
Abstract
Phosphatidylserine (PtdSer) is a phospholipid that is abundant in eukaryotic plasma membranes. An ATP-dependent enzyme called flippase normally keeps PtdSer inside the cell, but PtdSer is exposed by the action of scramblase on the cell’s surface in biological processes such as apoptosis and platelet activation. Once exposed to the cell surface, PtdSer acts as an ‘eat me’ signal on dead cells, and creates a scaffold for blood-clotting factors on activated platelets. The molecular identities of the flippase and scramblase that work at plasma membranes have long eluded researchers. Indeed, their identity as well as the mechanism of the PtdSer exposure to the cell surface has only recently been revealed. Here, we describe how PtdSer is exposed in apoptotic cells and in activated platelets, and discuss PtdSer exposure in other biological processes.
Similar content being viewed by others


Facts
-
ATP11A and ATP11C, members of the P4-ATPase family, act as flippases at the plasma membrane and are cleaved by caspase during apoptosis.
-
Of the TMEM16-family proteins, which carry 10 transmembrane segments, TMEM16F and four other family members function as Ca2+-dependent scramblases at the plasma membrane.
-
TMEM16F exposes phosphatidylserine (PtdSer) and release microparticles on activated platelets for blood clotting. It is also involved in releasing hydroxyapatite in osteoblasts for bone mineralization.
-
XK-related protein 8 (Xkr8) and two other Xkr-family proteins are cleaved during apoptosis and promote apoptotic PtdSer exposure.
Open Questions
-
What are the physiological roles of the plasma membrane flippases that are present only in the brain and testis?
-
What are the physiological roles of TMEM16-family members expressed only in the brain and intestine? Do their Ca2+-dependent scramblase activity at the plasma membrane plays a specific role there?
-
What are the physiological roles of Xkr-family members expressed only in the brain and intestine? Do their caspase-dependent scramblase activities play a specific role?
-
Is PtdSer exposure in activated lymphocytes, pyrenocytes, aged reticulocytes, capacitated sperm, tumor-associated endothelial cells and enveloped viruses regulated by the P4-type ATPase, TMEM16 and Xkr families?
-
How the flippases and scramblases translocate phospholipids between inner and outer leaflets of plasma membranes?
PtdSer is Distributed Asymmetrically in the Plasma Membrane
In eukaryotic cells, phospholipids in the plasma membrane are distributed asymmetrically.1, 2 The amine-containing phospholipids PtdSer and phosphatidylethanolamine (PtdEtn) are confined to the cytoplasmic leaflet of the plasma membrane, while phosphatidylcholine (PtdCho) and sphingomyelin (SM) are more concentrated in the exoplasmic leaflet. Phospholipid distribution in the plasma membrane is regulated by three types of phospholipid translocases.3 Flippase (or aminophospholipid translocase) specifically translocates PtdSer and PtdEtn from the outer to the inner leaflet of the lipid bilayer, in an ATP-dependent manner. Floppase is ATP-dependent, and is thought to translocate phospholipids, especially PtdCho, from the inner to outer leaflet. Scramblase non-specifically translocates or scrambles phospholipids between the lipid bilayers in both directions, without consuming ATP.
Plasma Membrane Flippases and their Down-regulation by Ca2+ and Caspase
Red blood cells can incorporate aminophospholipids in an ATP-dependent manner,4 and this ability was termed ‘flippase’ activity. Subsequently, flippase was abundantly detected in the chromaffin granules of bovine adrenal glands5 and was partially purified. Since the purified granule flippase had an ATPase activity that was biochemically similar to ATPase II, a major ATPase in chromaffin granules,6 ATPase II was proposed to be the flippase. The molecular cloning of bovine chromaffin flippase/ATPase (ATPase II) cDNA indicated that ATPase II is a type IV P-type ATPase (P4-ATPase),7 and ATPase II was thus designated ATP8A1.8, 9 The P4-ATPases, which exist only in eukaryotic cells, comprise a large family, with 5, 6, 15 and 14 members in yeast, Caenorhabditis elegans, mice and humans, respectively.10, 11, 12 P4-ATPases have 10 transmembrane segments and 2 large cytoplasmic loops that contain nucleotide-binding-site and ATPase domains. P4-ATPases are chaperoned to their proper subcellular location by CDC50, which carries two transmembrane segments with cytoplasmic N- and C-termini.13 CDC50A seems to be necessary for the flippase and lipid-transport activity of these ATPases.14
ATP8A1 and its yeast ortholog Drs2p localize mainly to intracellular vesicles such as granules and trans-Golgi networks,5, 15, 16 and a Drs2p deficiency in yeast has no effect on the PtdSer-flippase activity at plasma membranes.17 Thus, Drs2p, and probably mammalian ATP8A1, appear to be flippases that function in intracellular vesicles. Dnf1p and Dnf2p, two yeast P4-ATPases present in the plasma membrane, have been proposed to be plasma-membrane flippases,18 but their function to flip PtdSer is controversial.19 TAT-1, one of five P4-type ATPases found in C. elegans, was reported to function as a plasma-membrane flippase;20 however, it is also intracellularly localized21 suggesting that it may act as a flippase at intracellular membranes, too. Thus, the molecular identity of the plasma-membrane PtdSer flippase remained uncertain.
