
Abstract
Chromosomal abnormalities are implicated in a substantial number of human developmental syndromes, but for many such disorders little is known about the causative genes. The recently described 1q41q42 microdeletion syndrome is characterized by characteristic dysmorphic features, intellectual disability and brain morphological abnormalities, but the precise genetic basis for these abnormalities remains unknown. Here, our detailed analysis of the genetic abnormalities of 1q41q42 microdeletion cases identified TP53BP2, which encodes apoptosis-stimulating protein of p53 2 (ASPP2), as a candidate gene for brain abnormalities. Consistent with this, Trp53bp2-deficient mice show dilation of lateral ventricles resembling the phenotype of 1q41q42 microdeletion patients. Trp53bp2 deficiency causes 100% neonatal lethality in the C57BL/6 background associated with a high incidence of neural tube defects and a range of developmental abnormalities such as congenital heart defects, coloboma, microphthalmia, urogenital and craniofacial abnormalities. Interestingly, abnormalities show a high degree of overlap with 1q41q42 microdeletion-associated abnormalities. These findings identify TP53BP2 as a strong candidate causative gene for central nervous system (CNS) defects in 1q41q42 microdeletion syndrome, and open new avenues for investigation of the mechanisms underlying CNS abnormalities.
Similar content being viewed by others


Main
Chromosomal deletions that cause rare genetic disorders usually affect more than one gene and cause multiple phenotypic features. Identification of causative genes could extend our understanding of molecular pathways that are responsible for common diseases. Deletions in the chromosomal 1q41−q44 region are significantly associated with central nervous system (CNS) defects including neural tube defects (NTDs), agenesis of corpus callosum, microcephaly and hydrocephalus.1, 2 In particular, small interstitial deletions in the 1q41q42 region are implicated in the 1q41q42 microdeletion syndrome with features of severe developmental delay, intellectual disability, and brain morphological abnormalities. A critical region comprising the genes FBXO28, TP53BP2, CAPN2, and CAPN8 has been proposed for the 1q41q42 microdeletion syndrome based on the smallest region of overlap (SRO).3 FBXO28 (encoding the F-box only protein 28, a ubiquitin ligase) has been proposed as a candidate gene responsible for seizures and intellectual disability in microdeletion patients.4, 5 However, the genes responsible for other phenotypes, in particular the brain morphological abnormalities, of the syndrome are unknown.
TP53BP2, which is located at the boundary of 1q41 and 1q42, encodes the apoptosis-stimulating protein of p53 2 (ASPP2). It is also an ankyrin repeat, SH3 domain and proline-rich containing protein (ASPP), originally identified as a positive regulator of p53-mediated transcription of apoptotic genes.6, 7 In mice, ASPP2, encoded by Trp53bp2, is essential for embryonic development:8, 9 an ASPP2-null genotype in mice is lethal prior to embryonic day E7 (ref. 8) and mice with homozygous deletion of exon 3 exhibit loss of neuroepithelial cell polarity and hydrocephalus.10 ASPP2 is involved in regulating apoptosis through binding to p53 (and its sibling proteins p63 and p73) via its C-terminus and in regulating cell polarity through binding to the tight-junction protein PAR3 via its N-terminus.10, 11 Both p53 and PAR3 have been implicated in NTDs,12, 13, 14, 15 and the process of neural tube closure − which involves convergent extension and apical constriction to enable folding of a neuroepithelial sheet into the neural tube − involves apicobasal polarity and apoptosis.16, 17, 18 Therefore, we hypothesized that TP53BP2 might contribute to the developmental abnormalities associated with 1q41q42 deletions. Through examination of ASPP2 deficiency in mice and human patients, we show that TP53BP2 is a strong candidate for being a gene responsible for CNS defects and, potentially, other 1q4 deletion-associated phenotypes in humans.
Results
TP53BP2 deletions are associated with brain abnormalities in 1q41q42 microdeletion syndrome
To identify the gene(s) responsible for structural brain abnormalities in 1q41q42 microdeletion syndrome, we first established the SRO in 1q41q42 microdeletion cases with structural brain abnormalities. To date there are 31 reported 1q41q42 microdeletion cases and 26 of them have brain MRI/CT recorded. Among the 26 cases with deletions in the 1q41q42 region for which neuroradiology reports and/or MRI scans were available, 19 were from published reports and 7 are newly identified cases (Supplementary Table 1).3, 4, 19, 20, 21, 22, 23, 24, 25, 26 Of these 26 cases, 19 patients showed detectable structural brain abnormalities and their SRO (chr1:223 828 382−224 034 322; hg19) contains the entire coding regions of CAPN2 and TP53BP2, plus exons 1 and 2 of CAPN8 (Figure 1), whereas in 4 of the remaining 7 cases without brain abnormalities the TP53BP2 locus is not affected (Figures 1a and b).

TP53BP2 is deleted in 1q41q42 microdeletion patients with brain abnormalities. (a) Genomic locations of 1q41q42 deletions from new and published patients. Red indicates brain morphological abnormalities were reported for the patient, green indicates absence of reported brain abnormalities. The positions of the CAPN8, CAPN2, TP53BP2, and FBXO28 genes are marked with black arrows; black line marks the position of TP53BP2. *FBXO28 is not contained in the smallest region of overlap (SRO) of patient deletions with brain abnormalities. Panel generated using UCSC genome browser (http://genome.ucsc.edu), hg19 assembly. (b) The proportions of cases with and without TP53BP2 deletion with and without brain abnormalities. (c) Focus on the SRO of patient deletions with brain morphological abnormalities. Patients with abnormal ventricles are marked in dark purple
CAPN2 and CAPN8 encode Ca2+-dependent proteases of the calpain family.27 Mice deficient in the orthologous genes Capn2, Capn8, and Trp53bp2 have been generated previously: Trp53bp2-deficient mice exhibit abnormalities in the CNS,10 whereas Capn2-deficient and Capn8-deficient mice show no abnormalities in the brain or nervous system.28, 29, 30 Publicly available data also show that TP53BP2 is expressed more highly in the brain than all other examined tissues, whereas CAPN2 is expressed at very low levels in the brain compared to other tissues, and CAPN8 expression is restricted to the stomach (Supplementary Figure 1a, www.gtexportal.org)31 Additionally, TP53BP2 is most highly expressed in the ventricular and subventricular zones in the human embryonic brain throughout gestation, consistent with it having a key role in embryonic brain development (Supplementary Figure 1b, http://brainspan.org, http://human.brain-map.org).32, 33 We also found no deletions of TP53BP2 in publically available copy number data from two large cohorts of healthy individuals (19 584 healthy individuals;34, 35 or in an independent paediatric control group of 2026 (ref. 36)), suggesting that TP53BP2 deletions may be pathogenic. In contrast, two deletions, nsv520609 and nsv521758, spanning multiple exons of CAPN2, were detected in the cohort of 2026 disease-free individuals.36 These observations suggest that TP53BP2 is the strongest candidate for being the causative gene responsible for brain morphological abnormalities associated with 1q41q42 microdeletions.
