
Abstract
Mesenchymal stem cells (MSCs) protect tissues against cell death induced by ischemia/reperfusion insults. This therapeutic effect seems to be controlled by physiological cues released by the local microenvironment following injury. Recent lines of evidence indicate that MSC can communicate with their microenvironment through bidirectional exchanges of mitochondria. In particular, in vitro and in vivo studies report that MSCs rescue injured cells through delivery of their own mitochondria. However, the role of mitochondria conveyed from somatic cells to MSC remains unknown. By using a co-culture system consisting of MSC and distressed somatic cells such as cardiomyocytes or endothelial cells, we showed that mitochondria from suffering cells acted as danger-signaling organelles that triggered the anti-apoptotic function of MSC. We demonstrated that foreign somatic-derived mitochondria were engulfed and degraded by MSC, leading to induction of the cytoprotective enzyme heme oxygenase-1 (HO-1) and stimulation of mitochondrial biogenesis. As a result, the capacity of MSC to donate their mitochondria to injured cells to combat oxidative stress injury was enhanced. We found that similar mechanisms – activation of autophagy, HO-1 and mitochondrial biogenesis – occurred after exposure of MSC to exogenous mitochondria isolated from somatic cells, strengthening the idea that somatic mitochondria alert MSC of a danger situation and subsequently promote an adaptive reparative response. In addition, the cascade of events triggered by the transfer of somatic mitochondria into MSC was recapitulated in a model of myocardial infarction in vivo. Specifically, MSC engrafted into infarcted hearts of mice reduced damage, upregulated HO-1 and increased mitochondrial biogenesis, while inhibition of mitophagy or HO-1 failed to protect against cardiac apoptosis. In conclusion, our study reveals a new facet about the role of mitochondria released from dying cells as a key environmental cue that controls the cytoprotective function of MSC and opens novel avenues to improve the effectiveness of MSC-based therapies.
Similar content being viewed by others


Main
The use of mesenchymal stem cells (MSCs) represents a promising approach to reduce tissue damage following pathophysiological events such as cardiac ischemia/reperfusion or stroke.1, 2, 3 However, the positive results obtained in animal studies are not consistently reproduced in clinical trials,4 suggesting that a better understanding of the mechanisms underlying the therapeutic actions of MSC may help to maximize their efficacy for therapeutic applications. Although it was initially thought that MSC repair injured tissues through direct differentiation, the latest reports indicate that MSCs aid tissue recovery via paracrine processes involving the secretion of soluble factors and shedding of extracellular vesicles like exosomes and microvesicles.5 To enhance the therapeutic potential of MSC, the pharmacological or genetic manipulation of protective genes has been attempted. For example, MSC overexpressing heme oxygenase-1 (HO-1), the inducible defensive protein that degrades heme to biliverdin, iron and carbon monoxide, exhibit improved repairing ability in the ischemic heart.6, 7 This may relate to enhanced survival of stem cells once injected into the myocardium and to the intrinsic protective properties of HO-1 and its products, which exhibit antioxidant, anti-apoptotic and anti-inflammatory activities.8, 9, 10 In addition, HO-1 may be critical in the recovery of cardiac tissue after ischemia, as it is involved in autophagy and mitochondrial biogenesis, leading to elimination of dysfunctional mitochondria11, 12 and replenishment with new healthy organelles.13, 14
Intriguingly, it has been recently discovered that MSCs rescue damaged somatic cells by transferring their own mitochondria through membrane thin channels referred to as ‘tunneling nanotubes’ (TNTs).15, 16, 17, 18, 19, 20 Mitochondrial transfer preserved aerobic respiration and ATP production of recipient cells, harboring mitochondria dysfunction in in vitro and in vivo protocols mimicking ischemia/reperfusion injury,16, 18 oxidative stress or inflammatory damage.17, 21 Importantly, Miro-1, a calcium-binding mitochondria Rho GTPase, was identified as a key mediator of MSC-derived organelle trafficking that enables the movement of mitochondria along microtubules present in TNTs.22 The environmental cues that stimulate MSC to donate their own mitochondria to suffering cells are unknown. However, it is conceivable that stress signals originating from the recipient cells trigger MSC to effectuate mitochondrial transfer.21, 23
Interestingly, a new role for mitochondria as ‘danger signals’ following ischemia/reperfusion injury and severe tissue damage has been proposed.24 In particular, mitochondria are liberated from dying cells in the surrounding environment and in the bloodstream where multiple mitochondrial products, such as mtDNA and N-formyl peptides, alert the whole organism of a ‘danger’ situation.24, 25, 26 Thus, mitochondria and mitochondrial products are recognized as damage-associated molecular patterns (DAMPs) by specific receptors on immune and other cells, eliciting functional changes such as paracrine alteration of macrophages or accelerated maturation of dendritic cells.27 Importantly, we and others found that suffering somatic cells, including cardiomyocytes, renal tubular cells or smooth muscle cells, release entire mitochondria inside MSC,19, 28, 29 although the significance of this phenomenon is unknown.
Here we hypothesized that mitochondria from damaged cells are delivered to MSC, which respond to this danger signal by activating their reparative capacities. To test this hypothesis we used human multipotent adipose-derived stem cells (hMADS) as an MSC model, and co-cultured MSCs with endothelial or cardiac cells subjected to oxidative challenge with hydrogen peroxide (H2O2). We demonstrate that mitochondrial uptake and degradation within MSC are crucial for activating their rescuing properties in vitro and in a myocardial infarction model in vivo. We also delineate a molecular mechanism whereby mitochondria engulfed by MSC increase expression of HO-1, which directly controls the anti-apoptotic function of MSC by promoting mitochondrial biogenesis.
Results
Encounter with mitochondria from damaged somatic cells triggers the anti-apoptotic functions of MSC
We first analyzed the transfer of mitochondria from RL14 cardiomyocytes or human umbilical vein endothelial cell (HUVEC) endothelial cells toward MSC in co-cultures where MSC and somatic cells were previously labeled with Wheat Germ Agglutinin (WGA) or MitoTracker Green, respectively. We found that the amount of somatic mitochondria up-taken by WGA+ MSC was higher in co-cultures with stressed cells compared to naive counterparts (Figures 1a and b). This phenomenon did not occur in co-cultures performed with somatic cells devoid of respiratory function through depletion of mitochondrial DNA (ρ0 cells). We then assessed the survival of RL14 or HUVEC cells after H2O2 exposure in the presence of MSC through Annexin V/7AAD staining. We confirmed that MSC enhanced the survival of suffering somatic WGA− cells (Figures 1c and d). In contrast, MSC did not protect H2O2-injured RL14 or HUVEC cells in co-cultures made with somatic ρ0 cells or in the presence of transwell inserts (0.4 or 1 μm) that prevented the bidirectional transfer of mitochondria between MSC and somatic cells28 (Figures 1c and d). These observations indicate that the exchange of ‘respiring’ mitochondria from somatic cells to MSC is critical in the control of MSC anti-apoptotic function.