To identify plasma-membrane PtdSer flippases in mammalian cells, we performed a forward genetic screen using KBM7,22 a human myeloid-cell line with a near-haploid karyotype.23 KBM7 cells were randomly mutagenized with gene-trap retroviruses,24 and cells that could not efficiently incorporate fluorescently labeled PtdSer were collected by repeated cell sorting. We sequenced virus-insertion sites in the sorted population using a next-generation sequencer, and identified ATP11C and CDC50A as flippase candidates (Figure 1). ATP11C, a P4-ATPase, localized to the plasma membrane in a CDC50A-dependent manner, and an ATP11C deficiency severely reduced the PtdSer-flippase activity (by ~80%) at the plasma membranes. On the other hand, CDC50A-deficient cells completely lost the ability to flip PtdSer at the plasma membrane, and constitutively exposed PtdSer on the cell surface.22 These results identified ATP11C as the major plasma-membrane PtdSer flippase, but also indicated that other CDC50A-dependent P4-ATPases may contribute to the flipping of PtdSer at the plasma membrane. In fact, by establishing stable transformants expressing each member of human P4-ATPase family in ATP11C-null cells, not only ATP11C but also ATP11A and ATP8A2 were found to be localized at plasma membranes, and to flip PtdSer and PtdEtn25 (Table 1). ATP11A and ATP11C were ubiquitously expressed in various cells, while ATP8A2 is expressed only in the brain and testis.

Structure of flippases and its cleavage by caspase. The structure of ATP11A/ATP11C and CDC50A is schematically shown. ATP11A and ATP11C carry 10 transmembrane segments. The ATPase domain in the cytoplasm is divided into A, actuator; N, nucleotide-binding; P, phosphorylation domains. CDC50A carrying two transmembrane regions functions as a chaperone for proper localization of ATP11A and ATP11C at plasma membranes. CDC50A forms a complex with ATP11A or ATP11C in the plasma membrane, and may be necessary for the flippase activity. ATP11A and ATP11C contain 2 and 3 caspase-recognition sites (Red) that flank a α-helix (Blue), and are cleaved during apoptosis for the PtdSer exposure. TAT-1 and TAT-3, P4-ATPases in C. elegans also carry putative caspase-recognition sites in the corresponding positions. DTKT, the conserved phosphorylation site, is underlined
Increasing the intracellular Ca2+ in human erythrocytes inhibits their ability to incorporate aminophospholipids,26 suggesting that Ca2+ can regulate the flippase activity at plasma membranes. In fact, ATP11A- or ATP11C-mediated PtdSer flipping at the plasma membrane was inhibited by increasing the intracellular Ca2+ concentration.25 Their PtdSer-dependent ATPase activity was also inhibited by a high concentration of Ca2+, suggesting that Ca2+ directly binds to ATP11A and 11C to inhibit their activity.
Apoptosis is mediated by caspases, which cleave >400 cellular substrates to induce cell death.27 Apoptosis is almost universally accompanied by PtdSer exposure, which also requires caspase activation.28 Flippase is inactivated during apoptosis,29, 30 suggesting that flippase might be a caspase target. In fact, ATP11A and ATP11C carry two or three evolutionarily well-conserved caspase 3 recognition sites in its large cytoplasmic domains22 (Figure 1). Point mutations in these caspase-recognition sites generate a caspase-resistant flippase. Cells expressing this caspase-resistant flippase fail to expose PtdSer on the cell surface during apoptosis and are not engulfed by macrophages, indicating that the caspase-mediated cleavage of the flippase is essential to expose PtdSer during apoptosis. In this regard, it is noteworthy that the cell transformants expressing ATP8A2 that does not carry a caspase-recognition site fail to expose PtdSer during apoptosis, suggesting that ATP8A2 may have a specific function in the brain and testis.