Brain MRI reveals common ventricular abnormalities among patients with TP53BP2 deletions
We compared structural brain phenotypes among 1q41q42 microdeletion patients with and without heterozygous TP53BP2 deletions. Among the 22 TP53BP2 deletion cases, the most frequently observed brain abnormality, seen in 16 out of 22 patients (73%) with heterozygous TP53BP2 deletions, was dilation of ventricles and/or ventricular dysmorphism, frequently associated with hypoplasia or agenesis of corpus callosum (Supplementary Table 2). All four 1q41q42 microdeletion patients without TP53BP2 deletions showed normal ventricles (Supplementary Table 2, Figure 1b). Corpus callosum defects were observed in 10/22 (45%) of TP53BP2-deletion patients (5 with agenesis and 5 with hypoplasia), but in none of the patients without TP53BP2 mutations. Cerebellar hypoplasia, encephalocele, hypomyelination, plagiocephaly, holoprosencephaly, and malformations of sulcation were also observed in some TP53BP2 deletion patients at a lower frequency (Supplementary Table 2).
For the identified 26 cases of 1q41q42 microdeletion patients, only 14 brain MRI scans were available to us and 12 of them had morphometry-compatible MRI scans (9 with TP53BP2 deletion and 3 without) (Figure 2a). We used probabilistic morphometry to quantify the lateral ventricle (LV) volume of the 12 1q41q42 microdeletion patients (Figure 2b). 28 normal children’s brain MRIs from the NIH Paediatric MRI Data Depository were used as controls (Figure 2b). As expected, LV volume was significantly larger in the TP53BP2 deletion group (mean 33.4 ml) (Figure 2b) compared to control group (mean 10.0 ml, S.D.=4.5 ml) (P<0.001) and the LV volumes of microdeletion patients without TP53BP2 deletions were within normal range (Figure 2b).

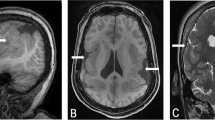
Enlarged ventricles and other abnormalities in patients with TP53BP2 deletions. (a) Summary of the patient records analysed in this study. (b) Quantification of lateral ventricle volume by probabilistic morphometry. Volumes were computed by ALVIN on all suitable scans in each MRI series, from 1q41q42 microdeletion patients with and without TP53BP2 deletion. Left panel – T1 and T2 sagittal, axial and coronal scans were used for computations of LV volumes, bars showing mean±S.D.; right panel – T1 axial scans used only, each data point showing the mean from T1 axial scans for each patient. MRI scans of 12 patients and a group of 28 healthy paediatric individuals (NIH controls) were used. Median age of microdeletion cohort=19 months, median of NIH controls=15 months. For statistical comparison of LV volumes in patient groups, values were first tested for normality by the D’Agostino and Pearson omnibus test, then compared to NIH controls using the two-tailed Student’s t-test and corrected for multiple hypothesis testing by multiplying the P-value by the number of tests. Mean±S.D. are shown. (c) Axial and sagittal MRI images of brains of 1q41q42 microdeletion patients. T1 non-contrast images are shown, except for cases where only T2 scans were available (Rosenfeld Subject 14, Spreiz et al.24 and Case 7). Double white arrowheads: hypoplasia of corpus callosum; single white arrowheads: asymmetry in lateral ventricle size; asterisk: mega cisterna magna
We observed defects in the ventricles of cases with TP53BP2 deletions but not in microdeletion cases without TP53BP2 deletions. For example, in four cases with TP53BP2 deletions the bodies of the LVs appeared more parallel due to hypoplasia of the corpus callosum (Figure 2c, Supplementary Table 2: patients Case 4; Shaffer 3; Rosenfeld 8; Rosenfeld 13). This is similar but more subtle than the ventricular dysmorphism seen in association with agenesis of corpus callosum. In three cases the LVs were asymmetric with one side larger than the other (Figure 2c: patients Shaffer 3; Rosenfeld 12 and Wat et al.26). These data show that TP53BP2 deletions are associated with ventricular abnormalities that are frequently accompanied by hypoplasia of corpus callosum and/or cerebellum.
ASPP2 deficiency in mice causes CNS abnormalities with similar features to 1q41q42 microdeletion patients
We used mice with deletion of Trp53bp2 exon 3 (Trp53bp2Δ3) to examine how ASPP2 deficiency affects CNS development. We showed previously that the Trp53bp2Δ3/Δ3 genotype results in severe developmental defects in a strongly background-dependent manner: BALB/c Trp53bp2Δ3/Δ3 mice are born at the expected rate but most develop severe hydrocephalus before weaning, whereas only 30% of mixed C57BL/6-129 Sv background (further referred to as ‘mixed’) Trp53bp2Δ3/Δ3 mice survive postnatally.10 On a pure C57BL/6 (referred to as B6) background, which we established by back-crossing over 10 generations, no homozygous Trp53bp2Δ3/Δ3 mice survived after birth. The surviving BALB/c Trp53bp2Δ3/Δ3 mice have a 100% incidence of hop gait. Most severe cases of neonatal hydrocephalus develop prior to birth and likely result from early defects in brain development.
