
Abstract
The question of how neural progenitor cells maintain its self-renewal throughout life is a fundamental problem in cell biology with implications in cancer, aging and neurodegenerative diseases. In this work, we have analyzed the p73 function in embryonic neural progenitor cell biology using the neurosphere (NS)-assay and showed that p73-loss has a significant role in the maintenance of neurosphere-forming cells in the embryonic brain. A comparative study of NS from Trp73−/−, p53KO, p53KO;Trp73−/− and their wild-type counterparts demonstrated that p73 deficiency results in two independent, but related, phenotypes: a smaller NS size (related to the proliferation and survival of the neural-progenitors) and a decreased capacity to form NS (self-renewal). The former seems to be the result of p53 compensatory activity, whereas the latter is p53 independent. We also demonstrate that p73 deficiency increases the population of neuronal progenitors ready to differentiate into neurons at the expense of depleting the pool of undifferentiated neurosphere-forming cells. Analysis of the neurogenic niches demonstrated that p73-loss depletes the number of neural-progenitor cells, rendering deficient niches in the adult mice. Altogether, our study identifies TP73 as a positive regulator of self-renewal with a role in the maintenance of the neurogenic capacity. Thus, proposing p73 as an important player in the development of neurodegenerative diseases and a potential therapeutic target.
Similar content being viewed by others


Main
The mammalian neocortex develops from a pseudostratified epithelium composed by neuroepithelial cells.1 During development these cells and their derivative progenitor will give rise to neurons, astrocytes and oligodendrocytes while maintaining their self-renewal capacity. During development of the mammalian central nervous system (mCNS), the self-renewal of neural stem cells (NSCs) and progenitors occurs either by symmetric cell divisions, which generate two undifferentiated cells with the same fate,2 or by asymmetric divisions, giving rise to one progenitor and one cell that differentiates into a neuron.3 NSCs are multipotential and self-renewing, whereas neural progenitor cells (NPC) have more restricted renewal and differentiation ability.4 Here, the term NPC will be used to include both neural stem and progenitor cells. Self-renewal and cell fate choice of NPC are coordinately controlled in a stage-dependent manner, but the mechanisms underlying such coordination remain poorly understood. Signals that regulate stem cell maintenance and fate specification will influence the final number of neurons and glia formed during mCNS development.5 Such signals would be determinant for the appropriate architecture of the neurogenic-niches in the adult brain and thus, the maintenance of the neurogenic capacity.
In adult mammalian brain NSCs are localized in two regions: the subventricular zone (SVZ) and the subgranular-cell layer zone (SGZ) of the dentate-gyrus in the hippocampus. NPC from the embryonic and mature mCNS can be propagated in vitro as clonal aggregates denoted neurospheres (NS).6, 7 The NS assay represents a serum-free, selective culture in which most cells within the differentiation process rapidly die, whereas NPC respond to mitogens, divide and form NS that can be dissociated and replated to generate secondary spheres.8 In a similar way, NPC from olfactory bulb (OB) of E14.5 embryos have been demonstrated to be derived from the neuroepithelium, and form NS that preserve their self-renewal ability and multipotency.9, 10
Some of the pathways that are necessary for self-renewal appear to regulate processes like proliferation, apoptosis or differentiation.2, 11 In somatic cells, p53-family members are deeply involved in the regulation of these processes. This family is constituted by the transcription factors p53, p73 and p63. The dual nature of these genes resides in the existence of TA and DN variants. The TA-proteins are transactivation competent, inducing cell cycle arrest, apoptosis, senescence and differentiation.12 Oppositely, the DN isoforms lack the transactivation domain and can act as dominant-negative repressors of p53 and TAp73, abrogating their ability to induce growth suppression in many cell types.13, 14 This family has been implicated in regulation of the survival and maintenance of CNS mature neurons and neuronal precursors, with p53 functioning as a cell death protein, and DNp73 and DNp63 as major survival proteins preventing apoptosis in post-mitotic neurons and embryonic cortical precursors, respectively.15, 16, 17 Deficiency of p73 in the Trp73−/− mice, which lacks all p73 isoforms, leads to multiple neurological abnormalities including hippocampal dysgenesis,18 denoting the relevance of p73 function in neural development and suggesting a possible role in neurogenesis. However, the role of p73 in the biology of NPC has never been addressed.
In this work, we analyzed p73 function in embryonic NPC biology using the NS assay, and demonstrate that p73 is a positive regulator of self-renewal in a p53-independent manner. This self-renewal impairment is later on reflected in a developmental retardation of the neurogenic niches and a significant depletion of the precursor pool of the SVZ and SGZ of P15-Trp73−/− mice, ultimately resulting in defective neurogenic niches.