Mitochondria released from dying somatic cells trigger the rescue properties of MSC. (a and b) Representative flow cytometry histogram (upper panels) and relative quantification (lower panels) of transfer of MitoTracker Green-labeled mitochondria from H2O2 insulted RL14 or RL14 ρ0 cardiomyocytes (a) or -HUVEC endothelial cells (b) to MSC (CC-H2O2 or CC-ρ0 H2O2, red line) by reference to co-cultures (CCs) with untreated somatic cells (CC-NT or CC-ρ0 NT, black line). Gray histograms correspond to unstained MSC cultured alone. *P<0.05. (c and d) Flow cytometry analysis of Annexin V/7ADD staining in WGA-gated cardiac or endothelial cells cultured alone (RL14 NT, RL14 ρ0 NT, HUVEC NT, HUVEC ρ0 NT), after an H2O2 insult (RL14 H2O2, RL14 ρ0 H2O2, HUVEC H2O2, HUVEC ρ0 H2O2) or in CC with MSC after an H2O2 insult (CC-RL14 H2O2, CC-RL14 ρ0 H2O2, CC-HUVEC H2O2, CC-HUVEC ρ0 H2O2). Lower panels represent the relative survival of RL14 or HUVEC cells after an H2O2 insult following CC with naive MSC or when a transwell insert of 0.4 (CC-TW0.4) or 1 μm (CC-TW1) pore size was impeding the contact between MSC and RL14 or HUVEC, by reference to H2O2-injured somatic cells cultured alone, respectively. Relative survival of ρ0 somatic cells injured with H2O2 in CC (CC-RL14 ρ0, CC-HUVEC ρ0) by reference to single culture is also shown. (a–d) Data represent the mean±S.E.M. of at least n=5 independent experiments. *P<0.05; **P<0.01; ***P<0.001
Damaged somatic cell-derived mitochondria promote rescue by enhancing the mitochondrial donor capacity of MSC
To determine whether delivery of mitochondria from damaged somatic cells stimulate MSC to donate their own mitochondria, we co-cultured MSC previously labeled with MitoTracker Green with untreated or H2O2-treated RL14 or HUVEC cells. We observed that the amount of MSC-derived mitochondria accumulated in somatic cells was higher after an H2O2 insult, but that no accumulation occurred in co-cultures performed with ρ0 somatic cells (Figures 2a and b). Furthermore, mitochondria delivered by MSC to somatic cells appeared functional, as transfer of mitochondria from MSC depleted in mtDNA (ρ0) or rendered dysfunctional through rhodamine 6G (R6G) treatment30 failed to rescue the respiratory activity of H2O2-insulted cardiac or endothelial cells (Figure 2c) as assessed by measurements of pericellular oxygen (Figure 2d and data not shown). Taken together, these observations support the idea that mitochondria released by damaged somatic cells activate MSC to transfer healthy mitochondria toward dying cells. Consistently, increased transcriptional expression of Miro-1, a GTPase involved in mitochondrial trafficking,22 was detected in MSC co-cultured with H2O2-injured HUVEC cells (Figure 2c). However, Miro-1 mRNA did not increase in co-cultures with normal MSC in the presence of transwell inserts or ρ0 HUVEC cells (Figure 2c).

Mitochondria released from dying somatic cells improve mitochondria donor capacity of MSC. (a and b) Representative flow cytometry histograms (left panels) and relative quantification (right panels) of transfer of MitoTracker Green-labeled mitochondria from MSC to H2O2-injured RL14 and -ρ0 RL14 (a) or H2O2-injured HUVEC and -ρ0 HUVEC cells (b; red line or CC—H2O2 and CC—H2O2 ρ0) by reference to CCs with untreated somatic cells (black line or CC-NT and CC-NT ρ0). Gray histograms correspond to unstained naive somatic cells. (c) Relative survival of RL14 or HUVEC cells after an H2O2 insult following CC with naive MSC (CC) or with MSC depleted in mtDNA (CC-MSC ρ0) or pretreated with rhodamine 6G (CC-R6G) by reference to single culture (RL14, HUVEC). (d) Oxygen consumption rate of ρ0 somatic cells in single culture (RL14 ρ0, blue line, HUVEC ρ0, green line) or that isolated from a CC with MSC (CC-RL14 ρ0, red line, or CC-HUVEC ρ0, purple line). (e) Relative transcriptional expression of miro-1 in MSC following CC with untreated HUVEC cells (CC-NT) or HUVEC cells treated with H2O2 (CC-H2O2); CCs of MSC with untreated (CC TW-0.4, CC TW-1) or H2O2-treated HUVEC cells (CC-H2O2 TW-0.4, CC-H2O2 TW-1) in the presence of a 0.4 or 1 μm pore size transwell insert, respectively; CCs of MSC with untreated HUVEC ρ0 (CC HUVEC ρ0) or H2O2-treated ρ0 HUVEC cells (CC-H2O2 HUVEC ρ0). Data represent the mean±S.E.M. of at least n=5 independent experiments. *P<0.1; **P<0.01; ***P<0.001
Degradation of somatic cell-derived mitochondria inside MSC is necessary for triggering rescue
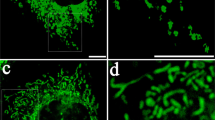
To investigate the fate of somatic-derived mitochondria once engulfed by MSC, we performed transmission electron microscopy (TEM) on co-cultures with injured somatic cells and observed in MSC an important number of mitochondria trapped into vesicles resembling phagosomes (Figure 3a). Immunocytochemistry experiments also revealed in MSC co-cultured with damaged somatic cells that a significant amount of mitochondria were co-localized with LC3B-stained autophagosomes (Figure 3a). This suggested that foreign mitochondria coming from damaged somatic cells are digested by MSC. To test this hypothesis, LC3B protein expression and autophagy activity were measured by immunocytochemistry, western blotting and Cyto-ID flow cytometry in different conditions. MSC mixed with H2O2-insulted RL14 or HUVEC cells expressed higher total LC3B and cleaved LC3-II protein levels and increased autophagic activity than those co-cultured with intact somatic cells (Figures 3b and c; Supplementary Figures 1a and b). However, these phenomena did not occur in co-cultures with transwell inserts or ρ0 somatic cells (Figure 3c). To determine whether autophagy was responsible for degradation of H2O2-treated somatic-derived mitochondria into MSC, we measured MitoTracker Green-labeled somatic mitochondria accumulated after co-culture with naive MSC or MSC pretreated with chloroquine to impede mitophagy. We observed that the degradation of somatic mitochondria into MSC was significantly higher in co-cultures with H2O2-insulted RL14 or HUVEC (Supplementary Figures 1c and d). We also detected increased levels of cytosolic heme, presumably derived from the digested mitochondria, in MSC following co-culture with H2O2-insulted somatic cells compared to untreated conditions (Supplementary Figures 1e and f).

Degradation of somatic-derived mitochondria by MSC is required for apoptotic cell rescue. (a) Left panel: transmission electron microscopy showing depolarized mitochondria (star) in a phagosome-like vesicle (arrow). Scale bar, 200 μm. Middle panel: fluorescence microscopy showing mitochondria (green signal) co-localized with autophagosome-LC3B marker (red signal) in WGA+ MSC (white signal) subjected to CC with H2O2-treated RL14 cells. Nuclei were counterstained with Hoechst 33342 (blue). Scale bar, 50 μm. Right panel: magnification of the square area shown in the middle picture. Scale bar, 50 μm. (b and c) Upper panels: LC3B immunostaining (red signal; left panels) and fluorescence quantification in WGA+ MSC (white signal) and WGA− RL14 or HUVEC cells following CC in the absence (CC-NT) or following an H2O2 treatment (CC-H2O2; right panels). Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. Lower panels: relative flow cytometry quantification of autophagy activity in MSC following CCs with H2O2-treated somatic cells (CC-H2O2) by reference to untreated ones (CC-NT) or CCs with H2O2-treated ρ0 somatic cells (CC-H2O2 ρ0) by reference to untreated ones (CC-NT ρ0) or CCs of H2O2-treated somatic cells (CC-H2O2 TW) by reference to untreated ones in the presence of a transwell insert (CC-NT TW). (d and e) Left panels: representative dot blots of flow cytometry analysis of Annexin V/7ADD staining on WGA− gated somatic cells in single culture after an H2O2 insult (RL14, HUVEC), in CC with naive MSC after an H2O2 insult (CC-RL14, CC-HUVEC) or in CC with MSC pretreated with chloroquine prior to the CC (CC-Chloro). Right panels: relative survival of RL14 or HUVEC cells after an H2O2 insult following CC with naive (CC) or chloroquine-treated MSC (CC-Chloro) by reference to single culture (Alone). (b–e) Data represent the mean±S.E.M. of at least n=4 independent experiments. *P<0.05; **P<0.01; ***P<0.001
To address whether degradation of somatic-derived mitochondria was essential for the rescuing efficacy of MSC, we determined the ratio of surviving somatic cells following an H2O2 insult in co-cultures with MSC pretreated with the autophagy inhibitor chloroquine. Under these conditions, MSC significantly lost their ability to protect RL14 or HUVEC cells against H2O2-induced apoptosis (Figures 3d and e), suggesting that mitochondria from damaged somatic cells have to be degraded inside MSC in order to trigger their anti-apoptotic effects.
