
Abstract
Chemoresistance often leads to the failure of breast cancer treatment. MicroRNAs (miRNAs) play an important role in the progression and chemoresistance of cancer. However, because of the complexity of the mechanisms of chemoresistance and the specificity of miRNA regulation in different cell types, the function of miR-20a in breast cancer chemoresistance is still unclear. Here, by using miRNA microarray and high-content screening techniques, we found that miR-20a/b were significantly downregulated in breast cancer tissues compared with normal breast tissues, and low miR-20a/b expression was correlated with poor survival in breast cancer patients. Ectopic overexpression of miR-20a sensitized breast cancer cells to a broad spectrum of chemotherapy drugs and suppress their proliferation both in vitro and in vivo. Further study demonstrated that miR-20a directly targeted the 3'untranslated region of MAPK1, and thus downregulated the expression of P-gp and c-Myc by inhibiting the MAPK/ERK signaling pathway, whereas c-Myc can bind to the promoter region of the miR-20a gene to promote the expression of miR-20a. Together, our study identified a novel miR-20a/MAPK1/c-Myc feedback loop that regulates breast cancer growth and chemoresistance. These findings suggest that miR-20a synergizing with anticancer drugs will be a promising treatment strategy, especially for chemoresistant patients.
Similar content being viewed by others


Main
Breast cancer is one of the most common female malignant adenocarcinomas in the world.1, 2 Clinically, anticancer drugs, including cytotoxic drugs and small molecule targeted drugs, remain one of the major treatments for cancers, including breast cancer. These chemotherapies are especially important for patients with advanced and metastatic tumors that cannot undergo surgery.3 Because of the high cost of targeted therapies, chemotherapy is clinically more preferred in many developing countries, especially in Asia.4 However, with the progress of treatment, patients often become resistant to chemotherapy and result in treatment failure.5, 6 Therefore, it is urgent to develop a new method of tumor therapy that can reduce the dose of chemotherapy drugs and improve the therapeutic effect.
In recent years, the role of a class of endogenous RNA molecules, microRNAs (miRNAs), in cell biology has received wide attention. MiRNAs participate in a series of biological processes, such as cell proliferation, differentiation and apoptosis, and so on.7, 8 It is estimated that at least one-third of the human coding genes are regulated by miRNAs.9 Many studies have shown that miRNAs are involved in the regulation of tumor chemoresistance.10, 11, 12, 13 However, because of the tissue specificity of transcriptome, the same miRNA could have different functions in different cell lines,14 and miRNAs that play a major regulatory role in different drug-resistant cells may also be different. Here, in our study, we used medullary breast cancer cell line BCap37 as a model to identify and study the miRNAs that can regulate its tolerance to paclitaxel (PTX). Medullary breast cancer is the third most pathological type of breast cancer, and there are few studies on it.15, 16, 17 PTX is a taxane first-line anticancer chemotherapy drug. However, patients usually acquire drug resistance during treatment.13 Nowadays, studies on how breast cancer gains PTX resistance are still rare and the mechanisms remain to be elucidated.
In order to find out miRNAs that regulate PTX resistance in breast cancer cells, we used the two chemoresistant cell lines, Bads-200 and Bats-72,18 that were obtained by long-term screening in our laboratory and their parental cell line BCap37 to compare the expression of miRNAs by the miRNA microarray technique, and found that multiple miRNAs in the miR-17-92 family, especially the miR-20a, were significantly downregulated in chemoresistant cell lines. The biological functions and molecular mechanisms of miR-20a in the progression and chemoresistance of breast cancer were comprehensively studied in vitro and in vivo.
Results
Identification of chemoresistance-related miRNAs by miRNA microarray and high-content screening system
In order to screen miRNAs that can enhance the response of breast cancer cells to chemotherapeutic drugs, two drug resistant cell lines, Bads-200 and Bats-72, were established using the first-line anticancer drug PTX by ‘time-increasing method’ and ‘dose-escalation method’, respectively. They represent two different types of drug resistance.18 Compared with the parental cells BCap37, the drug resistance of Bads-200 and Bats-72 to PTX increased significantly (Supplementary Figures S1a and B). We then used miRNA microarray technology to compare changes in miRNA expression profiles between the drug-resistant and the parental cells, and the results showed that the expression of a series of miRNAs changed (Figure 1a). Because the accuracy of miRNA microarray was limited and alerted miRNAs may not be relevant to regulatory function, we only selected those miRNAs that have >1.5-fold change between the parental cell line and the drug-resistant cell lines to construct a miRNA library (Supplementary Table S1). Then, the mimics or inhibitors of selected miRNAs were transfected into Bads-200 cells to detect their function in chemoresistant regulation, respectively. Table 1 lists some of the miRNAs that have the ability to control the drug resistance. Among all miRNAs that were detected as having an ability to regulate drug resistance, miR-20a-5p (miR-20a), which belongs to miR-17 family, had the strongest regulation ability. In addition, other members of this miRNA family, such as miR-93-5p, miR-20b-5p, miR-17-5p, miR-106a-5p and so on, also showed ability to reverse drug resistance in malignant cells. Moreover, the expression of miR-20a/b was significantly downregulated in both drug-resistant cells (Figure 1b).