To provide direct evidence that ASPP2 deficiency causes structural abnormalities in mouse embryonic brain, we used micro-computed tomography (microCT) and high-resolution episcopic microscopy (HREM) to examine Trp53bp2Δ3/Δ3 embryos. These methods allow reconstruction of three-dimensional images as well as individual sections, showing the morphology of anatomical defects.37 At embryonic stages E13.5 and E14.5, ~50% of Trp53bp2Δ3/Δ3 BALB/c embryos exhibited enlargement of brain ventricles (Figure 3a). Subcranial and intraventricular haemorrhages were also visible by naked eye and micro-CT in 50% of Trp53bp2Δ3/Δ3 BALB/c embryos (Figure 3a, Supplementary Figures 3a and b). Brains of virtually all Trp53bp2Δ3/Δ3 embryos examined by HREM had absent or severely hypoplastic pineal gland (Figure 3b). The spinal canal of all Trp53bp2Δ3/Δ3 embryos was abnormal with stenosis (Supplementary Figure 3c), the presence of blood (Supplementary Figure 3d) or small cyst-like structures in the caudal part of the neural tube (Supplementary Figure 3e). All Trp53bp2Δ3/Δ3 embryos examined had abnormalities in the cerebrospinal fluid space with blockage and/or haemorrhage in at least one part of the ventricular system and/or spinal canal. Heterozygous (Trp53bp2Δ3/+) embryos showed CNS abnormalities, such as spinal canal stenosis or asymmetric LVs, but with lower penetrance (Figure 3d). Twenty-five per cent of BALB/c Trp53bp2Δ3/+ embryos showed localized abnormalities in the distribution of neuroepithelial brain tissue associated with small patches of subcranial haemorrhage (Figure 3d). This shows that ASPP2 is required for early stages of CNS development. Interestingly, surface rendering of LVs of Trp53bp2Δ3/Δ3 embryos showed a similar shape to the dysmorphism observed in patients with TP53BP2 deletion, with bodies of the LVs being more parallel and bulbous (Figure 3c). This suggests that ASPP2 deficiency in the Trp53bp2Δ3/Δ3 mice might mimic effects of ASPP2 deficiency in humans resulting from the heterozygous deletion of TP53BP2.

Trp53bp2Δ3/Δ3 and Trp53bp2Δ3/+ mice have CNS defects. (a) Gross dilation of lateral ventricles is visible in Trp53bp2Δ3/Δ3 embryos, resulting in bulging foreheads. Intraventricular haemorrhage is frequently seen (white arrows). (b) Present and absent pineal gland (red arrows) in wild-type and Trp53bp2Δ3/Δ3 embryos, respectively, as seen in a single HREM section. (c) Surface rendering of lateral ventricles in 1q41q42 microdeletion patients (left, constructed from MRI scans) and Trp53bp2Δ3/Δ3 vs wild-type embryos (right, constructed from microCT scans). (d) Brain abnormalities in heterozygous Trp53bp2Δ3/+ embryos. Left: Asymmetry of lateral ventricles in a heterozygous Trp53bp2Δ3/+ embryo caused by a structural abnormality (red arrow). MicroCT image is shown. Right: Asymmetry in neuroepithelial tissue distribution in the brain (red arrow) and the associated subcranial haemorrhage (white and red arrows) in a BALB/c Trp53bp2Δ3/+ embryo
ASPP2 deficiency causes NTDs in mice
ASPP2 was reported previously to control the cell polarity and cell proliferation of neural epithelial cells.10 These are two important biological processes involved in neural tube closure, an early event in CNS development, the disruption of which causes NTDs. To further investigate the effect of genetic background on phenotype severity in ASPP2-deficient mice, we compared Trp53bp2Δ3/Δ3 embryos in mixed, B6 and BALB/c backgrounds. NTDs were detected in 15% (n=127) of embryos on the mixed background, 46% (n=13) on the pure B6 background and 8% (n=12) on the BALB/c background (Figure 4, Supplementary Table 3). The only NTD observed in BALB/c embryos was exencephaly (Figure 4, Supplementary Figure 4a). On the mixed background, NTDs were small at the early stages (E9.5−E10.5) but progressed to larger lesions at >E13.5, and featured mainly exencephaly and rostral spina bifida (Figure 4) but rarely craniorachischisis (Figure 4 − middle panel). In contrast, NTDs in Trp53bp2Δ3/Δ3 B6 embryos have a broad spectrum of phenotypes, ranging from spina bifida to craniorachischisis with some spina bifida phenotypes affecting a large section of the neural tube (Figure 4, Supplementary Figure 4b). MicroCT and HREM inspection of Trp53bp2Δ3/Δ3 embryos with NTDs revealed occasional overgrowth of neural tissue (Supplementary Figure 4a). The spinal column of the neural tube was often wrinkled, even in embryos without NTDs (Supplementary Figure 3e).

Trp53bp2Δ3/Δ3 mice have neural tube defects. Strain dependence of NTD penetrance in Trp53bp2Δ3/Δ3 mice: Embryo phenotypes in pure C57BL/6 (left), mixed 129 Sv-C57BL/6 (middle), and BALB/c (right) background embryos range in severity from small exencephaly (right, white arrow) to severe craniorachischisis (left upper panel, white arrows). Spina bifida is also observed in the mixed (middle panels, white arrows) and B6 (left lower panel, white arrow) backgrounds
Given the association of TP53BP2 deletions with brain structural abnormalities, we wondered whether TP53BP2 deletions might also be associated with NTDs in humans. We compared the genomic location of human genes orthologous to genes that have been implicated in NTDs in mice with chromosomal bands reported by Tyshchenko et al.1 as associated with NTDs in humans. Interestingly, TP53BP2 is one of only 16 human genes that lie in these significantly associated bands (Supplementary Figure 5). Deletions of the 1q41−q44 locus encompassing TP53BP2 are significantly associated with posterior encephalocele, generally considered a NTD.1, 38, 39 This suggests that TP53BP2 deficiency might also play a role in a subset of human NTDs.
Notably, we found no bias towards females in Trp53bp2Δ3/Δ3 embryos presenting NTDs, whereas NTDs in p53-null mice occur preferentially in females (Supplementary Figure 4c).14 This suggests that ASPP2 contributes to NTDs via a p53-independent mechanism. In summary, these results demonstrate that 100% of Trp53bp2Δ3/Δ3 mice have CNS defects but their phenotypic characteristics vary substantially with the genetic background.