Damaged somatic cell-derived mitochondria stimulate the anti-apoptotic functions of MSC though the cytoprotective HO-1 enzyme
Given that increased cytosolic heme was detected in MSC co-cultivated with H2O2-treated somatic cells, we examined whether the expression of HO-1, the inducible protein that degrades excess intracellular heme, was affected in MSC. We found that HO-1 expression and heme oxygenase enzymatic activity were significantly increased in MSC co-cultured with damaged RL14 or HUVEC cells than with intact somatic counterparts (Figures 4a–c). In contrast, HO-1 induction and heme oxygenase activity were markedly reduced in MSC co-cultured with injured ρ0 somatic cells or in MSC treated with chloroquine prior to co-culture (Figures 4a–c). These data suggest that digestion of somatic-derived mitochondria inside MSC leads to HO-1 upregulation.

Degradation of somatic-derived mitochondria stimulate the anti-apoptotic function of MSC through HO-1 induction. (a) HO-1 immunostaining (red signal; left panels) and fluorescence quantification in WGA+ MSC (white signal) and WGA− RL14 or HUVEC cells following CC in the absence (CC-NT) or following an H2O2 treatment (CC-H2O2; right panels). Nuclei were counterstained with Hoechst 33342 (blue). Scale bar, 50 μm. (b and c) Relative HO-1 mRNA expression (HUVEC cells only; c) and relative heme oxygenase activity (c) assessed in MSC co-cultured with H2O2-treated (CC-H2O2) by reference to untreated (CC-NT) RL14 or HUVEC cells, H2O2-treated (CC-H2O2 ρ0) by reference to untreated ρ0 RL14 or HUVEC cells (CC-NT ρ0) or CC of chloroquine-treated MSC with H2O2-treated (CC-H2O2 Chloro) by reference to untreated RL14 or HUVEC cells (CC-NT Chloro). (d and e) Representative dot blots of flow cytometry analysis of Annexin V/7ADD staining (left panels) and relative quantification of survival (right panels) on gated WGA− somatic cells after an H2O2 insult in CC with MSC in the absence (CC-RL14, CC-HUVEC) or presence of SnPPIX (CC-RL14+SnPPIX, CC-HUVEC+SnPPIX) by reference to single culture in the absence (RL14, HUVEC) or presence of SnPPIX (RL14+SnPPIX, HUVEC+SnPPIX), respectively. (b–e) Data represent the mean±S.E.M. of at least n=4 independent experiments. *P<0.1; **P<0.01; ***P<0.001
We then determined whether HO-1 was involved in the control of MSC-rescuing activities. We performed co-cultures in the presence of tin protoporphyrin IX (SnPPIX), an inhibitor of heme oxygenase activity, and found that MSC markedly diminished the rescue of H2O2-injured RL14 or HUVEC cells (Figures 4d and e). These results support that HO-1 mediates the anti-apoptotic function of MSC.
HO-1 promotes the anti-apoptotic response of MSC through stimulation of mitochondrial biogenesis
Because HO-1 is known to stimulate mitochondrial biogenesis,13, 31 we reasoned that this process might underlie the rescue efficiency of MSC by favoring the transfer of their healthy mitochondria toward damaged cells. Thus, we examined whether MSC mitochondrial mass was affected by co-culture with damaged cells. We observed an increased MitoTracker Green fluorescence, reflecting enhanced mitochondria content, in MSC co-cultured with H2O2-injured RL14 or HUVEC cells compared to healthy somatic counterparts (Figure 5a). No increase in MSC mitochondrial mass was detected in co-cultures performed with ρ0 RL14 or HUVEC cells or in MSC treated with chloroquine or SnPPIX to block autophagy or heme oxygenase activity, respectively (Figure 5a). Consistently, the mRNA expression of two master regulators of mitochondrial biogenesis, PGC1-α (peroxisome proliferator-activated receptor gamma coactivator-1-α) and mtTFA (mitochondrial transcription factor A), was elevated in MSC co-incubated with H2O2-insulted somatic cells (Figure 5b). mtTFA protein expression was also higher in MSC co-cultured with injured RL14 or HUVEC cells compared to healthy somatic cells (Figures 5c and d). Co-cultures conducted with H2O2-damaged ρ0 RL14 or HUVEC cells or with MSC, where autophagy or heme oxygenase activity was inhibited, prevented mitochondrial biogenesis stimulation in MSC (Figures 5a–d). Altogether, these results indicate the following: (1) mitochondrial biogenesis is stimulated in MSC in response to a crosstalk with damaged somatic cells; (2) this process is a consequence of phagocytosis of engulfed somatic-derived mitochondria; and (3) the process is dependent on HO-1 activity.

HO-1 induction following somatic-derived mitochondria degradation leads to stimulation of mitochondrial biogenesis in MSC. (a) Relative quantification of the MSC mitochondrial mass following CC with untreated (black line) or H2O2-treated RL14 or HUVEC cells (red line). Gray histograms: fluorescence of MSC grown alone. (b) Relative PGC-1α and mtTFA mRNA expression in co-cultivated MSC. (a and b) Data represent the mean±S.E.M. of at least n=4 independent experiments. ***P<0.001. (c and d) mtTFA immunocytochemistry (green signal, left panels) and fluorescence quantification in co-cultivated WGA+ MSC (white signal) and WGA− somatic cells (right panels). Nuclei were counterstained with Hoechst 33342 (blue). Scale bar, 50 μm. (a–d) CC-NT: CC with untreated somatic cells; CC-H2O2: CC with H2O2-treated somatic cells; CC-NT ρ0 and CC-H2O2 ρ0: CCs with ρ0 somatic cells; CC-NT Chloro and CC-H2O2 Chloro; CCs of MSC pretreated with chloroquine; CC-NT SnPPIX and CC-H2O2 SnPPIX: CCs in the presence of SnPPIX. (e and f) Representative dot blots of flow cytometry analysis of Annexin V/7ADD staining (left panels) and relative quantification of survival (right panels) on gated WGA− cardiac (e) or endothelial (f) cells after an H2O2 insult in CC in the presence of ddC (CC+ddC) by reference to single culture in the presence of ddC (RL14+ddC, HUVEC+ddC). Data represent the mean±S.E.M. of at least n=5 independent experiments. *P<0.05; **P<0.01
Finally, to determine whether mitochondrial biogenesis was required for eliciting the rescue properties of MSC, co-cultures of MSC with H2O2-damaged RL14 or HUVEC cells were performed in the presence of 2′,3′-dideoxycytidine (ddC), an inhibitor of DNA polymerase γ that controls mtDNA replication.32 We show that ddC significantly decreased the ability of MSC to protect damaged somatic cells from apoptosis (Figures 5e and f).
ROS and mitochondrial dynamic are involved in the rescue response of MSC toward doxorubicin-damaged somatic cells
To determine whether somatic mitochondria transfer into MSC is common to other stressful conditions or was specific to ROS (H2O2) injury, we performed co-cultures with somatic cells exposed to doxorubicin, another damaging agent. Similarly to H2O2, doxorubicin-damaged RL14 or HUVEC cells increased their mitochondria release toward MSC (Figure 6a) and were rescued by MSC (Figure 6b). An enhanced delivery of mitochondria also occurred from MSC toward suffering cells (Figure 6c). Furthermore, increased autophagic activity was detected in MSC (Figure 6d; Supplementary Figures 1a and b) together with enhanced degradation of somatic-derived mitochondria, cytosolic heme content (Supplementary Figures 1c–f) and expression of HO-1 and mtTFA proteins (Figures 6e and f).