Like many other processes in the cell-death pathway, apoptotic PtdSer exposure is phylogenetically well-conserved.31 Dying cells in C. elegans32 and Drosophila33 expose PtdSer downstream of caspase. Among the five P4-ATPases found in C. elegans, TAT-1 and TAT-3 have putative caspase-recognition sites in positions corresponding to those of human ATP11A and 11C (Figure 1).
Calcium-Dependent Scramblase
When platelets are activated, PtdSer is exposed on the cell surface and forms a scaffold for coagulation factors.34 This process is regulated by intracellular calcium, suggesting the involvement of one or more calcium-activated phospholipid scramblases.35 Although PLSCR1 (phospholipid scramblase 1) was once reported to be a scramblase,36, 37 its molecular properties and the phenotypes of PLSCR-deficient mice and Drosophila ruled PLSCR1 out as a phospholipid scramblase.38, 39
To identify phospholipid scramblases, we characterized calcium-dependent PtdSer exposure using the mouse cell line Ba/F3.40 Cells stimulated with the Ca2+ ionophore A23187 in the presence of 0.5 mM Ca2+ exposed PtdSer to the cell surface within 15 min and underwent necrotic cell death. However, cells treated with A23187 without extracellular Ca2+ transiently exposed PtdSer; the PtdSer was internalized again within 12 h. We used this property of transient, Ca2+-dependent PtdSer exposure to establish a Ba/F3 subline that strongly exposes PtdSer. Ba/F3 cells were stimulated with A23187 in the absence of Ca2+, and a population (1–5%) that strongly exposed PtdSer was collected by flow cytometry and expanded. This process was repeated 19 times, with a decreasing ionophore concentration each time (from 1 μM to 63 nM). This procedure produced Ba/F3-PS19, a Ba/F3 subline that strongly exposes PtdSer at an A23187 concentration (63 nM) too low to induce PtdSer exposure in the parental Ba/F3 cells.
To isolate scramblases, we constructed a cDNA library in a retrovirus-based mammalian expression vector using 2.5-6.0-kb cDNA prepared from the Ba/F3-PS19 cells. The original Ba/F3 cells were infected with the retroviral cDNA library, stimulated with A23187 and sorted by PtdSer exposure. After undergoing this process three times, a significant population of Ba/F3 cells was found to constitutively expose PtdSer. These cells carried transmembrane protein (TMEM) 16 F with a point mutation (Asp to Gly at amino-acid position 409) that appears to have been spontaneously introduced during the sorting procedure of the Ba/F3-P19 cells. Ba/F3 and other mouse cell lines transformed with the mutant TMEM16F constitutively exposed both PtdSer and PtdEtn, and internalized PtdCho and SM. Treating the transformants with an intracellular Ca2+ chelator, BAPTA-AM, blocked the PtdSer exposure. These results indicated that TMEM16F supports Ca2+-dependent phospholipid scrambling.40 A topological analysis of TMEM16F based on its amino-acid sequence suggested that it carries eight transmembrane regions. However, a recent structural analysis revealed that a TMEM16F ortholog from the fungus Nectria haematococca carries 10 transmembrane regions41 (Figure 2). Structural and chemical cross-linking analyses indicated that TMEM16F exists as a homodimer.41, 42

Structure of TMEM16F. (a) A schematic representation of TMEM16F. The scrambling domain (SCRD) between transmembrane regions IV and V, and the Ca2+-binding site comprised of Aspartate and Glutamate in transmembrane regions VI–VIII are shown by red asterisks. (b) A tertiary structure of TMEM16 from N. haematococca. From the study by Brunner et al.41
