, 50 μl of NED solution and then incubated for 30 min at RT. The OD of reactants was measured at 520 and 540 nm using an ELISA plate reader (Model: SpectraMAX 190, Molecular Devices).
Immunocytochemistry
Pretreated microglia growing on poly-D-lysine-coated coverslips at a density of 0.5 × 106 per well in 24-well plates were fixed with 4% PFA for 30 min, followed by the addition of blocking/permeabilization solution (10% normal goat serum, 0.2% Triton X-100 and 0.1 M glycine) for additional 30 min. At the end point, primary Abs, including rat monoclonal Ab Mac-1 (CD11b; 1/500; Serotec) and rabbit polyclonal Ab Kv1.3 (1 : 100, Alomone Labs, Jerusalem, Israel), were added to coverslips for 1 h at RT. Cells were subsequently incubated with Alexa Fluor 488 and Alexa Fluor 594-conjugated secondary Abs (1 : 1000, Invitrogen; Molecular Probes) for 1 h after wash. ProLong Gold antifade reagent with DAPI (Molecular Probes) was used for neuron nuclei staining. Images were obtained by a Zeiss LSM 510 META NLO microscope in a 40 × oil-immersion objective. A minimum of five images were taken from each slides.
Western blot analyses
Membrane or total proteins were prepared using a Membrane Protein Extraction Kit (BioVision, Mountain View, CA, USA) or a RIPA Buffer (Bio-Rad, Hercules, CA, USA). For membrane protein preparation, microglia were lysed in a Homogenize Buffer Mix supplemented with protease Inhibitor Cocktail (1 : 500, BioVision) for 30 min on ice. The lysates were centrifuged in 700 × g for 10 min at 4°C and the supernatants were then transferred to new vials and centrifuged at 10 000 × g for 30 min at 4°C. The pellets, total cellular membrane proteins, were re-suspended in 0.5% TritonX-100 in PBS before use. For total proteins extraction, microglia or neurons were suspended in RIPA buffer containing protease inhibitor and centrifuged at 8000 × g for 10 min at 4°C. The clarified lysates were transferred to fresh tubes on ice for analysis. Membrane proteins of 20 μg and 20 μg total proteins were separated by electrophoresis on 4–15% Tris-HCl gels and transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% dry milk in Tris-Buffered Saline (TBS) (all from Bio-Rad Laboratories) and probed overnight at 4°C with primary Abs including rabbit polyclonal Kv1.3 (1 : 200; Alomone Labs, Israel), phospho-p38 MAPK and p38 MAPK (1 : 1000; Cell Signaling Technology, Danvers, MA, USA), anti-mouse β-actin monoclonal antibody (1 : 10 000, Sigma-Aldrich). Membranes were washed (four times, 10 min each) in TBS with 0.2% Tween (TBS-T) and incubated with HRP-conjugated anti-rabbit, or anti-mouse secondary antibody (1 : 10 000, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 h at RT. Labeled proteins were visualized by Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL, USA). Band densities of phospho-p38 were normalized to total p38 in each sample.
Cytokine arrays
Cytokine assays were performed using Cytokine Antibody Array1 (Rat Antibody Array I; RayBiotech, Inc., Norcross, GA, USA) according to the manufacturer's instructions. Membranes pre-coated with cytokine antibody were blocked by a blocking buffer at RT for 30 min, then equal volumes of supernatants, collected from microglia pretreated with gp120 (Immunodiagnostic, Inc., Woburn, MA, USA) in the presence or absence of 4-AP or TEA, were added to replace the blocking buffer and incubated over night at 4°C. After washing three times with wash buffer I and two times with wash buffer II, each membrane was incubated with biotin-conjugated antibodies for 2 h and followed by a HRP-conjugated streptavidin at RT for 30 min. Membranes were developed by Detection buffer C and D mixture and visualized by exposure of the array to X-ray film. Densitometry analysis of the array was performed using the NIH Image J software.
Reverse transcription-PCR
Total RNA was isolated from microglia using TRIzol Reagent (Invitrogen) and purified by RNeasy Mini Kit (QIAGEN, Inc., Valencia, CA, USA). Total RNA of 10 μg was reverse transcribed at 65°C for 50 min according to SuperScript III reverse transcriptase (Invitrogen). PCR amplification was conducted using a Gene-Amp PCR 2400 system (PerkinElmer, Toronto, Canada) with Platinum PCR SuperMix (Invitrogen). The PCR mixture was incubated for 2 min at 94°C, subjected to 30 cycles consisting of a 30 s denaturing phase at 94°C, a 30 s annealing phase at 55°C, a 1 min extension phase at 72°C, and a final extension phase of 10 min at 72°C. The resulting DNA products were resolved on 1.5% agarose gels containing 0.5 mg ml–1 ethidium bromide. Densitometry analysis of products was measured using the Northern Eclipse 6.0 software (Bio-Rad) and normalized to each internal control β-actin. PCR primers were as follows: Kv1.3: forward primer was 5′-GTACTTCGACCCGCTCCGCAATGA-3′; reverse was 5′-GGGCAAGCAAAGAATCGCACCAG. β-Actin: forward primer was 5′-GTGGGGCGCCCCAGGCACCA-3′; reverse was 5′-CTTCCTTAATGTCACGCACGATTTC-3′.