ROS and mitochondrial dynamic are involved in the rescue of doxorubicin-damaged somatic cells by MSC. (a) Representative flow cytometry histogram (left panels) and relative quantification (right panels) of transfer of MitoTracker Green-labeled mitochondria from doxorubicin-insulted RL14 cardiomyocytes or -HUVEC endothelial cells to MSC (CC-Dox, red line) by reference to CCs with untreated somatic cells (CC-NT, black line). Gray histograms correspond to unstained MSC cultured alone. **P<0.01. (b) Representative dot blots of flow cytometry analysis of Annexin V/7ADD staining (left panels) and relative quantification of survival (right panels) on gated WGA− somatic cells after doxorubicin insult, in single culture (RL14 Dox, HUVEC Dox) or in CC with MSC (CC-Dox) without treatment or following exposure to NAC (+NAC), mdivi-1 (+mdivi-1) or cccp (+cccp). (c) Flow cytometry-relative quantification of transfer of MitoTracker Green-labeled mitochondria from MSC to doxorubicin-injured RL14 (upper panel) or -HUVEC cells (lower panel; CC-Dox) by reference to CCs with untreated somatic cells (CC-NT). (d) Relative flow cytometry quantification of autophagy activity in MSC following CCs with doxorubicin-treated somatic cells (CC-Dox) by reference to untreated ones (CC-NT). (e and f) Representative flow cytometry histogram (left panels) and relative quantification (right panels) of HO-1 (e) and mtTFA (f) in MSC following CCs with doxorubicin-treated somatic cells (CC-Dox) by reference to untreated ones (CC-NT). Gray histograms correspond to unstained naive somatic cells. (g) Representative flow cytometry histogram (left panels) and relative quantification (right panels) of transfer of Mitosox-labeled mitochondria from doxorubicin-insulted RL14 cardiomyocytes or -HUVEC endothelial cells to MSC (CC-Dox, red line) by reference to CCs with untreated somatic cells (CC-NT, black line). Gray histograms correspond to unstained MSC cultured alone. (a–f) Data represent the mean±S.E.M. of at least n=4 independent experiments. *P<0.05; **P<0.01; ***P<0.001. CC with MSC following exposure to NAC (+NAC), mdivi-1 (+mdivi-1) or cccp (+cccp)
As ROS are linked to stimulation of HO-1 and mitochondrial biogenesis,33 we investigated whether ROS from damaged somatic cells were involved in mitochondria signaling and MSC-mediated rescue. By using Mitosox dye staining we revealed that damaged somatic mitochondria transferred into MSC produced higher ROS levels compared to untreated conditions (Figure 6g). In doxorubicin-injured somatic cells pretreated with N-Acetyl-L-Cysteine (NAC), a ROS scavenger, mitochondria transfer to MSC was markedly attenuated (Figure 6a). Interestingly, pretreatment with NAC prevented the rescue properties of MSC (Figure 6b), as induction of mitochondria delivery to somatic cells (Figure 6c) and activation of autophagy, HO-1 expression and mitochondrial biogenesis (Figures 6d and f) were all impaired. These findings indicated that ROS produced by somatic cells following damage stimulate mitochondria delivery to MSC to trigger an adaptive cytoprotective response.
Recent findings associate mitochondrial fusion/fission with mitophagy, mitochondrial biogenesis and HO-1.34 Therefore, we investigated whether mitochondrial dynamic proteins participated in the MSC-mediated rescue once somatic-derived mitochondria were transferred and performed co-cultures with MSC treated with mdivi-1 or cccp to inhibit mitochondrial fission or fusion, respectively. Alteration of these two processes impaired the rescue properties of MSC (Figure 6b) and fusion inhibition in MSC, but not fission blockade, affected mitochondria delivery to suffering somatic cells and prevented induction of mitophagy, HO-1 or mitochondrial biogenesis in MSC (Figures 6c–f). Moreover, co-culture with suffering somatic cells stimulated the protein expression of mitofusin-1 and mitofusin-2 in MSC, and this process seemed to be dependent on HO-1 (Supplementary Figures 2a–d). Altogether, these results implicate mitochondrial fusion in MSC rescue activities, while mitochondrial fission seems to be involved by ensuring mitochondrial quality control to avoid the transfer of dysfunctional organelles.
Exposure of MSC to mitochondria isolated from somatic cells mimics the effects of co-culture
To strengthen the hypothesis that somatic-derived mitochondria are the organelles that stimulate MSC-rescuing capacities, we investigated whether the effects of co-culture could be mimicked by exposure of MSC to exogenous somatic mitochondria. Mitochondria previously labeled with MitoTracker Green were extracted from RL14 cardiomyocytes and then conjugated to the cell-penetrating peptide Pep-1 to favor their entry in MSC.35 After exposure for 24 h, increased MitoTracker Green fluorescence was detected in MSC, indicating the engulfment of exogenous mitochondria (Supplementary Figure 3). This engulfment process was also detected in naive and ρ0 somatic cells (Supplementary Figure 3). Furthermore, exposure to exogenous mitochondria increased MSC autophagic activity (enhanced LC3B staining) in MSC compared to untreated MSC, including in the presence of chloroquine or a heme oxygenase activity inhibitor (Figure 7a). Active autophagy was also supported by an increased expression of cleaved LC3II proteins and decreased p62 in MSC treated with exogenous mitochondria compared to the untreated ones (Figure 7b). Exogenous mitochondria raised HO-1 protein expression (Figure 7c) and heme oxygenase activity in MSC (Figure 7d) accompanied by activation of PGC1-α (mRNA) and mtTFA (mRNA and protein levels; Figures 7e and f). The increased heme oxygenase activity and mitochondrial biogenesis were impaired in MSC exposed to exogenous mitochondria with chloroquine or SnPPIX (Figures 7d–f). Thus, it appears that exogenous somatic mitochondria are sufficient to cause in MSC the same phenotypic changes stimulated by co-culturing with suffering somatic cells. Finally, exposure to exogenous ρ0 mitochondria reproduced the effects of respiring ones as MSC autophagy as well as HO-1 and mtTFA protein expression were still induced (Figure 7g). This suggests that mtDNA-deficient mitochondria, like healthy mitochondria, can trigger signaling-protective pathway once engulfed in MSC. Indeed, exogenous healthy or mtDNA-depleted mitochondria restored the rescue ability of MSC toward H2O2-insulted ρ0 somatic cells (Figure 7h), suggesting that the lack of rescue observed in co-cultures with ρ0 somatic cells was due to a defect of ρ0 cells in delivering mitochondria to MSC.

Exposure of MSC to exogenous mitochondria isolated from somatic cells mimics the effects of co-culturing. (a) LC3B immunocytochemistry (left panels) and fluorescence quantification (right panels) in MSC alone (untreated) or exposed to cardiac mitochondria (+Mitochondria) without treatment (naive) or after chloroquine (Chloro) or SnPPIX treatment (SnPPIX). (b) Representative flow cytometry histogram (left panels) and relative quantification (right panels) of cleaved LC3-II or p62 in MSC alone (black line) or MSC exposed to cardiac mitochondria (red line). Gray histograms: fluorescence of MSC stained with respective control IgG antibody. (c) HO-1 immunocytochemistry (left panels) and fluorescence quantification (right panels) in MSC alone (Mito−) or MSC exposed to cardiac mitochondria (Mito+) without treatment (naive) or after Chloro or SnPPIX. (d) Relative heme oxygenase activity in MSC exposed to cardiac mitochondria (Mito+) by reference to MSC grown alone (Mito−) without treatment (naive) or after Chloro or SnPPIX. (e) Immunocytochemistry showing mtTFA protein expression (green signal, left panels) and fluorescence quantification in MSC exposed to cardiac mitochondria (Mito+) by reference to MSC grown alone (Mito−) without any treatment (naive) or after Chloro or SnPPIX. (f) Relative PGC1-α and mtTFA mRNA expression in MSC exposed to cardiac mitochondria (Mito+) by reference to MSC grown alone (Mito−) without treatment (naive) or after Chloro or SnPPIX. (g) Relative flow cytometry quantification of autophagy activity and protein expression for HO-1 and mtTFA in MSC treated with cardiac ρ0 mitochondria (Mito ρ0+) by reference to untreated MSC (Mito ρ0−). (h) Relative flow cytometry quantification of Annexin V−/7ADD− cardiac or endothelial ρ0 cells after an H2O2 insult in CC with MSC previously treated with cardiac naive (CC+Mito) or ρ0 mitochondria (CC+Mito ρ0) by reference to single culture (Alone). (a, c and e) Nuclei were counterstained with Hoechst 33342 (blue). Scale bar, 50 μm. (b, d and f–h) Data represent the mean±S.E.M. of at least n=4 independent experiments. *P<0.05; **P<0.01; ***P<0.001
MSC rescue infarcted hearts through an endosomal acidification and HO-1-sensitive mechanisms
We investigated whether a crosstalk with damaged cells in vivo could also trigger MSC reparative capacities by delivering human MSC to either intact or infarcted mouse hearts and assessing the MSC phenotype after 24 h. We observed increased expression of HO-1 and PGC1-α genes in MSC infused in damaged hearts compared to MSC injected in intact myocardium while transcriptional expression of mtTFA was not changed (Figure 8a). Remarkably, upregulation of HO-1 and PGC1-α was not observed in MSC treated with chloroquine or SnPPIX prior to engraftment into infarcted hearts (Figure 8a). These results suggest that HO-1 expression and mitochondrial biogenesis are stimulated in engrafted MSC in response to myocardial injury and that MSC require proper autophagy activity to induce HO-1, which then activates mitochondrial biogenesis.