Non-CNS phenotypes overlap between Trp53bp2Δ3/Δ3 embryos and 1q41q42 microdeletion syndrome patients
Visual inspection of Trp53bp2Δ3/Δ3 embryos also revealed craniofacial and eye abnormalities (Figures 5a and b). We reported previously that ASPP2-deficient embryos have highly penetrant retinal abnormalities.10 Some B6 Trp53bp2Δ3/Δ3 embryos lacked one or both eyes (Figure 5a) while others showed coloboma (incomplete eye tissue) or microphthalmia (abnormally small eye(s)) (Figure 5b).

ASPP2-deficient mice have non-CNS phenotypes. (a) Craniofacial abnormalities in Trp53bp2Δ3/Δ3 E14.5 embryos include dolichocephaly and hypoplasia or agenesis of the lower jaw (white arrows). (b) Eye abnormalities observed in Trp53bp2Δ3/Δ3 mice feature coloboma (top, missing tissue indicated by red arrow) and microphthalmia (bottom) in E14.5 embryos. (c) Abnormal heart position is seen in all B6 embryos examined. The microCT-based 3D reconstruction shows the heart is twisted approximately 45° along the coronal axis (top panel), sometimes resulting in an altered direction of the ductus arteriosus with respect to the aorta (axial view, white arrow). Colour coding: yellow – right ventricle; pink – left ventricle; blue – trachea; red – aorta; light green – ductus arteriosus. (d) Ventricular septal defect in Trp53bp2Δ3/Δ3 E14.5 embryonic heart (red arrow), shown on a representative HREM section. (e) Palate (red arrows) showing cleft palate in Trp53bp2Δ3/Δ3 E14.5 embryo. (f) Largest-area HREM section of gonads (red arrows) showing abnormalities and unclear gender in a Trp53bp2Δ3/Δ3 E14.5 embryo versus male E14.5 wild-type embryo. (g) Single versus double ureter (red arrows) in wild-type versus Trp53bp2Δ3/Δ3 E14.5 embryos, respectively, as visible by HREM
We used microCT and HREM to examine Trp53bp2Δ3/Δ3 embryos comprehensively for developmental abnormalities. At E13.5 and E14.5, 67−100% of B6 and 33−56% of BALB/c Trp53bp2Δ3/Δ3 embryos had abnormal heart position involving a 30−45o twist along the coronal axis (Figure 5c), and 67% B6 and 33% BALB/c of Trp53bp2Δ3/Δ3 embryos had ventricular septal defect (VSD, Figure 5d). Heart function and chamber volumes of adult BALB/c Trp53bp2Δ3/Δ3 mice were normal (Supplementary Figure 6a). Trp53bp2Δ3/Δ3 embryos in B6 and mixed backgrounds showed various craniofacial abnormalities including dolichocephaly (an elongation of the head), micrognathia (smaller lower jaw) (Figures 5a and 6) and cleft palate (Figure 5e). A high percentage of embryos also showed gonadal abnormalities resulting in unclear gender at E14.5 (Figure 5f), urethral abnormalities (Figure 5g), abnormal trigeminal nerve and spinal ganglia (Supplementary Figure 6b and c), and multiple other defects of varying penetrance (Figure 6). Notably, 33% of BALB/c Trp53bp2Δ3/Δ3 embryos lacked a visible abducens nerve.

Summary of phenotypes in Trp53bp2Δ3/Δ3 and Trp53bp2Δ3/+ embryos. Abnormalities detected by naked eye (visual inspection), microCT, and HREM in E13.5 and E14.5 ASPP2-deficient embryos. Frequency and the number of embryos evaluated for each phenotype are given. Wild-type littermate embryos were used as reference controls. In the case of BALB/c embryos, eye abnormalities were difficult to determine visually and were therefore quantified for a small number of embryos
Embryos with heterozygous deletion of exon 3 (Trp53bp2Δ3/+) examined by microCT had 30% incidence of cleft palate in the B6 background, and 30% and 13% incidence of VSD in B6 and BALB/c backgrounds, respectively. While this manuscript was in preparation, the International Mouse Phenotyping Consortium (IMPC, www.mousephenotype.org) – who are performing broad phenotyping for 20 000 mouse genes (3154 to date) − released phenotypic data on mice with a Trp53bp2−exon4−deletion genotype, termed Trp53bp2tm1b(EUCOMM)Hmgu.40 Our analysis of this publicly available data showed that this homozygous deletion of Trp53bp2 exon 4 (Trp53bp2Δ4/Δ4) is lethal, with 100% penetrance prior to embryonic day 9 (Supplementary Figure 7a). Trp53bp2Δ4/+ mice were viable and males showed a significant hyperactive phenotype in the open-field test (Supplementary Figure 7b). The Trp53bp2Δ4/+ males showed no difference in the percentage of time spent in the centre of the open field, indicating the phenotype is highly specific to locomotor activation (Supplementary Figure 7b). Of note, these mice also showed lower grip strength normalized by body weight, short tibia, higher bone density, and an abnormal glucose response (Supplementary Figures 7c-e). These data from the heterozygotes demonstrate that ASPP2 is haploinsufficient in mouse development.
There is substantial overlap in the phenotypic spectrum – including brain, neurological, craniofacial, eye, heart, and urogenital defects − of ASPP2-deficient mice and patients with deletions involving 1q41q42. Fifty per cent of patients with TP53BP2 deletion had cleft palate, which was observed in 33% and 67% of BALB/c and B6 Trp53bp2Δ3/Δ3 embryos, respectively. Heart abnormalities, including VSD, were reported for 31% of patients with TP53BP2 deletion, and 30% of B6 Trp53bp2Δ3/+ embryos had VSD. 1q41q42 microdeletion patients also frequently present with hypotonia and hypoglycaemia with hypoglycaemic seizures, representing phenotypic overlap with the reduced grip strength and abnormalities in glucose response in Trp53bp2Δ4/+ mice (see above). Overall, the relative penetrance of defects in Trp53bp2Δ3/Δ3 embryos also broadly correspond to the frequency of their occurrence in 1q41q42 deletion patients: virtually all diagnosed cases with 1q41q42 deletions have intellectual disability and/or brain structural abnormalities and there is high penetrance of CNS abnormalities in Trp53bp2Δ3/Δ3 mice, with lower proportions of 1q41q42 deletion patients and Trp53bp2Δ3/Δ3 mice having the other phenotypes. These results suggest that TP53BP2 haploinsufficiency might be implicated in additional abnormalities associated with the 1q41q42 microdeletion syndrome and 1q41q42 deletions more generally.