TMEM16F is one of the TMEM16-family proteins, also known as anoctamins; this family has 10 members (TMEM16A-H, J and K; or Ano1-10).43 In 2008, three groups independently reported that TMEM16A, which is expressed ubiquitously, functions as a Ca2+-activated Cl− channel.44, 45, 46 Since TMEM16B, which is specifically expressed in the retina, also acts as a Ca2+-dependent Cl− channel, all of the TMEM16-family members were thought to be Cl− channels.43, 47, 48 Accordingly, several groups reported that some TMEM16-family members, including TMEM16F, act as ion channels.49, 50, 51, 52 The expression of the wild-type TMEM16F makes the cells sensitive to Ca2+-ionophore-induced scrambling of phospholipids.40 Mouse fetal-thymocyte and embryonic-fibroblast (MEF) cells lacking TMEM16F lose phospholipid-scrambling ability,53 clearly indicating that TMEM16F is indispensable for Ca2+-dependent phospholipid scrambling. On the other hand, while TMEM16A and 16B are robust Cl− channels under physiological conditions, we were not able to detect Cl−-channel activity for TMEM16F.53 Two groups recently confirmed that the ability of TMEM16F to support phospholipid scrambling, and concluded that TMEM16F’s ion-channel activity is a consequence of phospholipid translocation54 or a secondary activity triggered under extreme, not necessarily physiological, conditions.55
Meanwhile, we expressed the 10 TMEM16 members in a TMEM16F-deficient cell line and showed that not only TMEM16F, but also TMEM16C, 16D, 16G and 16J can scramble phospholipids at the plasma membrane53 (Table 2). TMEM16E, 16H and 16 K localize to intracellular membranes, where they might also function as phospholipid scramblases. The TMEM16 family is represented in all eukaryotes.56, 57 Saccharomyces cerevisiae and Aspergillus fumigatus carry a single TMEM16 ortholog, whose biochemical and physiological functions are unknown. Nematodes carry two genes encoding TMEM16 homologs, ANOH1 and ANOH2; ANOH1 was recently found to be involved in Ca2+-dependent PtdSer exposure,58 as we will discuss later.
A mutation analysis with TMEM16A indicated that six Glutamate and Aspartate residues located in transmembrane segments of VI–VIII are involved in direct binding of Ca2+.59 These amino-acid residues are evolutionally well-conserved, and the structure of TMEM16 from N. haematococca indicates that these Ca2+-binding sites are positioned close to the cavity between two subunits.41 Regarding the scrambling of phospholipids, Yu et al.54 compared the amino-acid sequences of TMEM16F paralogues from 24 animals using a program for Type II-divergence,60 and showed that a domain of 35 amino acids located between the transmembrane IV and V is sufficient to confer the lipid scrambling activity to TMEM16A. This domain was named as a scrambling domain (SCRD). It would be interesting to study whether any other members of the TMEM16 family have the SCRD or not.
PtdSer Exposure in Activated Platelets
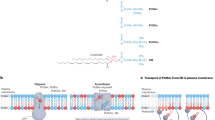
Normal, growing cells have a high intracellular ATP concentration, while the Ca2+ concentration is kept low by Ca2+-ATPase, a Ca2+ pump that removes Ca2+ from the cells.61 Under these conditions, ATP11A and ATP11C actively and specifically translocates PtdSer and PtdEtn from the outer to the inner leaflet of the lipid bilayer, creating their asymmetrical distribution in the plasma membrane. When platelets are activated with thrombin and collagen, the intracellular Ca2+ concentration increases locally and transiently,62 likely reaching several hundred micromolar near or beneath the plasma membrane.63 This local Ca2+ increase would transiently activate TMEM16F’s scramblase function and inactivate ATP11A’s and 11C’s flippase (Figure 3), thereby swiftly exposing PtdSer on limited regions of the cell’s surface. Intracellular Ca2+ levels decrease when the cell returns to a resting state, causing TMEM16F to stop scrambling, and ATP11A’s and 11C’s flippase activity to resume, thus re-establishing the asymmetrical distribution of PtdSer in the plasma membrane.