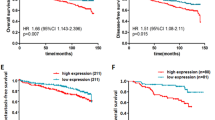
Identification of chemoresistance-related miRNAs by miRNA microarray and high-content screening system. (a) Comparison of miRNA expression in parental cell BCap37 and chemoresistant cell Bads-200 (left) and Bats-72 (right) by using the miRNA microarray. BC37, BCap37; DS200, Bads-200; TS72, Bats-72. Each cell was tested in triplicate. (b) MiR-20a/b expression in BCap37, Bads-200 and Bats-72 cell lines was detected using q-PCR. (c) The expression levels of miR-20a/b in cell lines BCap37, Bads-200, Bats-72, MDA-MB-453, MDA-MB-468, MDA-MB-231, MCF-7, Hs-578T, BT-474 and Sk-Br-3 were detected by q-PCR. The relationship between the expression level of miR-20a/b in each cell and their corresponding half-maximal inhibitory concentration (IC50) to PTX are shown. (d) MiR-20a expression levels were reduced in samples of patients who relapse after chemotherapy compared with patients who did not relapse (n=63). (e) MiR-20a/b expression levels were reduced in human breast cancer samples (n=30) compared with paired noncancerous tissues. (f) Retrospective analysis of Kaplan–Meier plots for miR-20a/b expression in association with overall survival. Patients were split into high and low expression groups by the mean expression of the miR-20a/b (n=66; log-rank test). ***P<0.001
In order to further verify the relationship between miR-20a and chemoresistance, we compared the expression of miR-20a/b in 10 breast cancer cell lines. The results showed that the expression level of miR-20a/b is negatively correlated with drug resistance (Figure 1c). In addition, we collected paraffin-embedded sections from 66 clinical breast cancer patients with PTX in treatment strategy. We divided the patients who relapse into the chemoresistant group, and otherwise into the chemosensitive group. The quantitative PCR (q-PCR) analysis showed that the expressions of miR-20a/b in cancer tissues from the chemoresistant group were lower than that in the chemosensitive group (Figure 1d and Supplementary Figure S1c).We obtained similar results in the specimens from the TCGA database (Supplementary Figure S1d). In addition, we compared the expression of miR-20a/b in 30 cases of the 66 breast cancer specimens and their matched adjacent normal breast tissues. The results showed that the expression of miR-20a and miR-20b were significantly decreased in cancer (Figure 1e). We analyzed another 40 pairs of breast cancer specimens that we collected and the breast cancer specimens in the TCGA database and obtained similar results (Supplementary Figures S1e and f). Correlation analysis revealed that low levels of miR-20a expression were significantly associated with malignancy of breast cancer (Supplementary Tables S4). Kaplan–Meier survival analysis showed that low levels of miR-20a/b were significantly associated with short overall survival in breast cancer patients (Figure 1f and Supplementary Figure S1g), and Cox regression analysis showed that low levels of miR-20a are independent prognostic factors for poor prognosis of patients with breast cancer (Supplementary Tables S5). We analyzed breast cancer specimens from the TCGA database and found that miR-20a/b can be used as a predictor of prognosis in breast cancer patients, especially when miR-20a and 20b are combined (Supplementary Figures S1h and i). In addition, we also found that among different histotypes of breast cancer, the prognosis of patients with luminal A were best suited to be predicted by miR-20a (Supplementary Figure S1j). These results suggest that miR-20a/b may play an important role in regulating the growth and drug resistance of breast cancer.
MiR-20a/b inhibits the proliferation and chemoresistance of breast cancer cells in vitro
In order to investigate the effect of miR-20a/b on breast cancer cell proliferation and resistance in vitro, we transfected mimics and inhibitors of miR-20a/b into BCap37, Bads-200 and Bats-72 cell lines, respectively (Supplementary Figures S2a–d). After overexpression of miR-20a/b in cells, we added IC50 concentrations of PTX to the corresponding cells according to the drug resistance of the cell lines. The results showed that overexpression of miR-20a/b did not only downregulate the drug resistance but also inhibited the cell proliferation (Figure 2a and Supplementary Figure S2e). Growth curve showed that miR-20a/b could significantly decrease the tolerance of cells to PTX. The inhibitory effect of miR-20a was more significant than miR-20b in both proliferation and chemoresistance of cells. Compared with the parental cell, the inhibitory effect is more obvious in the resistant cells (Figure 2b and Supplementary Figure S2f). In contrast, when miR-20a/b was inhibited by inhibitors, the chemoresistance of the three cell lines was increased (Figure 2b). However, it may be that because of the low abundance of miR-20a/b in breast cancer cells, the effect of inhibition of miR-20a/b by inhibitors on the regulation of drug resistance was not as significant as overexpression by mimics (Figure 2b).

MiR-20a/b inhibits the proliferation and chemoresistance of breast cancer cells in vitro. (a) Overexpression of miR-20a/b inhibits cell proliferation and chemoresistance. Cell proliferation was detected by MTT assay. (b) Overexpression of miR-20a/b increased the sensitivity of BCap37 and Bads-200 cell lines to PTX (up), and inhibition of miR-20a/b enhanced the resistance of BCap37 and Bads-200 cells to PTX (down). Cell growth rate was evaluated using MTT assay. (c) The apoptotic rate of the indicated cells transfected with miR-20a, or negative controls or together with PTX treatment. (d and e) MiR-20a inhibits cell colony formation. Colony formation (d) and soft agar (e) assays were performed in BCap37 cells (left) and Bads-200 cells (right) transfected with miR-20a or their negative controls or together with PTX treatment. Results from a representative experiment performed in triplicate. Bar, 500 μm. (f) Enhanced response to PTX by miR-20a overexpression is shown in multiple breast cancer cell lines, including HS-578bst, MCF-7, MDA-MB-231 and BT-474. (g) MiR-20a enhances the sensitivity of breast cancer cell lines Bcap37 (left) and Bads-200 (right) to multiple anticancer drugs. VNB, Vinorelbine; DOX, Doxorubicin; GEM, Gemcitabine; 5-FU, 5- Fluorouracil; CPT, Cisplatin. *P<0.05, **P<0.01, ***P<0.001
Because the regulation effect of miR-20a is stronger than miR-20b, and the abundance of miR-20a in cells is higher than miR-20b, and the their seed sequences are the same, we only study the effect of miR-20a on cell proliferation and drug resistance in the following experiments. We observed that overexpression of miR-20a promoted apoptosis, especially when combined with PTX treatment (Figure 2c and Supplementary Figure S2g). Colony formation experiment confirmed that miR-20a inhibited cell proliferation and drug resistance, and miR-20a combined with PTX treatment showed little cell growth in the plates (Figure 2d and Supplementary Figure S2h). Soft agar colony formation assays indicated that miR-20a also reduced the ability of breast cancer cells to grow in an anchorage-independent manner (Figure 2e and Supplementary Figures S2i and j). In addition, in breast cancer cell lines HS-578T, MCF-7, MDA-MB-231 and BT-474, miR-20a was also found to significantly enhance the sensitivity of these cell lines to PTX (Figure 2f). Next, we wanted to know whether miR-20a increases the sensitivity of cells to other chemotherapeutic agents. Significantly, we found that overexpression of miR-20a can improve the effectiveness of a variety of chemotherapeutic drugs, especially for vinorelbine (VNB), doxorubicin (DOX) and gemcitabine (GEM) (Figure 2g and Supplementary Figures S2k and l), echoing the clinical observation that cancer cells often adopt common mechanisms to resist different chemotherapy drugs.5 The expression level of miR-20a was also negatively correlated with the cell resistance (Supplementary Figure S2m). Furthermore, we want to know whether drug treatment will change the expression of miR-20a/b. The results showed that the expression of miR-20a/b was decreased after PTX treatment, and this effect is more significant in sensitive cells compared with chemoresistant cells (Supplementary Figure S2n). However, the change of miR-20a/b expression was not significant after treatment with other drugs (such as VNB, DOX, GEM and 5-FU) in other breast cancer cell lines (such as Hs-578T, MCF-7 and MDA-MB-231) (Supplementary Figure S2o).These results indicated that miR-20a/b can not only inhibit cell proliferation, but also strongly inhibit cell resistance.