Results
Loss of p73 impairs cellular self-renewal
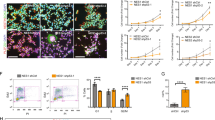
To address whether TP73 ablation affected the biology of embryonic NPC, we used the NS assay.7, 19 We utilized primary NS cultures obtained from OBs of Trp73−/− and WT E14.5 embryos. NPC in vivo are positive for the neuroepithelial-marker Nestin.20 In our in vitro assay, OBs cultures in the presence of mitogens form NS that express Nestin (Figure 1a). Analysis of p73-isoforms expression revealed that both, TA and DNp73, were expressed under proliferating conditions, with TAp73 being significantly more abundant than DNp73 (Figure 1b). These cell cultures were successively dissociated and reseeded at clonal density generating new NS capable of producing a differentiated progeny (Figure 1c, Supplementary Figure 1A), demonstrating their self-renewal and multipotency capacities. Moreover, TAp73 expression is upregulated during the differentiation process (Supplementary Figure 1B). The frequency of primary neurosphere-forming cells (NFC) represents the content of NPC in a given tissue.21, 22, 23 We observed that four times more cells were required to form primary NS from the Trp73−/− neurogenic tissue, than from WT (1/125 cells in WT (0.8%) versus 1/500 cells in Trp73−/− (0.2%), Supplementary Figure 1C), suggesting that lack of p73 produces a depletion of NFC in the neurogenic tissue. Interestingly, when we compared NS cultures, we consistently observed a decrease in the size and number in the Trp73−/− (named p73KO from now on) NS in 60% of the analyzed embryos (Figure 1c–e). The diameter of p73KO-NS was significantly reduced compared with that of WT cells (Figures 1c and d). To rule out the possibility that the observed phenotype was limited to a defect in the transient amplifying (TA) population, we performed long-term population analysis, for up to 10 passages, under clonal cultures conditions,8 and the size of the newly formed NS was evaluated at each subculturing step. The phenotype observed in the p73KO-NS was maintained, and even intensified, through the successive passages, indicating that the effect of p73 loss was not merely affecting the TA population, but rather the pool of NPC, the only capable of generating new NS (Figure 1c, NS-P8). The NS size reduction suggests that the net cell growth within the NS, which reflects the sum of cell divisions from self-renewing NSC and NPC, was impaired.

p73 deficiency impairs cellular self-renewal and increases cell death without affecting cell cycle progression. (a–f): Successive passages of E14.5 neurospheres of the indicated genotype cultured under clonal proliferation conditions. (a) Confocal microscopy images (10 × ) of NS of the indicated passage immunostained with anti-Nestin antibody. (b) qRT-PCR of TA- and DNp73 expression in WT-NS. (c) Bright-field photographs (10 × ) of NS passages (P2-P8). (d) Graphical representation of NS size (P2). (e) NS self-renewal assay. Quantification of new NS was performed after 4DIV at the indicated passages. We assigned 100% to the number of WT NS. (f) NS (P2) cellular kinetics after 5DIV. (g) FACS cell cycle analysis determined by propidium iodide (PI) staining, (h) qRT-PCR analysis of p21Cip1 and Bmi1expression, (i) cell death measured by the Annexin-V binding assay and (j) qRT-PCR analysis of Noxa in P2-NS cultures under proliferating conditions. Representative histograms and percentages from triplicate experiments are shown. The analyses were performed with data from two independent experiments with at least three embryos of each genotype (*P<0.05, **P<0.01, ***P<0.005)
The formation of new NS after each successive passage has been widely used as a quantitative measure of the self-renewing activity of neural stem cells.7, 24, 25 We used this in vitro assay to examine how p73 loss affects this parameter in our cultures. To that purpose, dissociated tissue cells were seeded under serial clonal dilution and the number of NS formed in each passage was quantified. We arbitrarily assigned 100% to the NS formed in the WT cultures. We observed a highly significant decrease in the number of NS formed in p73KO cultures (Figure 1e). This difference was maintained in consecutive passages. The decrease in the number and size of newly formed NS in each passage in the p73KO cultures suggests a successive loss of NFC, and signals p73 as a positive regulator of NPC self-renewal.
To further analyze the overall growth of the p73KO cells, we determined ratios of cell production after 5 days in vitro (DIV) relative to number of cells plated. Growth ratios in the p73KO cultures were significantly lower than in WT (Figure 1f). Analysis of cellular proliferation and death rates demonstrated that p73 loss affected the net cell growth of the culture, without altering the cell cycle progression (Figure 1g). Consistently, there were no significant differences in the expression levels of the cell cycle inhibitor and negative regulator of self-renewal, p21Cip1, neither in the expression of the positive self-renewal regulator and promoter of cell proliferation, Bmi1 (Figure 1h). We detected an increase in cell death (Figure 1i). Accordingly, levels of the proapoptotic p53 target gene Noxa were significantly upregulated in p73KO cultures (Figure 1j).