Discussion
Chromosomal deletion-associated human disorders often have variable phenotypes and severity among individuals, and the vast majority of pathogenic deletions include multiple genes, hampering the identification of causal genes. Experimental model systems are, therefore, needed to aid the identification of causal genes in human developmental disorders. Here we have identified TP53BP2 as a candidate gene responsible for brain abnormalities in 1q41q42 microdeletion syndrome and show that ASPP2-deficient mice also develop CNS abnormalities with 100% penetrance, with phenotypic differences depending on background. Notably, TP53BP2 is the only gene in the critical region for brain abnormalities of the syndrome for which the gene-deficient mouse model has a CNS phenotype.
Recently, a 1q41q42 microdeletion case with brain abnormalities was reported whose deletion included the last exon of FBXO28, suggesting that FBXO28 might also be responsible for these abnormalities.41 A mouse deficient in FBXO28 is currently unavailable. Importantly, Case 6 in our report (Figure 1, Supplementary Table 1) showed occipital encephalocele, yet harbours a deletion that includes TP53BP2 but excludes FBXO28. FBXO28 lies outside the existing SRO for brain abnormalities, but it is possible that there are multiple dosage-sensitive genes for brain development in the 1q41q42 region and future studies are needed to investigate this. For example, patients with non-overlapping DISP1 and FBXO28 deletions both showed seizures,4, 42 despite the fact that DISP1 is not contained in the critical region of the syndrome.
Few mouse models exist that are deficient in human 1q4-located genes and phenocopy the abnormalities associated with 1q4 deletions to a significant extent. The ASPP2-deficient mice described here show craniofacial, eye, heart and urogenital abnormalities in addition to CNS defects, all of which are associated with 1q41q42 deletions. Interestingly, among the 31% of patients with TP53BP2 deletions who presented with eye abnormalities, strabismus and optic nerve hypoplasia were common. Our HREM analysis of Trp53bp2Δ3/Δ3 embryos revealed hypoplastic or absent optic nerve and abducens nerve among 33% and 67% of B6 and BALB/c embryos, respectively. Given that the abducens nerve controls eye movement through the lateral rectus muscle, it would be interesting to test whether patients with strabismus and TP53BP2 deletions have any abducens nerve abnormalities. We note that our ability to determine penetrance precisely is limited by the low number of Trp53bp2Δ3/Δ3 embryos (n=3) on the BALB/c background analysed by HREM, due to the demanding nature of this technique. Overall, these observations might explain the significant phenotypic heterogeneity associated with 1q41q42 deletions: there are likely multiple dosage-sensitive genes in the region whose haploinsufficiency is both incompletely penetrant and variably expressed. The observation of NTDs in some of the ASPP2-deficient mice suggests that it would be informative to investigate whether mutations involving TP53BP2 might contribute to NTDs in humans.
To date, three different Trp53bp2-deficient mouse lines have been generated: exon 3 deletion mice reported by our group (Trp53bp2Δ3/Δ3),9 exon 4 deletion mice generated by the International Knockout Mouse Consortium (IKMC) (Trp53bp2Δ4/Δ4),43 and mice with deletion of exons 10−17 (Trp53bp2Δ10−17/Δ10−17).8 Homozygous deletion of exon 4 or exons 10−17 is lethal prior to embryonic day 9 with 100% penetrance, whereas deletion of exon 3 results in neonatal lethality with background-dependent penetrance. The exon 3 deletion is an in-frame deletion causing only the major isoform of Trp53bp2 to be deleted completely; low levels of shorter forms of Trp53bp2 can be expressed. Therefore Trp53bp2Δ3 acts as a hypomorphic allele, which has allowed us to discover developmental effects of ASPP2 deficiency that could not be observed with full-knockout alleles due to their early embryonic lethality.
Imaging of heterozygous Trp53bp2Δ3/+ embryos showed a lower frequency of structural abnormalities than observed in patients with heterozygous TP53BP2 deletions. One possible explanation is that the exon 3 deletion is a hypomorphic allele, compared to the complete deletion allele in the patients. In addition, more than one gene is deleted in each 1q41q42 microdeletion. Therefore, the concomitant deletion of multiple haploinsufficient genes might exacerbate the phenotype, as is the case in other known contiguous gene deletion syndromes.44, 45
At the protein level, the ability of ASPP2 to regulate apicobasal polarity and p53-dependent apoptosis is likely important for its role in neural tube closure. Recently, inappropriate activation of p53 during development was linked with the pathogenesis of CHARGE syndrome, a disorder characterized by abnormalities including developmental delay, heart defects, coloboma, cranial nerve abnormalities, choanal atresia, and inner ear abnormalities.44, 45 The phenotypic spectrum in the CHARGE syndrome mouse model shows substantial overlap with the ASPP2-deficient mice (including NTDs, short lower jaw, coloboma, and heart defects), suggesting that dysregulated p53 activity might be responsible for these phenotypes in ASPP2-deficient mice. However, while p53-deficient and p53-overactivated embryos show primarily exencephaly,14, 44 ASPP2-deficient embryos develop mainly spina bifida or craniorachischisis, pointing to the importance of p53-independent functions of ASPP2.
ASPP2 is also an important regulator of epithelial plasticity and apicobasal polarity.10, 46 Recent studies of Grainyhead-like 2 mutants have shown that dysregulated epithelial-to-mesenchymal transition can cause NTDs.47 It is possible that loss of epithelial character together with the loss of apicobasal polarity disrupts neural tube closure in ASPP2-deficient embryos. ASPP2-deficient embryos also showed overgrowth of neural tissue both in the brain and in the spinal cord. The surplus of neuroepithelial cells might also contribute to NTDs and potentially other CNS phenotypes in these embryos through mechanical obstruction. Regardless of the exact cellular mechanism, the observed phenotypical similarities between ASPP2 deficiency in mice and humans highlight the importance of ASPP2 in controlling CNS development in both mice and humans.