MiR-20a reverses chemoresistance in vivo
Because of the serious side effects of chemotherapy drugs, a more effective chemotherapy strategy with low dose is urgently needed.19 Therefore, we combined the miR-20a with a relatively low dose of PTX to investigate whether this combination has a synergistic effect in vivo (Figure 3a). In order to be close to clinical condition of chemoresistance, Bats-72 cells with moderate drug resistance were inoculated subcutaneously into immunodeficient mice BALB/c to form tumors. Approximately 1 week later, the tumor-bearing mice were treated with cholesterol-conjugated miR-20a and/or PTX. The dose of PTX was 10 mg/kg, approximately of the normal dose.18 After intratumor injection of cholesterol-conjugated miR-20a, the expression level of miR-20a in the breast cancer tissues increased by ∼15 times compared with that of PBS (Figure 3b). The results showed that miR-20a combined with PTX significantly inhibited tumor growth. MiR-20a alone also showed some antitumor effect, but the inhibitory effect of this low-dose of PTX on the tumor formed by drug resistant cells was not obvious (Figures 3c and d). Ki67 staining results showed that combination therapy significantly reduced Ki67-positive cells, and H&E staining implied the cell proliferation was inhibited, and apoptosis was significantly increased (Figure 3e). These results indicate that miR-20a can inhibit tumor growth and enhance the antitumor effect of PTX in vivo.

MiR-20a reverses chemoresistance in vivo. (a) Schematic outline of the combinational therapy in a subcutaneous tumor model. (b) q-PCR analysis of miR-20a expression in transplanted tumors (n=3, date are shown as mean±S.D.). (c and d) Effect of miR-20a restoration on transplanted tumor growth (n=5 for each group). Tumor volume was detected as indicated in (c) and tumor weight as indicated in (d). (e) Representative images of tumor samples that were stained with hematoxylin and eosin (H&E) and Ki67 by IHC (left). The percentages of Ki67-positive cells were measured (right). Bars: (main) 100 μm; (insets) 25 μm. *P<0.05, **P<0.01, ***P<0.001
MAPK1 is one of direct target genes of miR-20a
Next, we investigated the mechanism underling the inhibitory effect of miR-20a on cell proliferation and drug resistance. We first used TargetScan (http://www.targetscan.org) to predict the target of miR-20a, and obtained 1999 potential targets (Figure 4a). We submitted these targets to the Kyoto Encyclopedia of Genes and Genomes (KEGG) for signal pathway analysis. We found that these targets had the highest enriched gene number in cancer-related pathways and MAPK signaling pathways (43 and 40, respectively) (Figure 4b). We then performed a GO analysis of the genes involved in both pathways (Supplementary Table S11), and selected genes that regulate cell growth, proliferation and resistance (Figure 4c). Finally, we singled out 12 target genes (Figure 4d). We cloned the 3' untranslated region (UTR) of these 12 genes, and luciferase reporter assay was performed, respectively. Among the 12 genes, 5 genes, TGFBR2, PDGFRA, CCND1, E2F1 and MAPK1, were found to have a significant decrease in luciferase activity (Figure 4d). Among these five genes, the luciferase activity of MAPK1 was reduced most significantly, and its function and mechanism as the target gene of the miR-20a family in breast cancer growth and drug resistance have not been reported. Next, we used multiple target gene prediction algorithms to predict the target genes of miR-20a. It showed that MAPK1 was the target gene of miR-20a that was predicted by all software we used (Supplementary Figure S3a and Supplementary Table S14).

MAPK1 is one of the direct target of miR-20a. (a) A total of 1999 miR-20a target genes were predicted by TargetScan. (b) Enrichment analysis of predicted miR-20a targets as indicated in (a) in KEGG cell signaling pathway database. (c) Gene ontology (GO) analysis of the genes involved in the pathways in cancer and MAPK signaling pathway in (b). (d) Dual-luciferase assays showing that repression of candidate genes by miR-20a was measured as ratios of Renilla and Firefly luciferase activity in BCap37 cells. Mean±S.E.M. are shown from at least three independent experiments. (e) Predicted sequences between wild-type (WT) or mutant (mut) MAPK1 3′ UTR and miR-20a. The underscore portions of the sequences represent the mutant miR-20a binding sites in MAPK1 3′ UTR (up). Luciferase reporter assay showed the decreased luciferase activity in miR-20a-overexpressed cells for 3′ UTR constructs. The luciferase activity was normalized to Renilla luciferase. (f) q-PCR analysis of the expression of MAPK1 mRNA in BCap37, Bads-200 and Bats-72. (g and h) Western blot (g) and q-PCR (h) analysis of MAPK1 protein and mRNA levels after the transfection of miR-20a mimic, miR-20a inhibitor (anti-miR-20a) or their negative controls (mimic NC and inhibitor NC) in BCap37 and Bads-200 cells. (i) q-PCR analysis of MAPK1 expression levels from 30 breast cancer samples and their adjacent normal tissues. (j) Plotting the paired difference of tumor and normal samples expression for each marker (miR-20a versus MAPK1). The exact McNemar’s test indicates a significant association between the upregulation of expression in MAPK1 and the downregulation of miR-20a expression (n=60). *P<0.05, **P<0.01, ***P<0.001
The binding sites of miR-20a on MAPK1 3' UTR are shown in Figure 4e. We compared the miR-20a and MAPK1 binding sites and found that the miR-20a sequence was highly conserved among different species (Supplementary Figure S3b). To investigate whether MAPK1 is a direct target of miR-20a, we cloned wild-type and mutant 3' UTR fragments of miR-20a binding site of MAPK1 into luciferase reporter vector. The vector without miRNA binding site was treated as negative control. After the vector was transferred into BCap37 cells, miR-20a could decrease the luciferase activity of wild-type MAPK1 vector by ∼50% compared with the empty vector. If the binding site of miR-20a was mutated, the luciferase activity of the vector was restored (Figure 4e).
To further investigate the relationship between miR-20a and MAPK1, we investigated the expression level of MAPK1 in BCap37, Bads-200 and Bats-72, respectively. The amount of MAPK1 in these three cell lines was found inversely related to the amount of miR-20a (Figures 1b and 4f). Overexpression or inhibition of miR-20a would downregulate or upregulate both mRNA and protein expressions of MAPK1, respectively (Figures 4g and h). Furthermore, we collected 30 pairs of matched clinical specimens and found that MAPK1 levels were significantly elevated in breast cancer tissues compared with related normal breast tissues from the same patient (Figure 4i). Meanwhile, in all specimens, the amount of MAPK1 was significantly negatively correlated with the amount of miR-20a (Figure 4j). Importantly, TCGA breast cancer cohort (n=1096) also shared the same expression pattern (Supplementary Figure S3c). These results indicate that MAPK1 is targeted and regulated by miR-20a. However, the expression level of MAPK1 ca not be used to predict the prognosis of patients (Supplementary Figure S3d).