MSC delivery in mice protects infarcted hearts through autophagy- and HO-1-dependent mechanisms. (a) Transcriptional expression of HO-1, PGC1-α and mtTFA in MSC following their engraftment into infarcted hearts of mice (MI) either alone (naive) or in the presence of chloroquine (MSC Chloro) or SnPPIX (MSC SnPPIX) treatment. Data represent the mean±S.E.M. of at least n=5 mice per group and are expressed compared to sham-treated hearts (Sham). **P<0.01; ***P<0.001. (b) Quantification of plasmatic cardiac troponin 24 h after myocardial infarction in mice treated with HBSS, naive MSC or MSC in the presence of chloroquine or SnPPIX. Data represent the mean±S.E.M. of at least n=10 mice per group. *P<0.05; **P<0.01; ***P<0.001. (c) Ratio of cleaved/total caspase 3 protein expression in infarcted hearts of mice treated with HBSS, naive MSC or MSC in the presence of chloroquine or SnPPIX. Western blots were performed 24 h after myocardial infarction and cell delivery. Data represent the mean±S.E.M. of at least n=5 mice per group. *P<0.05; ***P<0.001
Notably, we demonstrated that human MSC protected cardiac tissue from cell death as plasma cardiac troponin t levels and intracardiac caspase-3 protein expression were significantly reduced in infarcted hearts of mice engrafted with naive MSC compared to hearts treated with saline Hank’s Balanced Salt Solution (HBSS) solution (Figures 8b and c). Conversely, when phagocytosis or heme oxygenase activity was inhibited, engrafted MSC markedly lose their ability to protect cardiac tissue following ischemic damage (Figures 8b and c).
Discussion
What are the signaling cues emitted by dying cells that activate the reparative functions of resident stem/progenitor cells to initiate wound-healing processes?36, 37 The identity of these cues is largely unknown and several ‘danger’ signals are likely responsible for MSC activation. In this study we addressed this question by considering preliminary observations from our laboratory, showing that MSC engulfed foreign mitochondria when co-cultured with suffering somatic cells.28 We reasoned that mitochondria from damaged cells could be one of the cues, in line with emerging evidence indicating that mitochondrial products act as DAMPs to alert the organism of a danger situation.24, 25, 26 By analyzing the interaction of mitochondria with MSC and the subsequent molecular events, we demonstrate that mitochondria transferred from dying somatic cells and, once digested in MSC, activated the cytoprotective protein HO-1. This led to increased mitochondrial biogenesis that allowed MSC to donate healthy mitochondria, thereby enhancing survival of suffering cells. Thus, mitochondria released by damaged somatic cells appear to act as master regulators of MSC functions.
Severe tissue injury following many types of stress causes rapture of cell membranes and release of intracellular components in the surrounding environment. Therefore, although our data clearly show that mitochondria from damaged cells accumulated in MSC, other environmental cues may be sensed by MSC and activate their anti-apoptotic properties. However, our experiments with direct exposure to mitochondria isolated from somatic cells prove that mitochondria alone are sufficient to activate the MSC-rescuing ability. We previously reported that mitochondria from damaged cells were mainly conveyed to MSC via TNTs.28 As apoptosis induced by oxidative stress was shown to promote TNT formation,38 this could explain why injured cells delivered higher amounts of mitochondria to MSC in our co-cultures. Our experiments also indicate that mitochondrial ROS produced upon cell injury control the release of mitochondria from damaged somatic cells, as ρ0 somatic cells, which were unable to produce ROS and thus to adapt their mitochondrial flux following stress exposure, failed to elicit a cytoprotective response from MSC.
Capture of mitochondria by MSC stimulated autophagy, supposedly because MSC recognized somatic organelles as foreign bodies, and digestion of somatic-derived mitochondria was necessary for HO-1 induction and for eliciting MSC adaptive response. As mitochondria are rich in heme-containing proteins and heme is an inducer and substrate of HO-1 enzymatic activity, we were not surprised to detect increased MSC cytosolic heme content after co-cultures with damaged somatic cells, which likely led to HO-1 induction. Consequently, HO-1 initiated mitochondrial biogenesis in MSC as previously demonstrated by others.13, 14 This mechanistic scenario is plausible as blockade of authophagy failed to upregulate HO-1, thus preventing mitochondrial biogenesis and MSC activation. Similarly, inhibition of heme oxygenase activity prevented mitochondrial biogenesis and attenuated the protective response of MSC. Interestingly, this cascade of events exemplifies how MSC adapt their mitochondrial mass to ensure the rescue of suffering cells through donation of their own mitochondria and to simultaneously preserve their own energetic homeostasis. Importantly, mitochondria dynamic appeared to have a role in the rescue mediated by MSC by ensuring the quality control and functionality of the mitochondria transferred toward damaged somatic cells.34 That the above mechanisms have physiological relevance was confirmed by our in vivo experiments in mouse myocardial infarction, where human MSC engrafted in the heart enhanced autophagy activity, HO-1 expression and mitochondrial biogenesis, and inhibition of these processes impaired their tissue-protective effects. Even though we identified that HO-1 is directly linked to enhancing the capacity of MSC to donate their mitochondria, further biological actions of this enzyme and its products, such as its pro-angiogenic39, 40 and anti-inflammatory properties,41, 42, 43 could contribute to this effect. Furthermore, other stress proteins and pro-survival systems are likely engaged by MSC to execute their reparative action. Collectively, our findings highlight that mitochondria, released from somatic cells as a result of tissue injury, act as signaling organelles capable of awakening MSC from a dormant state into a pro-healing phenotype and uncover novel molecular mechanisms that illustrate how the crosstalk between somatic mitochondria and MSC stimulates a coordinated rescue response.
Importantly, exploitation of the mitochondria/HO-1 axis may help to develop innovative strategies to improve the therapeutic potential of MSC-based approaches.
Materials and Methods
Cell isolation and culture
HMADS cells used as human MSC model were isolated from adipose tissues obtained from young donors after informed parental consent as previously reported.44 The human fetal ventricular RL14 cell line and primary HUVEC were purchased from the American Type Cell Culture (ATCC, LGC Standards S.a.r.l. Molsheim France) and PromoCell (Heidelberg, Germany), respectively.
HMADS cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), 1 g/l glucose, containing 10% heat-inactivated fetal bovine serum (FBS; Dominique Dutscher, Issy Les moulineaux, France), 100 U/ml penicillin, 100 μg/ml streptomycin and 10 mM HEPES (Invitrogen, Carlsbad, CA, USA). As described earlier,44 HMADS cells exhibited the following phenotype: CD44+, CD49b+, CD105+, CD90+, CD13+, Stro-1−, CD34−, CD15−, CD117−, Flk-1−, Gly-A−, CD133−, HLA-DR− and HLA-Ilow.