p73 regulates self-renewal independently of p53, but it is necessary for the enhanced self-renewal caused by p53 deficiency
The role of p73 in the developing nervous system has been defined on the basis of DNp73 function as an antiapoptotic protein that antagonizes p53 proapoptotic actions, and potentially TAp63.15, 16, 17 As the Trp73−/− mice lack all the isoforms,18 the absence of the pro-survival DNp73 function could result in a compensatory p53 activation, which may account for the enhanced apoptosis as well as Noxa enhanced expression in the p73KO cells, and even the impaired self-renewal observed in the p73KO-NS cultures. To address this issue, we analyzed the role of p53 in NS cultures, in the context of p73 deficiency, using the double mutant: p53KO; Trp73−/−mice (named DKO from now on). We observed that, although p53KO cultures presented larger NS (Figures 2a and b, P<0.0005), reflecting its enhanced self-renewal,26, 27 and the p73KO-NS were smaller (P<0.0005), the DKO cells generate NS with a size closer to WT-NS. Up to this point, the data suggested that the reduced size in p73KO-NS could be indeed because of a p53-compensatory activation, as elimination of p53 almost rescues the size phenotype. Nevertheless, the DKO NS were considerably smaller than those formed by p53KO cells (P<0.0005, Figures 2a and b), indicating that p73 loss hampered the effect of p53 deficiency, and implying that p73 function was necessary for the enhanced growth of the p53KO-NS. Cell cycle analysis of these cultures revealed that in DKO cells p53 deficiency significantly inhibited the basal apoptosis detected in the p73KO cultures, whereas enhancing cellular proliferation (significant increase in S-phase and decrease in G1, compared with WT and p73KO) (Figure 2c). Consistently, p21Cip1 and Noxa expression were repressed in the DKO cells, demonstrating that the regulation of these genes in NPC was p53 dependent (Figure 2d). Furthermore, the mitotic index of clonally formed NS quantified by anti-phospho-Histone-3 staining supported these results, as p53KO and DKO-NS had a significantly elevated mitotic index than WT, whereas the p73KO-NS had not (Figure 2e and f). It is important to underline that in the absence of p53, lack of DNp73 does not result in apoptosis. However, despite the enhanced proliferation rate and low apoptosis of DKO cultures, they showed an overall growth rate similar to the p73KO-NS, failing to attain p53KO growth rate (Figure 2g). This data reinforces the idea that p73KO effect in NPC was not simply due to a decrease in the proliferation rate or enhanced apoptosis.

p73 regulates self-renewal independently of p53, but it is necessary for the enhanced self-renewal observed in p53KO NS cultures. (a–h): NS (P2) cultures of the indicated genotypes under proliferating conditions. (a) Bright-field microphotography (10 × ) and (b) diameter quantification, (c) FACS-PI cell cycle analysis, (d) qRT-PCR of p21Cip1 and Noxa expression, (e) confocal microscopy images (20 × ) of p-His-3 immunostaining and (f) its quantification. (g) Cellular kinetics of NS (P2) of the indicated genotype after 5DIV. (h) Self-renewal assay: NS (P2) cells were seeded at different clonal cell densities in proliferating media from 0.6–5 cells per μl, and the formed NS were counted after 4DIV. The Trp73−/−, p53+/− littermates showed the same phenotype as the Trp73−/− mice (data not shown). The analyses were performed with data from two independent experiments with at least three embryos of each genotype (*P<0.05, **P<0.01, ***P<0.005)
The in vitro self-renewal assay revealed that p53KO cells formed significantly more NS than their WT counterparts, whereas p73KO cultures had significantly less NS, in all conditions analyzed (Figure 2h). However, p73 loss in the DKO cells abrogated the effect of p53 deficiency, as the number of NS formed in the DKO was similar to those of WT cultures, but significantly less than the observed in the p53KOs with functional p73. This was surprising, as NS obtained from the OB of p53KO mice embryos are enriched in NFC and have enhanced self-renewal.26 Therefore, our data indicates that these features in p53KO cultures were dependent upon p73 functional integrity.
p73 deficiency results in premature neuronal differentiation under proliferating conditions and faster kinetics of the differentiation process
Self-renewal and differentiation are cardinal features of stem cells. A tight regulation of the facultative use of symmetric versus asymmetric divisions by stem cells is crucial for maintaining self-renewal capacity. It has been documented that under proliferating culture conditions, NSCs in vitro are fast dividing Nestin-positive cells that clonally give rise to great numbers of Nestin-positive NPC and divide symmetrically to produce equivalent progeny.28 As shown in Figures 3a and b, p73 deficiency resulted in a reduction of Nestin-positive cells in NS under proliferating conditions, suggesting that these NS contained a reduced number of undifferentiated NPCs. Intriguingly, a closer observation of p73KO and DKO cells (Figure 3c) revealed pairs of cells that resembled daughter cells with different nuclei size and asymmetric distribution of Nestin (p73KO: 1.4%±0.002 and DKO: 4.1%±0.16, P≤0.003 and P≤0.02 compared with WT, respectively). However, WT and p53KO cultures presented few asymmetric cell pairs (0.6% ±0.009 and 0.5% ±0.1, respectively), consistent with the notion that under proliferating culture conditions, WT-NPC divide symmetrically. Thus, impaired NS formation in the p73KO cells could reflect fewer symmetric divisions in these cells.