The molecular mechanism for PtdSer exposure in cells with high Ca2+-concentration. The flippase comprised of P4-ATPase (ATP11A or ATP11C) and CDC50A, and a Ca2+-dependent scramblase (TMEM16F) are schematically shown. In activated platelets, the intracellular Ca2+ concentration increases and activates TMEM16F to scramble phospholipids, while it inactivates P4-ATPases and reduces their flipping activity. When the Ca2+ concentration returns to normal level, TMEM16F stops scrambling phospholipids, while P4-ATPases resume flipping PtdSer and PtdEtn. Thus, PtdSer is only transiently exposed to the cell surface in this process, and likely depends on the intracellular concentration of ATP and Ca2+. The constant flipping of PtdSer prevents the PtdSer-exposing cells to be engulfed by macrophages78
Caspase-Dependent Scramblases
In contrast to the Ca2+-induced PtdSer exposure, TMEM16F-deficient cells have normal apoptotic PtdSer exposure,53 confirming previous suggestions that two independent scrambling systems exist in human lymphocytes or mouse platelets.64, 65 To identify the scramblases responsible for apoptotic PtdSer exposure, we again performed expression cloning using the Ba/F3-PS19 cDNA library. Assuming that scramblases, like channels, should be large membrane proteins, we first screened a library of long cDNAs (>2.5 kb) and obtained the TMEM16F cDNA, encoding 911 amino acids. However, this library did not appear to contain the cDNA for a scramblase for apoptotic PtdSer exposure, so we switched to a library of smaller cDNAs (1–2.5 kb). The library was introduced into Ba/F3 cells, and their transformants were screened for cells that strongly exposed PtdSer when treated with A23187. By repeated sorting, we found a cell population that constitutively exposes PtdSer. These cells carried the cDNA for Xkr8, which consists of 401 amino acids and has 6–10 transmembrane regions (depending on the topography-prediction program) with cytoplasmic N- and C-termini66 (Figure 4). Except for Ba/F3 cells, transformation with Xkr8 did not cause constitutive PtdSer exposure in the cell lines we tested (mouse WR19L T-cell leukemia, and human Raji lymphoma and PLB985 leukemia), but strongly enhanced the apoptotic PtdSer exposure. MEF and mouse fetal-thymocyte cell lines lacking Xkr8 lost the ability to scramble phospholipids (to expose PtdSer and PtdEtn and internalize PtdCho and SM) on apoptosis, while the Ca2+-dependent phospholipid scrambling was not affected. It was also found that human Raji and PLB985 cells that are defective apoptotic PtdSer exposure67, 68 do not express Xkr8 due to hypermethylation in the Xkr8 promoter region. Xkr8 carries a well-conserved caspase 3 recognition site in its C-terminal tail region, and its cleavage by caspases 3/7 is essential for its scramblase activity66 (Figure 4).

Structure of Xkr8, and its caspase-cleavage site. (a) The structure of mouse Xkr8, a caspase-dependent phospholipid scramblase is schematically shown. The caspase-recognition site at C-terminal region is indicated. In addition to Xkr8, Xkr4 and Xkr9 also carry the caspase-recognition site at the C-terminal tail region, and work as a caspase-dependent scramblase. (b) The amino-acid sequences of mouse Xkr8 (mXkr8), CED8 of C. elegans and CG32579 of Drosophila are aligned to obtain the maximal homology. The caspase-recognition sites at the C-terminal region of mXkr866 and at the N-terminal region of CED871 are highlighted in green. Transmembrane regions are underlined. reproduced from Suzuki et al.66 with permission
Xkr8 belongs to the Xkr family, which has nine and eight members in humans and mice, respectively.69 To examine whether any other Xkr-family proteins function as apoptotic scramblases, we expressed all of the Xkr-family members in human PLB985 or Xkr8−/− mouse fetal-thymocyte cells. Our results showed that not only Xkr8, but also Xkr4 and Xkr9 support apoptotic PtdSer exposure70 (Table 3). Like Xkr8, Xkr4 and Xkr9 carry a caspase-recognition site in their C-terminal region, and this site is cleaved during apoptosis to activate the scramblase and expose PtdSer. Xkr8 is ubiquitously expressed in various tissues, and is expressed strongly in the testes. Xkr4 is ubiquitously expressed at low levels, but is strongly expressed in the brain and eyes. Xkr9 is strongly expressed in the intestines. Whether Xkr4 and Xkr9 are redundant to Xkr8 or play specific roles in the brain/eyes and intestines remains to be studied. PtdSer is exposed in apoptotic cells not only in mammals, but also in Drosophila and C. elegans.31 Accordingly, flies and nematodes carry an Xkr8 orthlog (CG32579 in D. melanogaster, and CED8 in C. elegans)66 (Figure 4). CED8 has a caspase (CED3)-recognition site in its N terminus71 and is indispensable for CED3-dependent PtdSer exposure.66 On the other hand, a Drosophila ortholog (CG32579) has no apparent caspase-recognition site, and it is not clear how this molecule is activated or whether it is involved in apoptotic PtdSer exposure.