siRNA transfection
Pre-designed siRNA on target plus SMART pool against Rat KCNA3 (Kv1.3, NM-019270) mRNA was purchased by Dharmacon, Inc. (Chicago, IL, USA). Microglia at a density of 2 × 106 cell per well in 6-well plates were transfected with 100 μl of 2 μM siRNA for 48 or 72 h in the presence of Dharma FECT Transfection Reagent (Dharmacon, Inc.) according to the manufacturer's instructions. A nonspecific control siRNA, on-target plus GAPD control pool (rat) (Dharmacon, Inc.) was also transfected at the same concentration as the control. After transfection, microglia were stimulated with gp120 (500 pM) or without gp120 stimulation for 24 h, then supernatants were collected for TUNEL and MTT assay and microglia were harvested for preparation of RNA and protein.
Statistical analysis
Experimental data are expressed as mean±S.E. unless otherwise indicated. Statistical analyses were performed by Student t-tests. A minimum P-value of 0.05 was estimated as the significance level for all tests.
Abbreviations
- 4-AP:
-
4-aminopyridine
- CNS:
-
central nervous system
- HIV-1:
-
human immunodeficiency virus type 1
- gp120:
-
HIV-1 glycoprotein 120
- HAND:
-
HIV-1-associated neurocognitive disorders
- IFN-γ:
-
interferon-γ
- MgTx:
-
margatoxin
- SB:
-
SB203580
- TEA:
-
tetraethylammonium
References
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007; 69: 1789–1799.
Glass JD, Wesselingh SL . Microglia in HIV-associated neurological diseases. Microsc Res Tech 2001; 54: 95–105.
Dheen ST, Kaur C, Ling EA . Microglial activation and its implications in the brain diseases. Curr Med Chem 2007; 14: 1189–1197.
Wickenden A . K(+) channels as therapeutic drug targets. Pharmacol Ther 2002; 94: 157–182.
Judge SI, Bever Jr CT . Potassium channel blockers in multiple sclerosis: neuronal Kv channels and effects of symptomatic treatment. Pharmacol Ther 2006; 111: 224–259.
Flet L, Polard E, Guillard O, Leray E, Allain H, Javaudin L et al. 3,4-diaminopyridine safety in clinical practice: an observational, retrospective cohort study. J Neurol 2010; 257: 937–946.
Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD . K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci 2004; 25: 280–289.
Eder C . Ion channels in microglia (brain macrophages). Am J Physiol 1998; 275 (2 Part 1): C327–C342.
Norenberg W, Gebicke-Haerter PJ, Illes P . Voltage-dependent potassium channels in activated rat microglia. J Physiol 1994; 475: 15–32.
Fischer HG, Eder C, Hadding U, Heinemann U . Cytokine-dependent K+ channel profile of microglia at immunologically defined functional states. Neuroscience 1995; 64: 183–191.
Schilling T, Quandt FN, Cherny VV, Zhou W, Heinemann U, Decoursey TE et al. Upregulation of Kv1.3 K(+) channels in microglia deactivated by TGF-beta. Am J Physiol Cell Physiol 2000; 279: C1123–C1134.
Walz W, Bekar LK . Ion channels in cultured microglia. Microsc Res Tech 2001; 54: 26–33.
Farber K, Kettenmann H . Physiology of microglial cells. Brain Res Brain Res Rev 2005; 48: 133–143.
Gendelman H, Ding S, Nan G, Liu J, Ramirez S, Persidsky Y et al. Monocyte chemotactic protein-1 regulates voltage-gated K+ channels and macrophage transmigration. J NeuroImmune Pharmacol 2009; 4: 47–59.
Fordyce CB, Jagasia R, Zhu X, Schlichter LC . Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci 2005; 25: 7139–7149.
Nutile-McMenemy N, Elfenbein A, Deleo JA . Minocycline decreases in vitro microglial motility, beta1-integrin, and Kv1.3 channel expression. J Neurochem 2007; 103: 2035–2046.
Xu C, Liu J, Chen L, Liang S, Fujii N, Tamamura H et al. HIV-1 gp120 enhances outward potassium current via CXCR4 and cAMP-dependent protein kinase a signaling in cultured rat microglia. Glia 2011; 56: 997–1007.
Kawanokuchi J, Mizuno T, Takeuchi H, Kato H, Wang J, Mitsuma N et al. Production of interferon-gamma by microglia. Mult Scler 2006; 12: 558–564.
Suzuki Y, Claflin J, Wang X, Lengi A, Kikuchi T . Microglia and macrophages as innate producers of interferon-gamma in the brain following infection with Toxoplasma gondii. Int J Parasitol 2005; 35: 83–90.