p73 deficiency results in premature neuronal differentiation under proliferating conditions. (a–e): NS (P2) cultures of the indicated genotypes under proliferating conditions (PM). (a) Confocal microscopy images (60 × ) immunostained with the antibodies: Nestin (a–c), Tuj1-TxRed (neurons) and p-His-3-FITC (mitotic marker) (d). (b) Quantification of Nestin-positive cell in (a). (c) Magnification of boxes marked in (a) showing different nuclei size and asymmetric Nestin distribution. The p53KO pair depicted represents a symmetric distribution with similar nuclei size. (e) Quantification of Tuj1+ cells in (d). The analyses were performed with data from two independent experiments with at least three embryos of each genotype (**P<0.01, ***P<0.005)
Increasing the number of asymmetric divisions would lead to a decrease in the pool of undifferentiated NPC and to an increase in the number of differentiated cells, with the concomitant impairment of self-renewal, whereas promoting a symmetric mode of division, like in p53KO cultures,26 would result in enhanced self-renewal by keeping the NPC in an undifferentiated state. In accordance with this model, the analysis of the spontaneous neuronal differentiation in NS after 3DIV under proliferating conditions revealed that the p73KO and DKO NSs frequently contained multiple Tuj-1-positive cells (Figure 3d, Supplementary Figure 2A). These cells showed various long and branched neurite extensions, congruent with a more mature neuronal phenotype (Supplementary Figure 2B). This was surprising, as the clonally newly-formed NS were kept under proliferating conditions and analyzed after only 3DIV, conditions at which the WT and p53KO NSs showed seldom Tuj-1-positive cells. Furthermore, few Tuj-1-positive cells detected in the latter cultures had short lamellipodia or short unbranched processes. This data indicates that p73 deficiency was prompting a premature neuronal differentiation and thus hindering the maintenance of the embryonic NPC undifferentiated population.
Our data suggest, but does not conclude, that p73 deficiency may lead to a bias towards asymmetric cell divisions, increasing the population of NPC, ready to differentiate into neurons, and thus exhausting the pool of undifferentiated NFC. If this hypothesis is correct, we would expect an enhanced rate and a faster kinetics of the differentiation process in p73KO-NS grown under conditions supporting progressive neuronal differentiation. Under these conditions,7 WT and p73KO cells differentiated into astrocytes and neurons after 8DIV, and oligodendrocytes were detected after 10DIV (Supplementary Figures 3A and 3B) with no significant differences in the total Tuj1+ cells after 5DIV or 8DIV (Figure 4b upper panel). However, despite the identical conditions, we found a significantly higher percentage of large branching neurons (Tuj1+ cells with long neurite extensions (Tuj1+ NE)) at all the time points analyzed in the p73KO-NS, suggesting an advanced stage in the differentiation process, (Figure 4a and b; lower panel and Supplementary Figure 3C). This supports the idea that p73 deficiency induces a premature onset of differentiation. When we compared the differentiation potential of p73KO, p53KO, DKO or WT-NS after 3- or 5DIV with differentiation media, we observed that either p53- or p73-deficient NS generated higher number of Tuj1+-NE cells compared with WT, confirming that p73 and p53 single-deficiency results in premature neuronal differentiation. Nevertheless, the combined deficiency in the DKO-NSs produced more neurons with mature phenotype than p53 or p73 single loss (Figures 4c and d), suggesting an additive effect. It is important to highlight that p53 control of neurogenesis and gliogenesis has been proposed to be independent of its regulatory pathways of self-renewal.29 We analyzed if there was a cross-talk in the regulation of cell decision fate between p73 and p53. Strikingly, although lack of p53 hinders astrocytic differentiation, DKO cells showed a stronger GFAP staining and a mature astrocytic phenotype in a number similar to WT or p73KO cell, but with significantly higher number of differentiated cells than the p53KO cultures (Figure 4d), suggesting that absence of p73, rescues p53 uncoupling of neuronal fate. At a further stage of differentiation, p73KO and DKO cells displayed neurons with a clear mature phenotype with multiple branched long neurites (Figure 4e), whereas the Tuj1+-WT, and even the p53KO, displayed yet an immature phenotype, corroborating that p73 deficiency resulted in enhanced rate and faster kinetics of the differentiation process independently of p53.