Human RL14 cells were grown in DMEM/F-12 (Life Technologies, Carlsbard, CA, USA) supplemented with 12.5% heat-inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 10 mM HEPES.45 HUVEC cells were expanded on gelatin (2%)-coated dishes with the growth medium recommended and commercialized by the manufacturer (PromoCell). All cell types were maintained in a 5% CO2 atmosphere at 37 °C.
Generation of cells deficient in mtDNA (ρ0 cells) or in functional mitochondria
Depletion of mitochondrial DNA in MSC (hMADS cells), RL14 or HUVEC cells was achieved by growing cells in a medium additioned with ethidium bromide (Sigma-Aldrich, St. Louis, MO, USA) at 0.5, 2.5 or 5 μg/ml, respectively, and supplemented with 50 μg/ml uridine (Sigma-Aldrich) and 100 μg/ml pyruvate (Sigma-Aldrich) for at least 1 month.46, 47, 48, 49 To quickly impair mitochondrial function, MSCs were treated with Rhodamine 6G (1 μg/ml, Invitrogen) during 48 h as previously reported.30
Oxidative stress treatments and co-culture settings
To induce apoptosis, RL14 or HUVEC cells were exposed to 600 μM H2O2 or with 1 μM doxorubicin (Sigma-Aldrich) for 2 or 24 h, respectively. Co-cultures were performed with MSC cells and naive or injured somatic cells at a density of 3500 cells/cm2 each (ratio 1 : 1) in DMEM supplemented with 10% FBS. To distinguish stem cells from somatic cells in mixed cultures, MSC cells were labeled with WGA conjugated to Alexa Fluor 647 (5 μg/ml; Life Technologies) prior to co-culture. Single cultures of RL14, HUVEC or MSC were performed in the same medium and at the same concentration. Separation of co-cultures was performed using cell culture inserts containing polycarbonate membrane (0.4 or 1 μm size pore, Millicell, Millipore). To scavenge mitochondrial ROS, somatic cells were treated with 1 mM NAC (Sigma-Aldrich) for 24 h prior to the co-culture. To inhibit mitochondrial fusion and fission, MSCs were treated with 10 μM Carbonyl Cyanide m-Chlorophenyldrazone (cccp, Merck Millipore, Molsheim, France)50 or 50 μM Mitochondrial Division Inhibitor-1 (mdivi-1, Merck Millipore)51 for 24 h prior to and during the co-culture.
Inhibition of stem cell autophagy, HO-1 and mitochondrial biogenesis during co-culturing
To inhibit autophagy, MSCs were treated for 3 h with chloroquine (100 μM, Sigma-Aldrich) prior to the co-culture.52 Heme oxygenase activity or mitochondrial biogenesis was inhibited in MSC by adding SnPPIX (10 μM, Frontier Scientific, Franckfurt, Germany)53 or ddC (100 μM; Abcam, Paris, France),32 respectively, to the co-culture.
Bidirectional exchanges of mitochondria and mitochondrial biogenesis assessment by flow cytometry
To assess mitochondria transfer from MSC to somatic cells or from somatic to MSC, MSC or somatic cells were, respectively, labeled with MitoTracker Green FM (40 nM, Invitrogen) prior to the co-culture. To determine ROS level released by somatic mitochondria transferred to MSC, somatic cells were labeled with Mitosox (1.25 μM, Invitrogen) prior to the co-culture. Mitochondrial exchanges and ROS produced by transferred somatic mitochondria were evaluated by flow cytometry 24 h after co-culturing.
To measure the mitochondrial mass of MSC after 24 h co-culture with healthy or injured somatic cells, MitoTracker Green (20 nM) was added at the end of the co-culture and its fluorescence was analyzed by flow cytometry on the WGA+ cell population corresponding to MSC.
Oxygen consumption measurements in living somatic ρ0 cells
Pericellular measurements of oxygen were achieved by detection of the oxygen concentration in ρ0 somatic cells, after flow cytometry selective separation from co-cultivated MSC in real-time using a Sensor Dish Reader (PreSens, Regensburg, Germany). Briefly, somatic cells alone or following co-culture and selective separation from MSC were seeded in a 24-well plates (105 cells/well) coated with an oxygen detection probe (Oxodish), and changes in oxygen concentrations were recorded continuously for 16 h as previously reported.54
Flow cytometry detection of somatic cell apoptosis following an H2O2 insult
Twenty-four hours following H2O2 or Doxorubicin insult, somatic RL14 or HUVEC cells grown alone or in co-cultures were stained with Annexin V conjugated to Phycoerythrin and 7AAD (BD Pharmingen, Le Pont de Claix, France) according to the manufacturer’s protocol and were analyzed by flow cytometry. The number of living somatic cells was obtained by counting the double-negative-stained cells in the gated WGA− population and expressed as the percentage of the total cell count.
Flow cytometry quantification of MSC autophagy activity and expression of HO-1 and mtTFA
The autophagy activity of MSC was quantified 24 h after co-cultures with either naive or injured somatic cells with H2O2 or doxorubicin by staining with × 500-diluted CytoID green solution (Enzo Life Sciences, Villeurbane, France) for 30 min and analysis of fluorescence by flow cytometry in the gated WGA+ population corresponding to stem cells. Cleavage of cytosolic LC3-I from autophagosome-associated LC3II was evaluated by flow cytometry following staining with the Muse Autophagy LC3-antibody-based kit (Merck Millipore) according to the manufacturer’s instructions. Autophagosome clearance was assessed after cell fixation and permeabilization using the Cytofix/Cytoperm Fixation/Permeablization kit (BD Pharmingen) followed by staining with alexa Fluor 488-conjugated rabbit monoclonal antibody to SQSTM1/p62 (clone EPR4844, Abcam) and flow cytometry analysis.
Flow cytometry quantification of HO-1 and mtTFA protein expression
Protein expression of HO-1 and mtTFA was quantified 24 h after co-cultures of MSC with either naive or Doxorubicin-injured somatic cells after cell fixation and permeabilization using Cytofix/Cytoperm Fixation/Permeabilization kit (BD Pharmingen) followed by intercellular staining with phycoerytrin-conjugated mouse monoclonal anti-HO-1 antibody (Abcam) or Alexa fluor 488-conjugated mouse monoclonal anti-mtTFA antibody (clone18G102B2E11, Abcam) and flow cytometry analysis.
Transmission electron microscopy
MSCs co-cultured with H2O2-injured RL14 or HUVEC cells were fixed overnight in 3% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.4, post-fixed for 1.5 h with 1% osmium tetraoxide, dehydrated by successive ethanol washes (70%, 90%, 100%, 100%) and impregnated with epoxy resin. After polymerization, 80- to 90-nm sections were cut by use of a Reichert Ultracut S ultramicrotome, stained with 2% uranyl acetate plus Reynold’s lead citrate and visualized under a JEOL 1011 transmission electron microscope with GATAN Erlangshen CCD camera.
Measurement of heme oxygenase activity
MSCs co-cultured with naive or H2O2-treated RL14 or HUVEC cells were collected for heme oxygenase activity after 24 h. The assay is based on the spectrophotometric determination of bilirubin as the final product of heme degradation by heme oxygenase as already described by our group.8 Samples were incubated with the substrate hemin, NADPH, liver cytosol (a source of biliverdin reductase) and other co-factors to sustain heme oxygenase activity. The reaction was allowed to proceed for 1 h at 37 °C in the dark and was terminated by addition of chloroform to extract the bilirubin produced. Bilirubin was measured spectrophotometrically, calculated in picomoles bilirubin/mg protein/60 min and then expressed as percentage of control.