MiR-20a inhibits the proliferation and resistance of breast cancer cells by inhibiting MAPK1 and thereby inhibiting P-gp
To investigate the relationship between the expression of MAPK1 and the proliferation and resistance of breast cancer cells, we knocked down MAPK1 in breast cancer cells by siRNA (Supplementary Figure S4a). Consistent with the result of overexpressing miR-20a, silencing of MAPK1 can impair the proliferation of breast cancer cells and reduce their drug resistance (Figure 5a and Supplementary Figures S4b and c). In contrast, when MAPK1 was overexpressed by vector in breast cancer cells, cell proliferation and drug resistance were enhanced and this effect was partially inhibited by overexpressing miR-20a (Figures 5b and c and Supplementary Figure S4d). Furthermore, we found that the Ulixertinib (specific inhibitor of MAPK1) could mimic the inhibitory effect of miR-20a on MAPK1 to a great extent that inhibited cell proliferation, promoted cell apoptosis and improved the efficacy of chemotherapeutic drugs (Supplementary Figures S4e–h). In order to verify whether miR-20a inhibits cell proliferation and drug resistance by targeting MAPK1, we constructed MAPK1-expressing plasmids containing wild-type or mutant miR-20a binding sites in their 3′ UTRs and transfected them into cells. As expected, the ectopic expression of MAPK1 with mutant 3′ UTR rescued the function of MAPK1 and exhibited a stronger ability to promote cell proliferation and resistance than the expression of wild-type gene when contransfected with miR-20a (Figure 5d).

MiR-20a inhibits the proliferation and resistance of breast cancer cells by inhibiting MAPK1 and thereby inhibiting P-gp. (a) Cell proliferation assays of BCap37 (up) and Bads-200 (down) cells transfected with MAPK1 siRNA (siMAPK1) or control siRNA (siNC) and then treated with miR-20a mimic and miRNA mimic negative control (NC mimic). (b) Ectopic expression of MAPK1 restored resistance to PTX in miR-20a-overexpressing BCap37 (up) and Bads-200 (down) cells. (c) Cell proliferation assays of BCap37 (left) and Bads-200 (right) cells transfected with MAPK1-expressing plasmid or control plasmid and then treated with miR-20a mimic or miRNA mimic negative control (NC mimic). (d) Growth curves of BCap37 (left) and Bads-200 (right) cells transfected with MAPK1-expressing vectors containing wild-type or mutant 3′ UTR and then treated with miR-20a mimic and miRNA mimic negative control (NC mimic) at different PTX concentrations. (e) Expression of β-actin (internal control), ERK1/2, phosphorylated ERK1/2 and P-gp were detected by western blot in three cells. Data are shown as a representative of three independent experiments. (f and g) siRNA-mediated MAPK1 knockdown reduced P-gp levels (f) and expressing vector-mediated MAPK1 overexpression increased P-gp levels (g) as shown by western blot analysis. Data are shown as a representative of three independent experiments. (h) Representative images of tumor samples that were stained with MAPK1 and P-gp by IHC (left). The levels of MAPK1 and P-gp protein expression were measured (right). Bars: (main) 100 μm; (insets) 25 μm. *P<0.05, **P<0.01, ***P<0.001
Tumor cell drug resistance involves a variety of mechanisms, one of the most famous is the elevated expression of members of the ATP binding cassette (ABC) transporter superfamily. The family members mainly include P-gly-coprotein (P-gp/ABCB1), multidrug resistance-associated protein (MRP1/ABCC1) and breast cancer resistance protein (BCRP/ABCG2). They can efflux a wide range of chemotherapeutic agents out of cells and result in drug resistance.18 We used two substrates of ABC transporters, Rh123 and DOX, to treat cells. Rh123 and DOX are fluorescent whereas PTX is not.20 Therefore, if the ABC transporter is highly expressed in the cells, the decrease in accumulation of Rh123 and DOX in the cells can be directly observed by fluorescence microscopy. As expected, we found that the accumulation of Rh123 and DOX in drug-resistant cells was significantly reduced compared with chemosensitive BCap37 cells (Supplementary Figures S4i and j). This suggested that cell resistance of Bads-200 and Bats-72 might associate with an increase in drug efflux pump.18 Furthermore, we detected the ABC transporters of the three breast cancer cell lines by western blot analysis. The result indicated a significant increase of P-gp in drug-resistant cells, and the degree of increase is proportional to the level of drug resistance (Figure 5e). The expression level of MRP1 and BCRP were similar between the two chemoresistant cells and chemosensitive cells.
Studies have indicated that in gastric cancer,21 leukemia,22 ovarian cancer23 and so on, the upregulation and activation of MAPK pathway can increase the expression of P-gp that leads to drug resistance. Indeed, in the present study, MAPK1 expression levels were positively correlated with P-gp in all the three cell lines (Figure 5e). In three representative clinical specimens we collected, the expression of miR-20a was negatively correlated with the expression of MAPK1 and P-gp. Similar results were obtained in the specimens from TCGA database (Supplementary Figures S4k and l). To further investigate the relationship between P-gp and MAPK1, we knocked down MAPK1 by siRNA and found that the expression of P-gp was decreased (Figure 5f). In contrast, ectopic expression of MAPK1 increased P-gp expression (Figure 5g). In addition, we subcutaneous injected Bads-72 cells to BALB/c nude mice and detected the levels of MAPK1 and P-gp in the tumors by immunohistochemistry staining. Overexpression of miR-20a significantly inhibited the expression of MAPK1 compared with the negative control. Meanwhile, the level of P-gp was also significantly reduced (Figure 5h). In addition, we constructed a BCap37-derived drug-resistant cell called BCap37/ MDR that stably overexpressed ABCB1 (P-gp). Overexpression of miR-20a effectively decreased the expression of P-gp as well as the drug resistance of BCap37/MDR cells (Supplementary Figure S4m–o). These results indicate that miR-20a inhibits cell proliferation and resistance by suppressing MAPK1, and the inhibition of the drug resistance was related to the decrease of P-gp mediated by MAPK1 silencing.
MiR-20a promotes apoptosis by inhibiting the MAPK/ERK pathway
The ectopic expression of miR-20a significantly promoted apoptosis in chemosensitive cells and drug-resistant breast cancer cells, especially in combination with PTX (Figure 2c and Supplementary Figure S2g). To investigate the effect of MAPK1 on apoptosis, MAPK1 was knocked down by siRNA. The results showed that downregulation of MAPK1 could promote cell apoptosis (Figure 6a), similar to the effect of miR-20a ectopic expression. In contrast, ectopic expression of MAPK1 mRNA without 3′ UTR partially counteracted miR-20a-induced apoptosis (Figure 6b).