Lack of p73 results in enhanced rate and faster kinetics of the differentiation process. (a–e): NS (P2) cultures at the indicated DIV under differentiating conditions (DM) immunostained with the antibodies: Tuj1-TxRed (neurons) or GFAP-FITC (astrocytes). (a) Confocal microscopy images (40 × ) and (b) quantification of Tuj1+ cells in (a) either total Tuj1+(b, upper panel) or with neurite (Tuj1+-NE) extensions four times bigger than the cell body diameter (b, lower panel). (c) Confocal microscopy images (40 × , upper panel and 20 × , lower panel) and (d) quantification of Tuj1+-NE and GFAP-positive cells in (c). (e) Confocal microscopy images (60 × ). The analyses were performed with data from three independent experiments with at least three embryos of each genotype and representative images are shown (*P<0.05, **P<0.01, ***P<0.005)
If lack of p73 produces a premature differentiation and, in this way, depletes the pool of embryonic NPC during development, it is reasonable to expect that p73 deficiency will result in defective neurogenic-niches in the adult mice. To address this, we scored the 5-bromo-2-deoxyuridine (BrdU)-positive cells, which identify proliferating NSC and NPC in the SVZ, and found a significantly lower number of proliferating cells in Trp73−/− mice (Figures 5a and c). Quantification of p-His-3-positive cells in the same region confirmed this result (Figures 5b and d). Moreover, assessment of p-His-3-positive cells in the SGZ also showed a severe depletion of the proliferating compartment in Trp73−/− mice (Figures 5e and f). A morphological analysis of the neurogenic-niches at electronic microscopy level exposed profound abnormalities in the architecture along the SVZ of the Trp73−/− mice. Assessment of the different proliferating cellular subpopulations of the SVZ – astrocyte-like type-B cells (NSC), type-C cells (transit amplifying progenitors) and type-A (neuroblast)30 – confirmed our BrdU data, revealing that p73 deficiency resulted in a reduction of type-C and A cells (Figures 5g and h). Furthermore, type-B cells, as well as type-A and -C cells, presented an immature morphology similar to those in newborn mice; however, the number of type-B cells was comparable with WT.

Lack of p73 delays the development of the SVZ and SGZ and depletes the number of proliferating cell population. (a–f): Coronal sections (10 × ) of the SVZ (a–d) or SGZ (e and f) of P15 mice brains of the indicated phenotype, immunostained with antibodies against anti-BrdU (after repetitive pulses over 8 h period) (a and c), or anti-p-Histone-3 (b, d and f) and the positive cells scored (c, d and f). Magnifications of the area included in the white squares in e are depicted in the inserts (100 × ). (g) Panel of the cellular analysis of the Trp73−/− and WT mice by electron microscopy of SVZ showing the differential morphology of Type B (B), Type C (C), Type A (A) and: Ependymal cells (E). In the p73KO the cells corresponding to C and A are not marked as such as they both look alike with morphological features corresponding to more immature cells. (h) Bar graph of the average number of SVZ Type A/C cells identified per unit length in WT and Trp73−/− mice (*P<0.05, **P<0.01, ***P<0.005)
Discussion
Learning about the mechanisms that regulate the coordinated control of proliferation and differentiation of tissue-specific stem cells is critical to understand development, cancer and neurodegeneration. In this work, we have analyzed the effect of p73 deficiency, and its functional interaction with p53, in embryonic NPC using the Ns assay. Our data reveal that p73 loss results in a depletion of the cells capable of forming NS (NSC and NPC) contained in the neurogenic tissue of the embryos. Furthermore, this effect was maintained in vitro where lack of p73 resulted in impaired net cell growth, decreased capacity to form new NS under clonal conditions (self-renewal) and enhanced apoptosis without altering the cell-cycle progression.
The role of p73 in the developing nervous system has been defined as a rheostat for survival versus death.15, 16, 17 As the Trp73−/− mice lack all p73 isoforms, it was possible that an exacerbated p53 activity, due to the lack of the prosurvival DNp73 function, could be accounted for the enhanced apoptosis and the impaired self-renewal observed in p73KO-NS. In apparent agreement with this view, the elevated apoptotic rates of the p73KO-NS disappeared in the DKO cultures, demonstrating that, indeed, the apoptosis observed in the p73KO cells was p53 dependent, probably through the regulation of target genes like Noxa. Hence, we confirm that p53 negatively regulates NSC self-renewal through the regulation of proliferation and survival. In this context, DNp73 is required to downregulate p53 activation of these pathways and its absence results in elevated apoptosis. Surprisingly, our data indicated that the NFC enrichment and the enhancement of the self-renewal capacity of the p53-deficient NPC, were dependent upon p73 functional integrity. Therefore, lack of p73 results in two independent, but related, phenotypes: a smaller neurosphere size (related to the proliferation and survival of the NPC) and a decreased capacity to form new NS (self-renewal). The former seems to be the result of p53 compensatory activity, whereas the latter is p53 independent and not because of a defect in cell cycle or apoptosis regulation.
The reduction of Nestin-positive cells in p73KO and DKO-NS, and the observation that p73-deficient NS growing under proliferative conditions contained multiple Tuj1+ cells suggested a defective maintenance of the undifferentiated state, which is central to stem/progenitor cells. This, together with the decrease of newly formed NS in each passage in p73KO and DKO cultures, directed us to hypothesize that p73 deficiency may lead to a bias towards asymmetric cell divisions and, in this way, impair self-renewal. The role of p53 in the regulation of self-renewal divisions has been widely documented,31 however, there was no data on the possible function of p73 in this process. However, very recent reports have been published32, 33, 34 that are in-line with our results, providing an independent confirmation of p73 involvement in neuronal stemness.