Immunocytochemistry
Cells were fixed with 4% PFA and stained with antibodies (Ab) against HO-1 (rabbit polyclonal Ab (pAb), 1:50, Enzo Life Sciences), LC3B (rabbit pAb, 1:500, Abcam), mtTFA (mouse monoclonal Ab, 1:100, Abcam) or human mitochondria (mouse mAb, 1:100, Abcam). Donkey secondary anti-rabbit or -mouse Ab (FITC- or Cy3-conjugated, 1:100) were purchased from Jackson ImmunoResearch Laboratories Inc. (Montlucon, France) Nuclei were stained with Hoechst 33342 (Sigma-Aldrich). Fluorescence was analyzed with a Zeiss Axioplan 2 Imaging microscope (Carl Zeiss, Marly le Roy, France). Measurement of fluorescence was calculated using ImageJ according to the formula ‘corrected total cell fluorescence=integrated density−(area of selected cell × mean fluorescence of background readings)’.
Isolation and delivery of somatic mitochondria to MSC
Mitochondria were isolated from RL14 cells using the Pierce Mitochondria Isolation Kit (Pierce Chemical Co, Rockford, IL, USA) following the manufacturer's instructions. Mitochondria from 20 × 106 naive or ρ0 RL14 cells were mixed with 0.06 mg Pep-1 (Eurogentec, SERAING, Belgium) for 20 min at room temperature as previously reported.35 Pep-1-conjugated somatic mitochondria were then incubated with MSC or somatic naive or ρ0 cells for 24 h (mitochondria from 3 × 106 RL14 cells were used to treat 150 000 cells).
Real-time PCR assays
PCR assays were performed in samples from MSC co-cultured with HUVEC cells (followed by CD31 negative magnetic bead isolation) or treated with exogenous mitochondria or from healthy or infarcted mouse hearts collected 1 day after delivery of MSC or HBSS. RNA extraction was performed using TRIzol reagent (Invitrogen), according to the manufacturer’s instructions, and then reverse-transcribed using the Superscript First-Strand Synthesis System (Invitrogen) and random primers. Specificity of primer sets was assessed using mouse or human universal RNA (Clontech, Saint Germain en Laye, France). Quantitative RT-PCR reactions were performed in triplicate on a 7900 real-time PCR detection system (Applied Biosystems, Waltham, MA, USA) using Platinum SYBR Green qPCR SuperMix (Invitrogen) for transcriptional expression of human HO-1 (Forward 5′-TTCTTCACCTTCCCCAACATTG-3′; Reverse 5′-CAGCTCCTGCAACTCCTCAAA-3′) and human miro-1 (HP227589, Origene) or Taqman universal Mix (Applied Biosystem) for transcriptional expression of human PGC1α (Hs01082775-m1, Applied Biosystem) and human mtTFA (H 01016730-g1, Applied Biosystem). Human SFA3A1 (Forward 5′-TGCAGGATAAGACGGAATCCAAA-3′ and Reverse 5′-GTAGTAAGCCAGTGAGTTGGAATCTTTG-3′) and human GAPDH (Hs00266705-g1, Applied Biosystems) were used as reference genes for SYBR or Taqman amplifications, respectively.
Mouse myocardial infarction and cell injections
All procedures were performed in accordance with the principles and guidelines established by the European Convention for the Protection of Laboratory Animals and were approved by the Lyon 1 Claude Bernard University Committee for animal research. Myocardial infarction was induced in 10–12-week-old male C57BL6/J mice (Charles River Laboratories, L’arbresle, France) by occluding the left coronary artery for 45 min followed by reperfusion for 10 min. After surgery, a 20 μl HBSS solution containing ~4.0 × 105 naive MSC or MSC previously treated with chloroquine (100 μM) or SnPPIX (10 μM) was injected into the myocardium surrounding the infarcted site. Simultaneously to intracardiac delivery of MSC previously treated with SnPPIX, mice received an intraperitoneal injection of SnPPIX (15 mg/kg). In control experiments, similar cell injections were performed in intact mouse hearts or infarcted mouse hearts received the same amount of HBSS. Injections were performed under a Zeiss microscope.
Quantification of Troponin I in mouse serum
Measurement of troponin I in the serum of infarcted mice was used for specific evaluation of cardiomyocytes’ death according to the manufacturer’s instructions (Architect Stat Troponin-I Abbott), using anti-troponin I primary antibody and acrinidium-coupled secondary antibody.
Western blots
Mouse ischemic heart area was lysed with Precellys 24 (parameters: 6,000 r.p.m.: 3 × 10 s) in buffer containing 1% Triton X-100, 50 mM Tris, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM DTT, 5 mM EDTA and supplemented with a cocktail of protease inhibitors (Sigma-Aldrich) and phosphatase inhibitors (PhosphoStop, Roche Diagnostics, Meylan, France).
Lysate protein concentration was determined by the bicinchoninic acid method (Interchim, Montlucon, France). After migration in sodium dodecyl sulfate 15% polyacrylamide gel (SDS-PAGE), proteins (100 μg per sample) were blotted to polyvinylidene difluoride membranes by electrotransfer (Trans-Blot Turbo Transfer, Bio-Rad). Protein membranes were then incubated with a primary antibody recognizing both total and cleaved forms of caspase-3 (Cell Signaling, Saint Quentin Yvelines, France, 9662) followed by a secondary horseradish peroxidase antibody. Detection was carried out with an ECL kit (GE Healthcare) and band quantification was carried out using the ImageLab software (Bio-Rad, Mitry-Mori, France).
Statistical analysis
Data analysis was performed using GraphPad Prism software version 5.0 (San Diego, CA, USA). Data are expressed as mean±S.E.M., and for statistical analysis one-way ANOVA combined with Bonferroni multiple comparison tests were applied. P-values smaller than 0.05 were considered significant.
References
Pavlichenko N, Sokolova I, Vijde S, Shvedova E, Alexandrov G, Krouglyakov P et al. Mesenchymal stem cells transplantation could be beneficial for treatment of experimental ischemic stroke in rats. Brain Res 2008; 1233: 203–213.
Zeng L, Hu Q, Wang X, Mansoor A, Lee J, Feygin J et al. Bioenergetic and functional consequences of bone marrow-derived multipotent progenitor cell transplantation in hearts with postinfarction left ventricular remodeling. Circulation 2007; 115: 1866–1875.
Burns TC, Verfaillie CM, Low WC . Stem cells for ischemic brain injury: a critical review. J Comp Neurol 2009; 515: 125–144.
Rowart P, Erpicum P, Detry O, Weekers L, Grégoire C, Lechanteur C et al. Mesenchymal stromal cell therapy in ischemia/reperfusion injury. J Immunol Res 2015; 2015: 602597.
Rani S, Ryan AE, Griffin MD, Ritter T . Mesenchymal stem cell-derived extracellular vesicles: toward cell-free therapeutic applications. Mol Ther 2015; 23: 812–823.
Cai C, Guo Y, Teng L, Nong Y, Tan M, Book MJ et al. Preconditioning human cardiac stem cells with an HO-1 inducer exerts beneficial effects after cell transplantation in the infarcted murine heart. Stem Cells 2015; 33: 3596–3607.
Tsubokawa T, Yagi K, Nakanishi C, Zuka M, Nohara A, Ino H et al. Impact of anti-apoptotic and anti-oxidative effects of bone marrow mesenchymal stem cells with transient overexpression of heme oxygenase-1 on myocardial ischemia. Am J Physiol Heart Circ Physiol 2010; 298: H1320–H1329.
Clark JE, Foresti R, Green CJ, Motterlini R . Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem J 2000; 348: 615–619.
Fang J, Akaike T, Maeda H . Antiapoptotic role of heme oxygenase (HO) and the potential of HO as a target in anticancer treatment. Apoptosis 2004; 9: 27–35.
Motterlini R, Foresti R . Heme oxygenase-1 as a target for drug discovery. Antioxid Redox Signal 2013; 20: 1810–1826.
Wang Y, Shen J, Xiong X, Xu Y, Zhang H, Huang C et al. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. PloS ONE 2014; 9: e98834.
Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G, Wang L et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PloS ONE 2013; 8: e75927.