MiR-20a promotes apoptosis by inhibiting the MAPK/ERK pathway. (a) Knockdown of MAPK1 increased PTX-induced cell apoptosis as determined by Annexin V staining (n=3, mean±S.E.M.). (b) Overexpression of MAPK1 reduced miR-20a and PTX-induced apoptosis. BCap37 and Bads-200 cells stably transfected with MAPK1 expressing plasmid were transfected with miR-20a mimics. Cell apoptosis was detected and analyzed as in (a). (c) Transfection of miR-20a mimics or MAPK1 siRNA into BCap37 (left) and Bads-200 (right) cells inhibited expression of MAPK signaling pathway-associated protein. Expression of the indicated proteins was detected by western blot. (d) Transfection of miR-20a inhibitors or MAPK1-expressing plasmid into BCap37 cells activated MAPK signaling pathway. Expression of the indicated proteins was detected by western blot as in (c). (e) Ectopic expression of miR-20a promoted the expression of apoptosis-related proteins and inhibited the expression of anti-apoptotic related proteins. Expression of the indicated proteins was detected by western blot as in (c). *P<0.05, **P<0.01, ***P<0.001
MAPK1, also known as ERK2, is a member of the MAPK/ERK signaling pathway.24 In breast cancer cells overexpressing miR-20a, we found that total ERK2 and phosphorylated ERK2 were inhibited. In addition, overexpression of miR-20a also inhibited the phosphorylation of downstream factors MNK and RSK, whereas the phosphorylation of MEK was not affected (Figure 6c). In contrast, miR-20a inhibitor could increase the MAPK1 protein and activated the MAPK/ERK pathway in BCap37 (Figure 6d) and Bads-200 cell lines (Supplementary Figure S5a).
Some studies have shown that ERK inhibitors sensitize cells to drug-induced apoptosis,25 and thus we tried to identify whether miR-20a regulates apoptosis-related proteins. Indeed, after ectopic expression of miR-20a in parental and drug-resistant cells, the apoptosis marker caspase-3 was downregulated and the activated caspase-3 increased. In addition, the expression of anti-apoptotic protein Bcl-2 and transcription factor c-Myc was also decreased when miR-20a was overexpressed. Furthermore, overexpression of miR-20a in drug-resistant cells Bads-200 inhibited the expression level of P-gp. Interestingly, PTX may also promote P-gp expression to some extent (Figure 6e). These results indicate that miR-20a can regulate apoptosis-related proteins by inhibiting MAPK/ERK pathway, and thus enhance drug sensitivity and promote apoptosis.
Dysregulated histone methylation and miR-20a/MAPK1/c-Myc feedback loop involved in regulation of miR-20a expression
As miR-20a was significantly downregulated in breast cancer, we tried to investigate the mechanism of miR-20a inhibition. MiR-20a belongs to the miR-17- 92 family that is located in the genome Chr13q31.3.26 Studies have shown that the gene deletion occurs in ∼21.9% of breast cancer cases in this family.27 We wanted to know whether there were other mechanisms that downregulate miR-20a. It has been shown that histone methylation affects miRNA expression. The methylation of histone H3 at Lys4 (H3K4) is associated with transcriptional activation, whereas the methylation of H3 at K9 or K27 and H4 at K20 is associated with transcriptional repression.28 First, we compared the gene copy of miR-20a in three cells and found no deletions or amplification of genes in drug-resistant cells compared with parental cells. Next, we investigated the histone methylation of miR-20a. By chromatin immunoprecipitation (ChIP) technique, we found that the H3K4me3 level in the genomic regulatory regions of the miR-20a gene decreased whereas the H4K20me3 levels increased in drug-resistant cells compared with parental cells (Figures 7a and b). However, H3K9me3 and H3K27me3 did not change. This suggests that the histone methylation abnormalities of miR-20a gene site may also be one of the mechanisms for the downregulation of miR-20a expression in breast cancer cells.

Dysregulated histone methylation and miR-20a/MAPK1/c-Myc feedback loop involved in regulation of miR-20a expression. (a) A schematic diagram shows the genomic context of miR-20a and the black arrowheads represent miR-20a. ChIP analysis for H3K4me3, H3K9me3, H3K27me3 and H4K20me3 was done with two sets of primers spanning the reported genomic regulatory regions (shown as filled bars). (b) The q-PCR analysis of the regulatory regions of miR-20a by ChIP assay showed that H3K4me3 descended and H4K20me3 increased in drug-resistant cell lines compared with parental cell line. (c) Ectopic expression of miR-20a decreased c-myc levels as shown by western blot analysis. (d) Representative images of tumor samples that were stained with c-Myc by IHC (up). The levels of c-Myc protein expression were measured (down). Bars: (main) 100 μm; (insets) 100 μm. (e) Schematic representation of the genomic interval encompassing the miR-17/20a cluster. Putative c-Myc binding sites are indicated (CACGTG or CATGTG). q-PCR amplicons are represented by numbered lines. (f) q-PCR analysis of c-Myc chromatin immunoprecipitated DNA. Three fragments derived from chromosome 13q31 upstream of miR-17/20a cluster were examined. (g) Effects of c-Myc on the miR-17/20a promoter. Bcap37 cells were transfected with the empty pGL3-Basic vector or the Squ-1 to Squ-5 constructs as indicated along with the empty pcDNA vector or a construct expressing c-Myc. Squ-1, pGL3-Basic vector contain 3.9 kb (−3800 to + 100) fragment; Squ-2, Squ-1 deleted site 1; Squ-3, Squ-1 deleted site 2; Squ-4, Squ-1 deleted site 3; Squ-5, Squ-1 deleted sites 1–3. (h) siRNA-mediated c-Myc knockdown reduced miR-17 and miR-20a levels as shown by q-PCR analysis in BCap37 and Bads-200 cells, whereas the expression of miR-1249 was not affected. (i) Ectopic expression of c-Myc promoted the expression of miR-17 and miR-20a as shown by q-PCR analysis. *P<0.05, **P<0.01, ***P<0.001
The MAPK/ERK pathway regulates the expression of various downstream transcription factors, including c-Myc.29 In the breast cancer specimens from the TCGA database, the expression of c-Myc was positively correlated with MAPK1. Like MAPK1, the expression level of c-Myc protein is not sufficient to predict the prognosis of patients (Supplementary Figures S6a–d). In this study, we tried to identify whether c-Myc can also regulate the expression of the miR-17-92 family in breast cancer cells. In the specimens of breast cancer patients, the expression of MAPK1 and c-Myc was increased in breast cancer tissues compared with paracancerous tissues, whereas the expression of miR-20a was decreased (Supplementary Figure S4k). After ectopic expression of miR-20a in breast cancer cells, the expression of c-Myc was inhibited (Figure 7c). The expression of c-Myc also decreased after overexpression of miR-20a in the subcutaneous breast cancer of nude mice model (Figure 7d). We analyzed the sequence of ∼2 kb upstream of the miR-17-92 family transcriptional initiation site and found three c-Myc binding sites (Figure 7e).The c-Myc can bind to the canonical E-box sequence CACGTG and non-canonical E-box sequence CATGTG.30 By ChIP technique, we found that c-Myc was significantly enriched at the second binding sites (Figure 7f). We cloned the sequence of ∼4 kb upstream of the transcription initiation site into the pGL3-Basic vector, and constructed the vectors lacking the binding sites 1, 2 and 3, respectively. These vectors were transferred into BCap37, respectively, and then the luciferase activity was detected. The results showed that when the cells were transfected with the vector that contained the complete sequence, luciferase activity increased ∼72 times compared with negative control. Compared with the blank vector, c-Myc significantly enhanced luciferase activity. When the cell was transfected with the vector lack of binding site 2, luciferase activity decreased most obviously, whereas it was not as severe in the absence of binding sites 1 or 3 (Figure 7g). This demonstrated that binding site 2 is the most important for c-Myc transcriptional activation. To further investigate the effect of c-Myc on miR-17 and miR-20a expression, we knocked down c-Myc and found that miR-17/20a was significantly decreased whereas another unrelated miR-1249 was unaffected (Figure 7h and Supplementary Figure S6e). Conversely, overexpression of c-Myc resulted in a significant increase in miR-17/20a levels (Figure 7i and Supplementary Figure S6f). In summary, these data suggested that in breast cancer cell miR-20a inhibits c-Myc expression and c-Myc promotes miR-20a expression, forming a feedback loop.