Materials and Methods
Patient recruitment
Patients with 1q4 deletions were enrolled and de-identified data collected following the UK NHS Research Ethics Committee approval (09/H0606/78, Amendment 3), and in accordance with the Institutional Review Board protocols of the participating institutions.
Comparative genomic hybridization
DNA isolated from patients’ blood were analysed by chromosomal microarray following the participating institutions’ standard protocols. With the exception of Case 7, all new 1q microdeletion cases have been deposited on the DECIPHER database (https://decipher.sanger.ac.uk/).
Brain MRI and morphometry
De-identified brain MRI and/or CT scans were obtained in DICOM format from radiology departments where the scans were recorded. Normal MRI controls used in the preparation of this article were obtained from the Pediatric MRI Data Repository created by the NIH MRI Study of Normal Brain Development. To quantify LV volume, MRI scans were first converted to Nifti format using MRIConvert (University of Oregon, USA; http://lcni.uoregon.edu/downloads/mriconvert) and subsequently analysed by ALVIN.48 Multiple scans in each series were used to compute LV volume to produce Figure 2b − left. Axial T1 scans only were used to compute LV volumes for genotype comparison as shown in Figure 2b − right. Out of the fourteen 1q41q42 microdeletion MRI scans, two patient scans (Rosenfeld 9 and Case 7) were unsuitable for morphometry: Case 7 is a prenatal case and the scan for Rosenfeld 9 cannot be converted into Nifti. Therefore, the morphometric analysis of LV volume was performed with 12 patient scans. More information on the NIH cohort is given in Acknowledgments.
Mice
Experiments with mice received UK Home Office approval, Project licence number 30/2862 and fully complied with the UK Home Office guidelines. For this study, Trp53bp2Δ3 mice in a pure C57BL/6J, mixed C57BL/6Jx129SvJ, and pure BALB/c backgrounds were crossed to generate wild-type, Trp53bp2Δ3/+, and Trp53bp2Δ3/Δ3 embryos.10 Pregnant females were killed at the indicated time points during pregnancy and embryos were collected and genotyped using the following primers: 5′-CTCCACCCCAGGAAATTACA-3′ (intron 3), 5′-CGGTTTGGAAGTCAAAGGAA-3′ (exon 3), and 5′-GGACCGCTATCAGGACATA-3′ (neomycin resistance gene).
Embryo sexing
The following primers for ZFY were used for sexing by PCR: 5′-GACTAGACATGTCTTAACATCTGTCC-3′ and 5′-CCTATTGCATGGACAGCAGCTTATG-3′. The following GADPH primers were used as controls for the PCR reaction: 5′-TGGCTACAGTAACCGAGTGGT-3′ and 5′-CTTTGAGTGGAAGCCGAAGT-3′.
Mouse micro-computed tomography
Embryos generated from mice as described above were first killed on ice-cold PBS, washed in phenol red-free Hank’s salts with EDTA, removed from their yolk sac, their umbilical cord cut, and the freed embryo fixed in 4% formaldehyde for at least 24 h. Each embryo was then immersed in 2 ml Lugol solution (L6146, Sigma-Aldrich, St Louis, MO, USA) for 72 h at 4 °C. All incubations included the replacement of staining solution after the first 24 h. Four embryos were embedded in 1% agarose (Web Scientific, Crewe, UK) in a dedicated sample holder and scanned using a micro-CT scanner (SkyScan 1172, Bruker, Kontich, Belgium) at a nominal isotropic pixel size of 4.9 μm (scan parameters: X-ray source voltage – 48 kV; current – 208 mA; filter – a 0.5 mm Al filter; number of projections – 900). The total scan time was 8 h for four embryos.
The images' projections were reconstructed with the scanner-built-in reconstruction software ‘NRecon’ and downsampled offline by a factor of 2, resulting in a 9.8 μm isotropic voxel size. Image analysis (i.e. 3D segmentation and rendering) was performed in Amira (FEI Ltd, Hillsboro, OR, USA).
High-resolution episcopic microscopy
HREM images of E14.5 embryos were obtained as previously described.37 We used the episcopic procedures for sectioning and image capture using a previously described arrangement of microtome and magnification optic.49
Mouse echocardiography
Adult female Balb/c Trp53bp2Δ3/Δ3 mice (n=8) and age-matched wild-type controls (n=7) received echocardiographic examination at a mean age of 28 weeks. Mice were lightly anaesthetized using 1−1.5% isoflurane in 100% medical oxygen, kept warm on a homeothermic blanket, and imaged using a VisualSonics Vevo 2100 (Toronto, Canada) with 22−55 MHz transducer. B-mode trans-thoracic short-axis and long-axis images were obtained to assess left ventricular volumes and function, and pulmonary artery Doppler flow as a measure of right-sided function. Images were obtained and analysed using Vevo 2100 v1.6.0 software by a single operator blinded to genotype.
Trp53bp2 exon 4 deletion mice (Trp53bp2Δ4)
Publicly available phenotypic data for Trp53bp2Δ4/Δ4 and Trp53bp2Δ4/+ mice were mined from the International Mouse Phenotype Consortium (IMPC) URL (https://www.mousephenotype.org/data/genes/MGI:2138319) release 4.3, last updated 12 May 2016, and from Phenoview (https://www.mousephenotype.org/phenoview), updated 18 May 2016. Raw data were re-plotted using Graphpad Prism version 6.05 for Windows (GraphPad Software, La Jolla, CA, USA). Experimental design by IMPC followed the protocols of the International Mouse Phenotyping Resource of Standardized Screens (https://www.mousephenotype.org/impress). For statistical testing, results from IMPC’s Mixed Model framework, linear mixed-effects model, were adopted. P values below 0.05 were deemed significant.