Bachis A, Mocchetti I . The chemokine receptor CXCR4 and not the N-methyl-D-aspartate receptor mediates gp120 neurotoxicity in cerebellar granule cells. J Neurosci Res 2004; 75: 75–82.
Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL et al. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol 1998; 8: 595–598.
Hanisch UK . Microglia as a source and target of cytokines. Glia 2002; 40: 140–155.
Garden GA . Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 2002; 40: 240–251.
Kim SU, de Vellis J . Microglia in health and disease. J Neurosci Res 2005; 81: 302–313.
Kaul M, Garden GA, Lipton SA . Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 2001; 410: 988–994.
Makela J, Koivuniemi R, Korhonen L, Lindholm D . Interferon-gamma produced by microglia and the neuropeptide PACAP have opposite effects on the viability of neural progenitor cells. PLoS One 2010; 5: e11091.
Kotecha SA, Schlichter LC . A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci 1999; 19: 10680–10693.
Menteyne A, Levavasseur F, Audinat E, Avignone E . Predominant functional expression of Kv1.3 by activated microglia of the hippocampus after Status epilepticus. PLoS One 2009; 4: e6770.
Lee YB, Schrader JW, Kim SU . p38 map kinase regulates TNF-alpha production in human astrocytes and microglia by multiple mechanisms. Cytokine 2000; 12: 874–880.
Li Y, Liu L, Barger SW, Mrak RE, Griffin WS . Vitamin E suppression of microglial activation is neuroprotective. J Neurosci Res 2001; 66: 163–170.
Medders KE, Sejbuk NE, Maung R, Desai MK, Kaul M . Activation of p38 MAPK is required in monocytic and neuronal cells for HIV glycoprotein 120-induced neurotoxicity. J Immunol 2010; 185: 4883–4895.
Del Corno M, Liu QH, Schols D, de Clercq E, Gessani S, Freedman BD et al. HIV-1 gp120 and chemokine activation of Pyk2 and mitogen-activated protein kinases in primary macrophages mediated by calcium-dependent, pertussis toxin-insensitive chemokine receptor signaling. Blood 2001; 98: 2909–2916.
Lee C, Liu QH, Tomkowicz B, Yi Y, Freedman BD, Collman RG . Macrophage activation through CCR5- and CXCR4-mediated gp120-elicited signaling pathways. J Leukoc Biol 2003; 74: 676–682.
Stefoski D, Davis FA, Faut M, Schauf CL . 4-Aminopyridine improves clinical signs in multiple sclerosis. Ann Neurol 1987; 21: 71–77.
van Diemen HA, Polman CH, van Dongen MM, Nauta JJ, Strijers RL, van Loenen AC et al. 4-Aminopyridine induces functional improvement in multiple sclerosis patients: a neurophysiological study. J Neurol Sci 1993; 116: 220–226.
Shi R, Sun W . Potassium channel blockers as an effective treatment to restore impulse conduction in injured axons. Neurosci Bull 2011; 27: 36–44.
Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci USA 2006; 103: 17414–17419.
Vianna-Jorge R, Suarez-Kurtz G . Potassium channels in T lymphocytes: therapeutic targets for autoimmune disorders? BioDrugs 2004; 18: 329–341.
Goffe B, Papp K, Gratton D, Krueger GG, Darif M, Lee S et al. An integrated analysis of thirteen trials summarizing the long-term safety of alefacept in psoriasis patients who have received up to nine courses of therapy. Clin Ther 2005; 27: 1912–1921.
Esamai F, Tenge CN, Ayuo PO, Ong’or WO, Obala A, Jakait B . A randomized open label clinical trial to compare the efficacy and safety of intravenous quinine followed by oral malarone versus intravenous quinine followed by oral quinine in the treatment of severe malaria. J Trop Pediatr 2005; 51: 17–24.
Acknowledgements
We thank Ms Julie Ditter, Ms Robin Taylor and Ms Johna Belling for their excellent administrative supports. This work was supported by the NIH grants R01 NS041862-9, 2R56 NS041862 and 5 R01 NS063878 to HX.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by D Bano
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Liu, J., Xu, C., Chen, L. et al. Involvement of Kv1.3 and p38 MAPK signaling in HIV-1 glycoprotein 120-induced microglia neurotoxicity. Cell Death Dis 3, e254 (2012). https://doi.org/10.1038/cddis.2011.140
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2011.140
Keywords
This article is cited by
-
Progress in Pathological and Therapeutic Research of HIV-Related Neuropathic Pain
Cellular and Molecular Neurobiology (2023)
-
Microglial diversity along the hippocampal longitudinal axis impacts synaptic plasticity in adult male mice under homeostatic conditions
Journal of Neuroinflammation (2022)
-
HIV-Proteins-Associated CNS Neurotoxicity, Their Mediators, and Alternative Treatments
Cellular and Molecular Neurobiology (2022)
-
Co-receptor signaling in the pathogenesis of neuroHIV
Retrovirology (2021)
-
NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology
Cellular & Molecular Immunology (2020)