Discussion
Because of the cell and tissue-type specificity of the function of miRNA and the diversity of its target genes, it is still a challenge to obtain miRNAs with specific regulatory functions.19 In order to screen out the miRNAs regulating the drug resistance in breast cancer cells, two drug-resistant cells, Bads-200 and Bats-72, that were screened from BCap37 in our laboratory were used as the model. The two drug-resistant cells have the same source, but there are some differences in drug resistance.18 Thus, in theory, miRNAs that altered their expression level in the two chemoresistant cell lines compared with parental BCap37 cells may associate with drug resistance. After a series of experiments, finally, we found that miR-20a, which was downregulated in both drug-resistant cells, could markedly reduce cell resistance and inhibit cell proliferation. Besides, with the clinical specimens we collected (n=106) and the breast cancer cohort from TCGA (n=1096), we also found that miR-20a/b is a potential marker for predicting prognosis in breast cancer patients, especially for patients with luminal A breast cancer.
The role of miR-20a in the development of cancer remains to be elucidated. Most of the studies suggested that miR-20a has a promoting effect on cancer.31, 32, 33, 34, 35, 36 However, it has been reported that in oral squamous cell carcinoma,37 pancreatic cancer38 and esophageal squamous cell carcinoma,39 miR-20a acts as a tumor suppressor. In breast cancer, both the cancer-promoting and cancer-suppressing functions have been reported.40, 41, 42 These opposite conclusions may be because of the use of different cell models in different studies. In terms of regulation of cell resistance, studies have shown that miR-20a can improve drug resistance of colorectal cancer43 and leukemia.44 However, in osteosarcoma45 and NSCLC,46 miR- 20a was reported as reducing the resistance of cancer cells to chemotherapeutic drugs. These conflicted results fully illustrate the complexity of the miR-20a regulatory network and it requires further study. Indeed, a miRNA often targets multiple mRNAs, known as the ‘targetome’, to achieve different functions.47 Previous studies have shown that miR-17/20 may promote breast cancer cell line MCF-7 to be sensitive to doxorubicin-induced apoptosis by targeting Akt1.48 In light of the present study, miR-20a may be involved in the regulation of drug resistance by targeting different pathways. Moreover, there may be other miR-20a-associated molecular pathways in breast cancer resistance that may further reveal miR-20a regulation of drug resistance in the integrity of the pathway.
Impressively, we found that the dual roles of miR-20a in inhibiting cell proliferation and drug resistance were achieved through directly targeting of MAPK1 and inhibiting its expression. MAPK1, also known as ERK2, is a member of the Ras/Raf/ERK(MAPK/ERK) pathway and is thought to play a role in promoting cell proliferation.24 The Ras/Raf/ERK pathway is often overactivated in tumor cells and promotes cell proliferation and malignant transformation.24 Therefore, the pathway is considered an attractive drug target.49 In this study, on one hand, miR-20a inhibited tumor growth by directly targeting Ras/ Raf/ERK pathway, and on the other hand, miR-20a also downregulated the resistance protein P-gp by inhibiting ERK2. P-gp can pump a range of anticancer drugs out from cells, including anthracycline, vinblastine and taxanes.50 This may be one reason explaining how miR-20a can increase the sensitivity of breast cancer cells to multiple drugs. In summary, we identified MAPK1 as a new target for miR-20a. By inhibiting MAPK1, miR-20a plays a role in inhibiting tumor cell proliferation and drug resistance. This may be the mechanism by which miR-20a plays dual functions in this study.
The expression of miR-20a is affected by a variety of factors, including histone methylation dysregulation and PTX treatment. We found that PTX will reduce the expression of miR-20a in BCap37 cell line, a mechanism that remains to be further studied. In addition, the MAPK/ERK pathway also regulates the expression of miR-20a. MAPK/ERK promotes cell growth by activating a series of downstream transcription factors, including c-Myc.24 c-Myc, as an oncogene, is upregulated in a variety of cancer cells. For human breast cancer, ∼50% of the samples showed c-Myc gene amplification.51 The half-life of c-Myc is very short, and phosphorylated ERK can phosphorylate c-Myc, thereby increasing its stability.52 Previous studies have shown that the expression of miR-17/20a is induced by c-Myc in B cells,30 and this was also demonstrated in breast cancer cells by us. ChIP assay showed that c-Myc could bind to the promoter region of miR-17/20a gene. Overexpression of c-Myc increased the expression of miR-17/20a, whereas miR-17/20a was decreased after knockdown of c-Myc. Interestingly, studies have shown that in B cells, c-Myc mainly binds to the site 1 of the promoter region of miR-17/20a gene.30 However, in our study, we found that c-Myc mainly binds to site 2 (Figures 7d–f). This may be because of the different cell models used. We also demonstrated for the first time that miR-20a decreased the expression of c-Myc by inhibiting the MAPK/ERK pathway and formed a feedback loop, thus controlling the growth signal of the cell (Figure 8). In this loop, miR-20a is induced by c-Myc and c-Myc can be downregulated by miR-20a through targeting MAPK1. Thus, miR-20a/Myc loop forms a dynamic balance in the breast cancer cells, and final development of the tumor will depend to some extent on the trend of the balance in this study. It may be that this balance in tumor tissue tends to decrease the expression of miR-20a and increase the expression of c-Myc and MAPK1.