Piantadosi CA, Carraway MS, Babiker A, Suliman HB . Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res 2008; 103: 1232–1240.
Zheng M, Kim S-K, Joe Y, Back SH, Cho HR, Kim HP et al. Sensing endoplasmic reticulum stress by protein kinase RNA-like endoplasmic reticulum kinase promotes adaptive mitochondrial DNA biogenesis and cell survival via heme oxygenase-1/carbon monoxide activity. FASEB J 2012; 26: 2558–2568.
Acquistapace A, Bru T, Lesault P-F, Figeac F, Coudert AE, Le Coz O et al. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells 2011; 29: 812–824.
Han H, Hu J, Yan Q, Zhu J, Zhu Z, Chen Y et al. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol Med Rep 2016; 13: 1517–1524.
Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y et al. Mitochondrial transfer of induced pluripotent stem cell–derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke–induced damage. Am J Respir Cell Mol Biol 2014; 51: 455–465.
Liu K, Ji K, Guo L, Wu W, Lu H, Shan P et al. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia–reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 2014; 92: 10–18.
Plotnikov EY, Khryapenkova TG, Galkina SI, Sukhikh GT, Zorov DB . Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp Cell Res 2010; 316: 2447–2455.
Caicedo A, Fritz V, Brondello J-M, Ayala M, Dennemont I, Abdellaoui N et al. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci Rep 2015; 5: 9073.
Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K et al. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 2012; 18: 759–765.
Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Rehman R et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 2014; 33: 994–1010.
Yasuda K, Park H-C, Ratliff B, Addabbo F, Hatzopoulos AK, Chander P et al. Adriamycin nephropathy: a failure of endothelial progenitor cell-induced repair. Am J Pathol 2010; 176: 1685–1695.
Galluzzi L, Kepp O, Kroemer G . Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 2012; 13: 780–788.
Nakahira K, Hisata S, Choi AM . The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid Redox Signal 2015; 23: 1329–1350.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464: 104–107.
Maeda A, Fadeel B . Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis 2014; 5: e1312.
Figeac F, Lesault P-F, Coz O, Damy T, Souktani R, Trébeau C et al. Nanotubular crosstalk with distressed cardiomyocytes stimulates the paracrine repair function of mesenchymal stem cells. Stem Cells 2014; 32: 216–230.
Vallabhaneni KC, Haller H, Dumler I . Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev 2012; 21: 3104–3113.
Cho YM, Kim JH, Kim M, Park SJ, Koh SH, Ahn HS et al. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012; 7: e32778.
MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE et al. Activation of mitochondrial biogenesis by heme oxygenase-1–mediated NF-E2–related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med 2012; 185: 851–861.
Amiri M, Hollenbeck PJ . Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol 2008; 68: 1348–1361.
Bartz RR, Suliman HB, Piantadosi CA . Redox mechanisms of cardiomyocyte mitochondrial protection. Front Physiol 2015; 6: 291.
Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016; 1: e85817.
Chang J-C, Liu K-H, Chuang C-S, Su H-L, Wei Y-H, Kuo S-J et al. Treatment of human cells derived from MERRF syndrome by peptide-mediated mitochondrial delivery. Cytotherapy 2013; 15: 1580–1596.
Li F, Huang Q, Chen J, Peng Y, Roop D, Bedford JS et al. Apoptotic cells activate the ‘phoenix rising’ pathway to promote wound healing and tissue regeneration. Sci Signal 2010; 3: ra13.
Ranganath SH, Levy O, Inamdar MS, Karp JM . Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 2012; 10: 244–258.
Wang Y, Cui J, Sun X, Zhang Y . Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ 2011; 18: 732–742.
Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Calzi SL et al. Stromal cell–derived factor 1 promotes angiogenesis via a heme oxygenase 1–dependent mechanism. J Exp Med 2007; 204: 605–618.
Fayad-Kobeissi S, Ratovonantenaina J, Dabiré H, Wilson JL, Rodriguez AM, Berdeaux A et al. Vascular and angiogenic activities of CORM-401, an oxidant-sensitive CO-releasing molecule. Biochem Pharmacol 2016; 102: 64–77.
Foresti R, Motterlini R . CO-releasing molecules: avoiding toxicity and exploiting the beneficial effects of CO for the treatment of cardiovascular disorders. Future Med Chem 2013; 5: 367–369.
Ryter SW, Choi AM . Regulation of autophagy in oxygen-dependent cellular stress. Curr Pharm Des 2013; 19: 2747–2756.
Wenzel P, Rossmann H, Müller C, Kossmann S, Oelze M, Schulz A et al. Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur Heart J 2015; 36: 3437–3446.
Rodriguez A-M, Pisani D, Dechesne CA, Turc-Carel C, Kurzenne J-Y, Wdziekonski B et al. Transplantation of a multipotent cell population from human adipose tissue induces dystrophin expression in the immunocompetent mdx mouse. J Exp Med 2005; 201: 1397–1405.
Maayah ZH, Elshenawy OH, Althurwi HN, Abdelhamid G, El-Kadi AO . Human fetal ventricular cardiomyocyte, RL-14 cell line, is a promising model to study drug metabolizing enzymes and their associated arachidonic acid metabolites. J Pharmacol Toxicol Methods 2015; 71: 33–41.
King MP, Attardi G . Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 1989; 246: 500–504.
Miller SW, Trimmer PA, Parker WD, Davis RE . Creation and characterization of mitochondrial DNA-depleted cell lines with ‘neuronal-like’ properties. J Neurochem 1996; 67: 1897–1907.
Spees JL, Olson SD, Whitney MJ, Prockop DJ . Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 2006; 103: 1283–1288.
Tan AS, Baty JW, Dong L-F, Bezawork-Geleta A, Endaya B, Goodwin J et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 2015; 21: 81–94.
Legros F, Lombès A, Frachon P, Rojo M . Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 2002; 13: 4343–4354.
Rosdah AA, K Holien J, Delbridge LM, Dusting GJ, Lim SY . Mitochondrial fission–a drug target for cytoprotection or cytodestruction? Pharmacol Res Perspect 2016; 4: e00235.
Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ et al. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradationnovelty and significance. Circ Res 2013; 112: 1159–1170.
Otterbein L, Sylvester SL, Choi AM . Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am J Respir Cell Mol Biol 1995; 13: 595–601.
Haas B, Chrusciel S, Fayad-Kobeissi S, Dubois-Randé J-L, Azuaje F, Boczkowski J et al. Permanent culture of macrophages at physiological oxygen attenuates the antioxidant and immunomodulatory properties of dimethyl fumarate. J Cell Physiol 2015; 230: 1128–1138.
Acknowledgements
We are grateful to Régine Cartier (Hospices Civils de Lyon, France) for helpful technical assistance in measuring mouse plasmatic cardiac troponin, Alain Schmitt (Institut Cochin, Paris, France), for TEM experiments, and Patricia Zadigue (INSERM U955, Créteil, France) for her technical contribution in cell culture experiments. This work was supported by funding from French National Institute of Health and Medical Research (INSERM) and Association pour la Recherche et l’Etude des Maladies Cardiovasculaires (AREMCAR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Mahrouf-Yorgov, M., Augeul, L., Da Silva, C. et al. Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ 24, 1224–1238 (2017). https://doi.org/10.1038/cdd.2017.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.51
This article is cited by
-
Exploring the limitations of mitochondrial dye as a genuine horizontal mitochondrial transfer surrogate
Communications Biology (2024)
-
Stem Leydig cells support macrophage immunological homeostasis through mitochondrial transfer in mice
Nature Communications (2024)
-
Mitochondrial Transportation, Transplantation, and Subsequent Immune Response in Alzheimer’s Disease: An Update
Molecular Neurobiology (2024)
-
Mitochondria in Mesenchymal Stem Cells: Key to Fate Determination and Therapeutic Potential
Stem Cell Reviews and Reports (2024)
-
Mitochondrial transplantation as a novel therapeutic strategy for cardiovascular diseases
Journal of Translational Medicine (2023)