Visualization and statistical analysis
Data for panels in Figures 1a and c were generated using the UCSC Genome Browser (http://genome.ucsc.edu/),50 genome release hg19. Plots in panels Figures 1b and 2b, Supplementary Figure 1b, and Supplementary Figures 7a-d were generated using Graphpad Prism version 6.05 for Windows (GraphPad Software, La Jolla, CA, USA). Chromosome plots in Supplementary Figure 5 were generated using Phenogram (http://visualization.ritchielab.psu.edu/phenograms/plot).51 Statistical tests were performed in Graphpad Prism (see above) and R.52
Abbreviations
- ASPP2:
-
apoptosis-stimulating protein of p53, 2
- NTD:
-
neural tube defect
- CNS:
-
central nervous system
- CAPN2:
-
calpain 2
- CAPN8:
-
calpain 8
- FBXO28:
-
F-box only protein 28
- SRO:
-
smallest region of overlap
- MRI:
-
magnetic resonance imaging
- LV:
-
lateral ventricle
- NIH:
-
National Institutes of Health
- microCT:
-
micro-computed tomography
- HREM:
-
high-resolution episcopic microscopy
- B6:
-
C57BL6/J
- VSD:
-
ventricular septal defect
- IMPC:
-
International Mouse Phenotyping Consortium
- CHARGE:
-
Coloboma, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, Ear abnormalities
- EMT:
-
epithelial-to-mesenchymal transition
References
Tyshchenko N, Lurie I, Schinzel A . Chromosomal map of human brain malformations. Hum Genet 2008; 124: 73–80.
Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D . A chromosomal deletion map of human malformations. Am J Hum Genet 1998; 63: 1153–1159.
Rosenfeld JA, Lacassie Y, El-Khechen D, Escobar LF, Reggin J, Heuer C et al. New cases and refinement of the critical region in the 1q41q42 microdeletion syndrome. Eur J Med Genet 2011; 54: 42–49.
Au PYB, Argiropoulos B, Parboosingh JS, Micheil Innes A . Refinement of the critical region of 1q41q42 microdeletion syndrome identifies FBXO28 as a candidate causative gene for intellectual disability and seizures. Am J Med Genet A 2014; 164: 441–448.
Cepeda D, Ng HF, Sharifi HR, Mahmoudi S, Cerrato VS, Fredlund E et al. CDK‐mediated activation of the SCFFBXO28 ubiquitin ligase promotes MYC‐driven transcription and tumourigenesis and predicts poor survival in breast cancer. EMBO Molec Med 2013; 5: 1067–1086.
Gorina S, Pavletich NP . Structure of the p53 tumor suppressor bound to the Ankyrin and SH3 domains of 53BP2. Science 1996; 274: 1001–1005.
Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh J-K, Zhong S et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 2001; 8: 781–794.
Kampa KM, Acoba JD, Chen D, Gay J, Lee H, Beemer K et al. Apoptosis-stimulating protein of p53 (ASPP2) heterozygous mice are tumor-prone and have attenuated cellular damage–response thresholds. Proc Natl Acad Sci USA 2009; 106: 4390–4395.
Vives V, Su J, Zhong S, Ratnayaka I, Slee E, Goldin R et al. ASPP2 is a haploinsufficient tumor suppressor that cooperates with p53 to suppress tumor growth. Gene Dev 2006; 20: 1262–1267.
Sottocornola R, Royer C, Vives V, Tordella L, Zhong S, Wang Y et al. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev Cell 2010; 19: 126–137.
Bergamaschi D, Samuels Y, Jin B, Duraisingham S, Crook T, Lu X . ASPP1 and ASPP2: common activators of p53 family members. Mol Cell Biol 2004; 24: 1341–1350.
Gao Y, Chen X, Shangguan S, Bao Y, Lu X, Zou J et al. Association study of PARD3 gene polymorphisms with neural tube defects in a Chinese Han population. Reprod Sci 2012; 19: 764–771.
Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR . High-frequency developmental abnormalities in p53-deficient mice. Curr Biol 1995; 5: 931–936.
Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T . A subset of p53-deficient embryos exhibit exencephaly. Nat Genet 1995; 10: 175–180.
Gu X, Xing L, Shi G, Liu Z, Wang X, Qu Z et al. The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ 2012; 19: 397–405.
Eom DS, Amarnath S, Agarwala S . Apicobasal polarity and neural tube closure. Dev Growth Differ 2013; 55: 164–172.
Monier B, Gettings M, Gay G, Mangeat T, Schott S, Guarner A et al. Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature 2015; 518: 245–248.
Yamaguchi Y, Shinotsuka N, Nonomura K, Takemoto K, Kuida K, Yosida H et al. Live imaging of apoptosis in a novel transgenic mouse highlights its role in neural tube closure. J Cell Biol 2011; 195: 1047–1060.
Shaffer LG, Theisen A, Bejjani BA, Ballif BC, Aylsworth AS, Lim C et al. The discovery of microdeletion syndromes in the post-genomic era: review of the methodology and characterization of a new 1q41q42 microdeletion syndrome. Genet Med 2007; 9: 607–616.
Mazzeu JF, Krepischi-Santos AC, Rosenberg C, Szuhai K, Knijnenburg J, Weiss JMM et al. Chromosome abnormalities in two patients with features of autosomal dominant Robinow syndrome. Am J Med Genet A 2007; 143A: 1790–1795.
Mazzeu JF, Vianna-Morgante AM, Krepischi ACV, Oudakker A, Rosenberg C, Szuhai K et al. Deletions encompassing 1q41q42.1 and clinical features of autosomal dominant Robinow syndrome. Clin Genet 2010; 77: 404–407.
Rice GM, Qi Z, Selzer R, Richmond T, Thompson K, Pauli RM et al. Microdissection-based high-resolution genomic array analysis of two patients with cytogenetically identical interstitial deletions of chromosome 1q but distinct clinical phenotypes. Am J Med Genet A 2006; 140 A: 1637–1643.
Filges I, Röthlisberger B, Boesch N, Weber P, Wenzel F, Huber AR et al. Interstitial deletion 1q42 in a patient with agenesis of corpus callosum: phenotype–genotype comparison to the 1q41q42 microdeletion suggests a contiguous 1q4 syndrome. Am J Med Genet A 2010; 152 A: 987–993.