Schematic model of miR-20a in regulating drug response. MiR-20a increases drug sensitivity in breast cancer cells through reducing drug efflux by inhibiting P-gp in a MAPK1-dependent manner. In drug-resistant cells, the dysregulated histone methylation inhibits the expression of miR-20a. In addition, miR-20a decreased the expression of c-Myc by inhibiting the MAPK/ERK pathway and formed a feedback loop, thus controlling the drug response and growth signal of the cells
Our findings provide a new understanding of miRNA in breast cancer growth and resistance, and provide new potential clues for the treatment of breast cancer. PTX, a first-line chemotherapeutic agent for breast cancer, can improve short-term survival rate, but is not beneficial in 5-year survival rate of patients. The main reason is that treatment with PTX will lead to drug resistance.53 Based on our research, miR-20a can inhibit the growth and resistance of breast cancer by targeting MAPK1. Our study also revealed the presence of miR-20a/MAPK1/c-Myc signaling loop in breast cancer that may represent a novel mechanism for breast cancer growth regulation. As chemotherapeutic agents are still the first choice for cancer treatment in developing countries,19 miR-20a combined with chemotherapeutic agents may maximize their synergistic effect and improve the therapeutic effect of breast cancer, especially for patients who are resistant to chemotherapy.
Materials and methods
Tissues and cell lines
The paraffin-embedded of primary breast cancer samples and paired adjacent normal tissues were obtained from patients who had undergone surgery at First Affiliated Hospital of Zhejiang University. The fresh breast cancer tissues and paired adjacent normal tissues were from Zhejiang Provincial Hospital of Traditional Chinese Medicine and First Affiliated Hospital of Zhejiang University. Surgically removed tissues were quickly frozen in liquid nitrogen until analysis. The details of all samples are shown in Supplementary Tables S2 and S3. The effect of chemotherapy on the tumors was assessed as the three-dimensional volume reduction rate or tumor response rate as described previously.54
The human embryonic kidney cell line 293T, the cervical cancer cells Hela and the human breast cancer cell lines BCap37, Hs-578T, MDA-MB-231, MDA-MB-468, MDA-MB-453, BT-474, Sk-Br-3 and MCF-7 were purchased from cell bank of the Chinese Scientific Academy (Shanghai, China). BCap37, BCap37/MDR, Bads-200, Bats-72, BT-474 and MCF-7 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Biological Industries, Cromwell, CT, USA). MDA-MB-231, MDA-MB-453 and MDA-MB-468 cells were cultured in Leibovitz’s L-15 (Gibco) with 10% FBS. Hs-578T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) and 10% FBS. SK-BR-3 cells were cultured in McCoy’s 5A (modified, Gibco) with 10% FBS. The cultures were kept at 37 °C with 5% CO2 in a water-jacketed incubator (Thermo Fisher, Carlsbad, CA, USA).
The miRNA expression microarray data are available in GEO database (www.ncbi.nlm.nih.gov/geo/) with accession number GSE87570.
Accession codes
References
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F et al. Cancer statistics in China, 2015. CA Cancer J Clin 2016; 66: 115–132.
Siegel RL, Miller KD, Jemal A . Cancer statistics, 2016. CA Cancer J Clin 2016; 66: 7–30.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG . Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13: 714–726.
Park JW, Amarapurkar D, Chao Y, Chen PJ, Geschwind JF, Goh KL et al. Consensus recommendations and review by an International Expert Panel on Interventions in Hepatocellular Carcinoma (EPOIHCC). Liver Int 2013; 33: 327–337.
Longley DB, Johnston PG . Molecular mechanisms of drug resistance. J Pathol 2005; 205: 275–292.
Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100: 672–679.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–233.
Ambros V . The functions of animal microRNAs. Nature 2004; 431: 350–355.
Kim AH, Reimers M, Maher B, Williamson V, McMichael O, McClay JL et al. MicroRNA expression profiling in the prefrontal cortex of individuals affected with schizophrenia and bipolar disorders. Schizophr Res 2010; 124: 183–191.
Park EY, Chang E, Lee EJ, Lee HW, Kang HG, Chun KH et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res 2014; 74: 7573–7582.
Hu Q, Gong JP, Li J, Zhong SL, Chen WX, Zhang JY et al. Down-regulation of miRNA-452 is associated with adriamycin-resistance in breast cancer cells. Asian Pac J Cancer Prev 2014; 15: 5137–5142.
Li G, Wu X, Qian W, Cai H, Sun X, Zhang W et al. CCAR1 5′ UTR as a natural miRancer of miR-1254 overrides tamoxifen resistance. Cell Res 2016; 26: 655–673.
Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y et al. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem 2010; 285: 21496–21507.
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007; 129: 1401–1414.
Foschini MP, Eusebi V . Rare (new) entities of the breast and medullary carcinoma. Pathology 2009; 41: 48–56.
Bertucci F, Finetti P, Cervera N, Charafe-Jauffret E, Mamessier E, Adélaïde J et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res 2006; 66: 4636–4644.
Vincent-Salomon A, Gruel N, Lucchesi C, Macgrogan G, Dendale R, Sigal-Zafrani B et al. Identification of typical medullary breast carcinoma as a genomic sub-group of basal-like carcinomas, a heterogeneous new molecular entity. Breast Cancer Res 2007; 9: R24.
Jiang D, Sui M, Zhong W, Huang Y, Fan W . Different administration strategies with paclitaxel induce distinct phenotypes of multidrug resistance in breast cancer cells. Cancer Lett 2013; 335: 404–411.
Mu W, Hu C, Zhang H, Qu Z, Cen J, Qiu Z et al. miR-27b synergizes with anticancer drugs via p53 activation and CYP1B1 suppression. Cell Res 2015; 25: 477–495.
Jiang D, Huang Y, Han N, Xu M, Xu L, Zhou L et al. Fulvestrant, a selective estrogen receptor down-regulator, sensitizes estrogen receptor negative breast tumors to chemotherapy. Cancer Lett 2014; 346: 292–299.
Zheng H, Liu Z, Liu T, Cai Y, Wang Y, Lin S et al. Fas signaling promotes chemoresistance in gastrointestinal cancer by up-regulating P-glycoprotein. Oncotarget 2014; 5: 10763–10777.
El Azreq MA, Naci D, Aoudjit F . Collagen/beta1 integrin signaling up-regulates the ABCC1/MRP-1 transporter in an ERK/MAPK-dependent manner. Mol Biol Cell 2012; 23: 3473–3484.
Zhao BX, YB Sun, Wang SQ, Duan L, Huo QL, Ren F et al. Grape seed procyanidin reversal of p-glycoprotein associated multi-drug resistance via down-regulation of NF-kappaB and MAPK/ERK mediated YB-1 activity in A2780/T cells. Plos ONE 2013; 8: e71071.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007; 1773: 1263–1284.
Lin JC, Chang SY, Hsieh DS, Lee CF, Yu DS . Modulation of mitogen-activated protein kinase cascades by differentiation-1 protein: acquired drug resistance of hormone independent prostate cancer cells. J Urol 2005; 174: 2022–2026.
Yan HL, Xue G, Mei Q, Wang YZ, Ding FX, Liu MF et al. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J 2009; 28: 2719–2732.
Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA 2006; 103: 9136–9141.
Esteller M . Epigenetics in cancer. N Engl J Med 2008; 358: 1148–1159.
Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 2003; 17: 1263–1293.
O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT . c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005; 435: 839–843.
Chang Y, Liu C, Yang J, Liu G, Feng F, Tang J et al. MiR-20a triggers metastasis of gallbladder carcinoma. J Hepatol 2013; 59: 518–527.
Wang M, Gu H, Qian H, Zhu W, Zhao C, Zhang X et al. miR-17-5p/20a are important markers for gastric cancer and murine double minute 2 participates in their functional regulation. Eur J Cancer 2013; 49: 2010–2021.
Huang G, Nishimoto K, Zhou Z, Hughes D, Kleinerman ES . miR-20a encoded by the miR-17-92 cluster increases the metastatic potential of osteosarcoma cells by regulating Fas expression. Cancer Res 2012; 72: 908–916.
Li X, Pan JH, Song B, Xiong EQ, Chen ZW, Zhou ZS et al. Suppression of CX43 expression by miR-20a in the progression of human prostate cancer. Cancer Biol Ther 2012; 13: 890–898.
Zhao S, Yao D, Chen J, Ding N, Ren F . MiR-20a promotes cervical cancer proliferation and metastasis in vitro and in vivo. PLoS ONE 2015; 10: e0120905.
Cheng D, Zhao S, Tang H, Zhang D, Sun H, Yu F et al. MicroRNA-20a-5p promotes colorectal cancer invasion and metastasis by downregulating Smad4. Oncotarget 2016; 7: 45199–45213.
Chang CC, Yang YJ, Li YJ, Chen ST, Lin BR, Wu TS et al. MicroRNA-17/20a functions to inhibit cell migration and can be used a prognostic marker in oral squamous cell carcinoma. Oral Oncol 2013; 49: 923–931.
Yan H, Wu J, Liu W, Zuo Y, Chen S, Zhang S et al. MicroRNA-20a overexpression inhibited proliferation and metastasis of pancreatic carcinoma cells. Hum Gene Therapy 2010; 21: 1723–1734.
Jing C, Ma G, Li X, Wu X, Huang F, Liu K et al. MicroRNA-17/20a impedes migration and invasion via TGF-beta/ITGB6 pathway in esophageal squamous cell carcinoma. Am J Cancer Res 2016; 6: 1549–1562.
Kim K, Chadalapaka G, Lee SO, Yamada D, Sastre-Garau X, Defossez PA et al. Identification of oncogenic microRNA-17-92/ZBTB4/specificity protein axis in breast cancer. Oncogene 2012; 31: 1034–1044.
Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol 2008; 182: 509–517.
Yu Z, Willmarth NE, Zhou J, Katiyar S, Wang M, Liu Y et al. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci USA 2010; 107: 8231–8236.
Chai H, Liu M, Tian R, Li X, Tang H . miR-20a targets BNIP2 and contributes chemotherapeutic resistance in colorectal adenocarcinoma SW480 and SW620 cell lines. Acta Biochim Biophys Sin (Shanghai) 2011; 43: 217–225.
Weng H, Huang H, Dong B, Zhao P, Zhou H, Qu L . Inhibition of miR-17 and miR-20a by oridonin triggers apoptosis and reverses chemoresistance by derepressing BIM-S. Cancer Res 2014; 74: 4409–4419.
Pu Y, Yi Q, Zhao F, Wang H, Cai W, Cai S . MiR-20a-5p represses multi-drug resistance in osteosarcoma by targeting the KIF26B gene. Cancer Cell Int 2016; 16: 64.
Jiang Z, Yin J, Fu W, Mo Y, Pan Y, Dai L et al. MiRNA 17 family regulates cisplatin-resistant and metastasis by targeting TGFbetaR2 in NSCLC. PLoS ONE 2014; 9: e94639.
Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 2011; 19: 232–243.
Yu Z, Xu Z, Disante G, Wright J, Wang M, Li Y et al. miR-17/20 sensitization of breast cancer cells to chemotherapy-induced apoptosis requires Akt1. Oncotarget 2014; 5: 1083–1090.
Santarpia L, Lippman SM, El-Naggar AK . Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets 2012; 16: 103–119.
Katayama K, Yoshioka S, Tsukahara S, Mitsuhashi J, Sugimoto Y . Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol Cancer Ther 2007; 6: 2092–2102.
Liao DJ, Dickson RB . c-Myc in breast cancer. Endocr Relat Cancer 2000; 7: 143–164.
Sears R, Leone G, DeGregori J, Nevins JR . Ras enhances Myc protein stability. Mol Cell 1999; 3: 169–179.
Ding S, Chamberlain M, McLaren A, Goh L, Duncan I, Wolf CR . Cross-talk between signalling pathways and the multidrug resistant protein MDR-1. Br J Cancer 2001; 85: 1175–1184.
Fang L, Li H, Wang L, Hu J, Jin T, Wang J et al. MicroRNA-17-5p promotes chemotherapeutic drug resistance and tumour metastasis of colorectal cancer by repressing PTEN expression. Oncotarget 2014; 5: 2974–2987.
Acknowledgements
We acknowledge Dr. YingNan LiTao for critical amendments on the manuscript. We thank Dr. Xinhua Feng for presenting the c-Myc expression plasmid and Dr. Su Zeng for presenting the BCap37/MDR cell line. We are also grateful to Youfa Zhu, Yanwei Li, JiaJia Wang and Qiong Huang, the teachers in Public Platform of Zhejiang University of Medicine, to provide the technical support in the manipulation of flow cytometry and IHC assay. This study was supported by Grants National Natural Science Foundation of China 81572987 and 81372462, Grant 2014C03012 from the Department of Science and Technology of Zhejiang province, National Key Project on New Drug Developmental Program 2014ZX09507009026 (to WF) and Grants National Natural Science Foundation of China 81302288 (to DJ).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Del Sal
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Si, W., Shen, J., Du, C. et al. A miR-20a/MAPK1/c-Myc regulatory feedback loop regulates breast carcinogenesis and chemoresistance. Cell Death Differ 25, 406–420 (2018). https://doi.org/10.1038/cdd.2017.176
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.176
This article is cited by
-
Landscape of NcRNAs involved in drug resistance of breast cancer
Clinical and Translational Oncology (2023)
-
Long noncoding RNA LINC00958 suppresses apoptosis and radiosensitivity of colorectal cancer through targeting miR-422a
Cancer Cell International (2021)
-
The network of non-coding RNAs and their molecular targets in breast cancer
Molecular Cancer (2020)
-
DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma
Molecular Cancer (2020)
-
MiR-93 suppresses tumorigenesis and enhances chemosensitivity of breast cancer via dual targeting E2F1 and CCND1
Cell Death & Disease (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}