Our differentiation studies demonstrated that loss of p73 did not abolish NS multipotency, but rather accelerated the appearance of Tuj1+ cells with long and branched neurite extensions, congruent with a mature neuronal phenotype. These data connote that p73 deficiency is releasing the constraints that keep the embryonic NPC undergoing symmetric, proliferative division, thus, hampering their self-renewal and producing a premature differentiation. Although p73 functional integrity seem to be required for the enhancement of the self-renewal capacity of p53-deficient NPCs, the effect of p53 and p73 deletion over premature differentiation seems to be additive. Therefore, p73 and p53 regulation of self-renewal and differentiation should imply independent, yet interconnected pathways. This is supported by the observation that p53 deficiency on cell fate determination is restored by p73 depletion in DKO cells, implying a cross-talk between these genes in the regulation of this process. Altogether, our work has revealed a novel p53-independent function of p73, which regulates NPC self-renewal through the regulation of cellular differentiation and proposes that this regulation is at the base of p73 function in neuronal development.
Alterations in the mechanism that promotes or inhibits the differentiation of the embryonic NPC, like p73 deficiency does, would eventually deplete the pool of embryonic NSC, rendering deficient neurogenic-niches in the adult mice. Supporting this hypothesis, the Trp73−/− and TAp73−/− mice models exhibited profound hippocampal dysgenesis, demonstrating that p73, and TAp73 in particular, was critical for the development and maintenance of the hippocampus, one of the neurogenic NSC pools.35 Our data reveals that p73 deficiency slows down the development of both the SVZ and the SGZ niches, depletes the number of proliferating cells, and thus, renders deficient neurogenic-niches in the adult mice. Impairment in neurogenesis may compromise the extent of plasticity of the hippocampus and their associated neural circuits leading to enhanced neuronal vulnerability and functional detriment. Recent evidence suggests that neurogenesis is impaired in animal models of Alzheimer's disease (AD), in both subventricular and subgranular zones.36 In AD, compromised neurogenesis has been proposed to take place earlier than the onset of hallmark lesions or neuronal loss, and may have a role in the initiation and progression of the disease. In this context, p73 loss could become a risk factor for these ailments. This is consistent with reports indicating that p73 is essential to prevent neurodegeneration during aging, and that p73 heterozygosity may be a susceptibility factor for AD.17 Thus, p73 could be an important player in the development of neurodegenerative diseases and therefore a relevant target for study.
Materials and Methods
Mice husbandry and dissection of brain tissues
Animal experiments were conducted in agreement with European and Spanish regulations for the protection of experimental animals (Council Directive 86/609/CEE and RD-1201/2005, respectively) with the appropriate institutional committee approval. Mice heterozygous for Trp73 on a mixed background C57BL/6 × 129/svJae18 were backcrossed to C57BL/6, at least five times, to enrich for C57BL/6 background. To obtain the double Trp73; p53 Knockout mice (DKO), heterozygous animals for Trp73 were crossed with p53 KO mice;37 then, Trp73+/−; p53+/− mice were inter-crossed to obtain the DKO animals. Mice were genotyped as described before.18, 38
In vivo analysis
Perfusion and BrdU incorporation analysis in postnatal 15-day-old mice (P15): BrdU (Sigma-Aldrich, St. Louis, MO, USA) was administered at 150 mg/kg intraperitoneally in pulse injections every 2 h during 8 h. Animals were anesthetized with ketamin/medetomidin before transcardial perfusion. Animals were perfused with PBS, 0.1 M and subsequently with 4% paraformaldehyde (Merck, Darmstadt, Germany). After perfusion, brains were dissected, post-fixed for 24 h in 4% PFA solution at 4°C, and immersed in 30% sucrose. At this point, tissues were stored at −80°C. Coronal sections of 30 μm were obtained using a Microm HM-450 sliding microtome, and p-His-3 and BrdU immunohistochemistry (IHC) was performed as described before.27
Electron microscopy analysis: P15 mice were perfused with 0.9% PBS, 1% heparin, followed by 2% paraformaldehyde/2.5% glutaraldehyde solution (PFA/GA, Electron Micoscopy Sciences, Hatfield, PA, USA) and post-fixed in PFA/GA overnight. Sections of 50 μm were cut on a vibratome (Leica VT-1000, Wetzlar, Germany), embedded in araldite (Durcupan, Fluka, Hatfield, PA, USA). Semi-thin sections (1.5 μm) were cut with a diamond knife and stained lightly with 1% toluidine blue. The block with semi-thin sections was cut in ultra-thin (0.05 μm) sections with a diamond knife, stained with led citrate and examined under a Fei Tecnai spirit electron microscopy. Pictures were taken with Soft Image System (Morada, Olympus, Tokyo, Japan) camera. For quantification purposes, three mice for each group were used and serial sections corresponding to the same SVZ region (separated by 250 μm) were chosen and examined under a FEI Tecnai Spirit electron microscopy using established protocols and criteria.30
Primary culture of NPCs
To initiate each independent embryonic culture, the OBs of at least three different E14.5 embryos were dissected out, and mechanically dissociated, as previously described.10 Cells were seeded (2 × 104 cells/ml) in NS complete medium (Supplementary Table 1). For each passage, spheres formed after 4 days in vitro (DIV) were enzymatically dissociated with Accutase (Stem Cell Technologies, Vancouver, BC, Canada) and reseeded in complete medium. Cellular viability was assessed by trypan blue exclusion. To determine the size and number of NS, cells were seeded at clonal density (2 cells per μl). For clonogenic and limiting dilution assays, NS were disaggregated and seeded in flat bottom 96-well plates at log2 dilutions with a cell concentration from 500–1 cell per well. After four DIV the number of NS was determined (three independent experiments with at least three embryos of each genotype for each one). Statistical analyses were performed using the Student's two-tailed t-test.