Spreiz A, Haberlandt E, Baumann M, Baumgartner Sigl S, Fauth C, Gautsch K et al. Chromosomal microaberrations in patients with epilepsy, intellectual disability, and congenital anomalies. Clin Genet 2014; 86: 361–366.
Christensen RD, Yaish HM . A neonate with the Pelger-Huet anomaly, cleft lip and palate, and agenesis of the corpus callosum, with a chromosomal microdeletion involving 1q41 to 1q42.12. J Perinatol 2012; 32: 238–240.
Wat MJ, Veenma D, Hogue J, Holder AM, Yu Z, Wat JJ et al. Genomic alterations that contribute to the development of isolated and non-isolated congenital diaphragmatic hernia. J Med Genet 2011; 48: 299–307.
Ono Y, Sorimachi H . Calpains — an elaborate proteolytic system. Biochim Biophys Acta 2012; 1824: 224–236.
Takano J, Mihira N, Fujioka R, Hosoki E, Chishti AH, Saido TC . Vital role of the calpain-calpastatin system for placental-integrity-dependent embryonic survival. Mol Cell Biol 2011; 31: 4097–4106.
Dutt P, Croall D, Arthur JS, Veyra TD, Williams K, Elce J et al. m-Calpain is required for preimplantation embryonic development in mice. BMC Dev Biol 2006; 6: 3.
Hata S, Abe M, Suzuki H, Kitamura F, Toyama-Sorimachi N, Abe K et al. Calpain 8/nCL-2 and Calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet 2010; 6: e1001040.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013; 45: 580–585.
Hawrylycz M, Ng L, Feng D, Sunkin S, Szafer A, Dang C. The Allen Brain Atlas. In: Kasabov N (ed.). Springer Handbook of Bio-/Neuroinformatics. Springer: Berlin, Heidelberg, 2014, pp. 1111–1126.
Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A et al. Transcriptional landscape of the prenatal human brain. Nature 2014; 508: 199–206.
Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 2014; 46: 1063–1071.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011; 43: 838–846.
Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res 2009; 19: 1682–1690.
Weninger WJ, Meng S, Streicher J, Mueller GB . A new episcopic method for rapid 3-D reconstruction: applications in anatomy and embryology. Anat Embryol 1998; 197: 341–348.
Rowland CA, Correa A, Cragan JD, Alverson CJ . Are Encephaloceles neural tube defects? Pediatrics 2006; 118: 916–923.
National Guideline CNeural Tube Defects. Agency for Healthcare Research and Quality (AHRQ): Rockville, MD, USA, 2013.
Brown SDM, Moore MW . The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mammalian Genome 2012; 23: 632–640.
Cassina M, Rigon C, Casarin A, Vicenzi V, Salviati L, Clementi M . FBXO28 is a critical gene of the 1q41q42 microdeletion syndrome. Am J Med Genet A 2015; 167: 1418–1420.
Jun KR, Hur YJ, Lee JN, Kim HR, Shin JH, Oh SH et al. Clinical characterization of DISP1 haploinsufficiency: a case report. Eur J Med Genet 2013; 56: 309–313.
The International Mouse Knockout C. A mouse for all reasons. Cell 2007; 128: 9–13.
Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 2014; 514: 228–232.
Sanlaville D, Verloes A . CHARGE syndrome: an update. Eur J Hum Genet 2007; 15: 389–399.
Wang Y, Bu F, Royer C, Serres S, Larkin JR, Soto Manuel S et al. ASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1. Nat Cell Biol 2014; 16: 1092–1104.
Ray HJ, Niswander LA . Grainyhead-like 2 downstream targets act to suppress EMT during neural tube closure. Development 2016; 143: 1192–1204.
Kempton MJ, Underwood TSA, Brunton S, Stylios F, Schmechtig A, Ettinger U et al. A comprehensive testing protocol for MRI neuroanatomical segmentation techniques: evaluation of a novel lateral ventricle segmentation method. NeuroImage 2011; 58: 1051–1059.
Mohun TJ, Leong LM, Weninger WJ, Sparrow DB . The morphology of heart development in Xenopus laevis. Dev Biol 2000; 218: 74–88.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM et al. The Human Genome Browser at UCSC. Genome Res 2002; 12: 996–1006.
Wolfe D, Dudek S, Ritchie M, Pendergrass S . Visualizing genomic information across chromosomes with PhenoGram. BioData Min 2013; 6: 18.
Team RDC R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2008.
Acknowledgements
This work was funded by the Ludwig Institute for Cancer Research. JZ is supported by the Skaggs-Oxford Scholarship (The Scripps Research Institute). JES acknowledges support from the BHF (FS/11/50/29038), and the BHF Centre of Research Excellence, Oxford (RE/08/004). We thank Mary Muers and Andrew Wilkie for critical reading of the manuscript, and Michael Schocke for radiological assistance. This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the DECIPHER project was provided by the Wellcome Trust. Normal MRI controls used in the preparation of this article were obtained from the Pediatric MRI Data Repository. This is a multi-site, longitudinal study of typically developing children, from ages newborn through young adulthood, conducted by the Brain Development Cooperative Group and supported by the National Institute of Child Health and Human Development, the National Institute on Drug Abuse, the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke (Contract Nos. N01-HD02-3343, N01-MH9-0002, and N01-NS-9-2314, -2315, -2316, -2317, -2319 and -2320). A listing of the participating sites and a complete listing of the study investigators can be found at http://www.bic.mni.mcgill.ca/nihpd/info/participating_centers.html.
Author contributions
JZ and XL designed the study; JZ, VV, DS, AV, JES, PM, ES, FP, CAL, and TJM performed the experiments; SJ, YL, EC, LFE, MT, ASA, HD, TAC, JA, ADC, EH, DK, DAS, MJP, ZZ, YSC, DW, AMI, KRJ, and SZ contributed clinical data; JZ, PP, and JAR analysed clinical data, JZ, VV, and XL wrote the paper; all authors analysed the data and reviewed the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Disclaimer
This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH.
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Zak, J., Vives, V., Szumska, D. et al. ASPP2 deficiency causes features of 1q41q42 microdeletion syndrome. Cell Death Differ 23, 1973–1984 (2016). https://doi.org/10.1038/cdd.2016.76
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.76

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}