Differentiation assays were performed seeding the NS in 15 mm coverslips covered with poli-L-ornitine (Sigma), in differentiation medium (Supplementary table 1) and incubated for 3, 5 or 8 days.
Flow cytometry assays
Cell cycle analysis was performed as described before26 using single cell suspensions obtained from enzymatically disaggregated NS. Samples were analyzed by flow cytometry in a CyAn ADPy (Beckman Coulter, Brea, CA, USA), and data were analyzed using Summit v4.3 (Dako, Glostrup, Denmark). For Annexin-V Binding Assay, NS were enzymatically disaggregated and labeled according to the manufacturer's instructions (BD, San Jose, CA, USA).
RNA isolation and real-time RT-PCR analysis
Total RNA from P2 NS was extracted with TRI reagent (Ambion, Austin, TX, USA) and cDNA was prepared using SuperScript II First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The expression of cell cycle and apoptosis markers was detected by real-time PCR in a StepOnePlus real-time PCR system (Applied Biosystems, Carlsbad, CA, USA) using FastStart Universal SYBR Green Master (ROX) (Roche, Basel, Switzerland). Primer sequences were obtained from Primer bank Database39, 40 (http://pga.mgh.harvard.edu/primerbank/index.html) and are indicated in the Supplementary Table 2. The PCR conditions were previously described.39, 40
Immunocytochemistry
IHC was performed as described before26 using the following antibodies: polyclonal-GFAP (1:400, Neomarkers, Labvision, Fremont, CA, USA), monoclonal-GFAP (1:100, Millipore, Billerica, MA, USA), III-β-Tubulin/Tuj-1 (1:1000, Covance, Princeton, NJ, USA), O4 (1:50), phospho-Histone-3 (1:100 Millipore) or Nestin (1:100, BD, Billerica, MA, USA). Images were obtained with Nikon EclipseTE2000 confocal microscope. Statistical analysis (Student's two-tailed t-test) was performed using triplicates from three independent experiments.
Abbreviations
- NSC:
-
neural stem cells
- NPC:
-
neural progenitor cells
- NS:
-
neurospheres
- NFC:
-
neurospheres-forming cells
- CNS:
-
central nervous system
- SVZ:
-
subventricular zone
- SGZ:
-
subgranular-cell layer zone
- OB:
-
olfactory bulb
- DIV:
-
days in vitro
- AD:
-
Alzheimer's disease
- BrdU:
-
5-bromo-2-deoxyuridine
References
Gotz M, Huttner WB . The cell biology of neurogenesis. Nat Rev Mol Cell Biol 2005; 6: 777–788.
Molofsky AV, Pardal R, Morrison SJ . Diverse mechanisms regulate stem cell self-renewal. Curr Opin Cell Biol 2004; 16: 700–707.
Wodarz A, Huttner WB . Asymmetric cell division during neurogenesis in Drosophila and vertebrates. Mech Dev 2003; 120: 1297–1309.
Seaberg RM, van der Kooy D . Stem and progenitor cells: the premature desertion of rigorous definitions. Trends Neurosci 2003; 26: 125–131.
Edlund T, Jessell TM . Progression from extrinsic to intrinsic signaling in cell fate specification: a view from the nervous system. Cell 1999; 96: 211–224.
Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A . EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron 2002; 36: 1021–1034.
Reynolds BA, Weiss S . Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 1996; 175: 1–13.
Vescovi AL, Galli R, Reynolds BA . Brain tumour stem cells. Nat Rev Cancer 2006; 6: 425–436.
Vergano-Vera E, Yusta-Boyo MJ, de Castro F, Bernad A, de Pablo F, Vicario-Abejón C . Generation of GABAergic and dopaminergic interneurons from endogenous embryonic olfactory bulb precursor cells. Development 2006; 133: 4367–4379.
Vicario-Abejon C, Yusta-Boyo MJ, Fernandez-Moreno C, de Pablo F . Locally born olfactory bulb stem cells proliferate in response to insulin-related factors and require endogenous insulin-like growth factor-I for differentiation into neurons and glia. J Neurosci 2003; 23: 895–906.
Kokovay E, Shen Q, Temple S . The incredible elastic brain: how neural stem cells expand our minds. Neuron 2008; 60: 420–429.
Collavin L, Lunardi A, Del Sal G . p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ 2010; 17: 901–911.
Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S . Possible oncogenic potential of DeltaNp73: a newly identified isoform of human p73. Cancer Res 2002; 62: 636–641.
Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M et al. DeltaNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J Exp Med 2002; 196: 765–780.
Dugani CB, Paquin A, Fujitani M, Kaplan DR, Miller FD . p63 antagonizes p53 to promote the survival of embryonic neural precursor cells. J Neurosci 2009; 29: 6710–6721.
Jacobs WB, Kaplan DR, Miller FD . The p53 family in nervous system development and disease. J Neurochem 2006; 97: 1571–1584.
Wetzel MK, Naska S, Laliberté CL, Rymar VV, Fujitani M, Biernaskie JA et al. p73 regulates neurodegeneration and phospho-tau accumulation during aging and Alzheimer's disease. Neuron 2008; 59: 708–721.
Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000; 404: 99–103.
Gritti A, Frölichsthal-Schoeller P, Galli R, Parati EA, Cova L, Pagano SF et al. Epidermal and fibroblast growth factors behave as mitogenic regulators for a single multipotent stem cell-like population from the subventricular region of the adult mouse forebrain. J Neurosci 1999; 19: 3287–3297.
Lendahl U, Zimmerman LB, McKay RD . CNS stem cells express a new class of intermediate filament protein. Cell 1990; 60: 585–595.
Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev 2005; 19: 1438–1443.
Martens DJ, Tropepe V, van Der Kooy D . Separate proliferation kinetics of fibroblast growth factor-responsive and epidermal growth factor-responsive neural stem cells within the embryonic forebrain germinal zone. J Neurosci 2000; 20: 1085–1095.
Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R . Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev 2005; 19: 1432–1437.
Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D . Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 1999; 208: 166–188.
Ferron S, Mira H, Franco S, Cano-Jaimez M, Bellmunt E, Ramírez C et al. Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 2004; 131: 4059–4070.
Armesilla-Diaz A, Bragado P, Del Valle I, Cuevas E, Lazaro I, Martin C et al. p53 regulates the self-renewal and differentiation of neural precursors. Neuroscience 2009; 158: 1378–1389.
Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J . p53 suppresses the self-renewal of adult neural stem cells. Development 2006; 133: 363–369.
Karpowicz P, Morshead C, Kam A, Jervis E, Ramunas J, Cheng V et al. Support for the immortal strand hypothesis: neural stem cells partition DNA asymmetrically in vitro. J Cell Biol 2005; 170: 721–732.
Nagao M, Campbell K, Burns K, Kuan CY, Trumpp A, Nakafuku M . Coordinated control of self-renewal and differentiation of neural stem cells by Myc and the p19ARF-p53 pathway. J Cell Biol 2008; 183: 1243–1257.
Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A . Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 1997; 17: 5046–5061.
Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 2009; 138: 1083–1095.
Agostini M, Tucci P, Chen H, Knight RA, Bano D, Nicotera P et al. p73 regulates maintenance of neural stem cell. Biochem Biophys Res Commun 2010; 403: 13–17.
Fujitani M, Cancino GI, Dugani CB . TAp73 acts via the bHLH Hey2 to promote long-term maintenance of neural precursors. Curr Biol 2010; 20: 2058–2065.
Talos F, Abraham A, Holembowski L . p73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell Death Differ 2010; 17: 1816–1829.
Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22: 2677–2691.
Lazarov O, Marr RA . Neurogenesis and Alzheimer's disease: At the crossroads. Exp Neurol 2010; 223: 267–281.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery Jr CA, Butel JS et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356: 215–221.
Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005; 7: 363–373.
Spandidos A, Wang X, Wang H, Dragnev S, Thurber T, Seed B . A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance. BMC Genomics 2008; 9: 633.
Wang X, Seed B . A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res 2003; 31: e154.
Acknowledgements
LGC is beneficiary of a predoctoral fellowship from Consejo de Educación de la Junta de Castilla y León and RFA from Spanish Ministerio de Ciencia e Innovación. This work was supported by Grants SAF2009-07897 from Spanish Ministerio de Ciencia e Innovacion (to MCM), Grant from Cajas de Ahorro de Castilla y León (to MCM), and Grants LE030A07 (to MMM) and LE015A10-2 (to MCM) from the Junta de Castilla y León.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Gonzalez-Cano, L., Herreros-Villanueva, M., Fernandez-Alonso, R. et al. p73 deficiency results in impaired self renewal and premature neuronal differentiation of mouse neural progenitors independently of p53. Cell Death Dis 1, e109 (2010). https://doi.org/10.1038/cddis.2010.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2010.87
Keywords
This article is cited by
-
p73 isoforms meet evolution of metastasis
Cancer and Metastasis Reviews (2022)
-
Non-oncogenic roles of TAp73: from multiciliogenesis to metabolism
Cell Death & Differentiation (2018)
-
p73 Regulates Primary Cortical Neuron Metabolism: a Global Metabolic Profile
Molecular Neurobiology (2018)
-
p73 is required for appropriate BMP-induced mesenchymal-to-epithelial transition during somatic cell reprogramming
Cell Death & Disease (2017)
-
Expression of the Parkinson’s Disease-Associated Gene Alpha-Synuclein is Regulated by the Neuronal Cell Fate Determinant TRIM32
Molecular Neurobiology (2017)