PtdSer Exposure on Apoptotic and Other Dying Cells
Although apoptosis has been considered to be responsible for all programmed cell death,72 recent studies indicate that cell death can also occur by programmed necrosis73 by necroptosis or pyroptosis, which are induced by tumor necrosis factor (TNF) and bacterial infection, respectively. Cells that undergo apoptosis, necroptosis or pyroptosis expose PtdSer on their surface and are engulfed by phagocytes in a PtdSer-dependent manner.74, 75, 76, 77
In the apoptotic caspase cascade, caspases 3 and 7 in the downstream of the cascade cleave the flippases of ATP11A and ATP11C and inactivate them. At the same time, caspases 3/7 cleave the Xkr8 scramblase, and activate it (Figure 5). Cleavage by caspases, which is irreversible, thus leading to irreversible PtdSer exposure. Living cells that expose PtdSer by the constitutive active forms of TMEM16 F are not engulfed by macrophages,78 but cells exposing PtdSer due to the lack of flippase activity are recognized and engulfed by macrophages.22 PtdSer-expressing cells by the constitutive-active scramblase, carry active flippase. They seem to have a low affinity to the PtdSer receptor on macrophages,78 probably because the PtdSer is constantly being flipped. In the absence of flippase, the PtdSer remains exposed and does not return to the inner leaflet, increasing the cell’s affinity for the PtdSer receptor. The difference between complete, irreversible flippase inactivation and partial, transient inactivation may explain why PtdSer-exposing apoptotic cells are engulfed by macrophages, but activated platelets are not.79 Once established, it takes days to disrupt the asymmetrical distribution of phospholipids because spontaneous translocation across the bilayer is extremely low.80, 81 In this regard, a scramblase(s) is essential for quick PtdSer exposure during apoptosis.

The PtdSer exposure in apoptotic cells. A caspase-dependent phospholipid scramblase of Xkr8 and flippase (ATP11A/ATP11C associated with CDC50A) are schematically shown. When cells undergo apoptosis, caspase 3 or caspase 7 in the downstream of the caspase cascade cleaves Xkr8 to activate its scramblase activity, while the same caspases cleave and inactivate ATP11A and ATP11C. This is the irreversible process, and the PtdSer exposed on the cell surface is recognized by macrophages for engulfment
Recently, a scramblase is reported to function in PtdSer exposure in necrotic cell death in nematodes. Dominant mutations in mec-4 (the core subunit of a mechanically gated sodium channel) in touch neurons in C. elegans cause hyper channel activity, which results in excess Ca2+ influx and necrotic cell death.82, 83 Li et al.58 recently showed that necrotic cells in mec-4 nematodes expose PtdSer by the action of ANOH1 (a TMEM16 homolog) and CED7 (an ABCA1-transporter homolog). TNF-induced necroptosis and bacteria-induced pyroptosis are mediated by calcium influx.84, 85 It will be interesting to investigate whether TMEM16F or other TMEM16 proteins play a role in exposing PtdSer in necroptosis or pyroptosis.
Defects in Flippases and Scramblases
Mouse WR19L cells that lack CDC50A or that express constitutively active TMEM16F expose PtdSer but grow normally,22, 78 suggesting that cell growth may not require an asymmetrical distribution of phospholipids at the plasma membrane. On the other hand, and ATP11C-null mice suffer from B-cell lymphopenia, cholestasis, anemia, dystocia and hepatocellular carcinoma.86, 87, 88, 89 In ATP11C-null B cells and erythrocytes, the flippase activity is reduced and the population of erythrocytes that expose PtdSer is slightly increased.88, 89 How this small effect on flippase activity in ATP11C−/− cells leads to the strong phenotypes seen in ATP11C-null mice is unclear. Lymphocytes or erythrocytes might express only the ATP11C flippase at a specific developmental stage; an ATP11C-null mutation may cause the PtdSer exposure in cells at this stage, leading macrophages to recognize and engulf these cells or causing the destabilization and abnormal assembly of plasma-membrane proteins.90, 91
Scott syndrome, a mild autosomal bleeding disorder, is caused by the failure of Ca2+-dependent PtdSer exposure in activated platelets.92 Patients in two families carrying Scott syndrome had a homozygous null mutation or compound heterozygous mutations in the TMEM16F gene.40, 93 Canine Scott syndrome, a naturally occurring bleeding disorder, is found in German shepherd dogs.94 As with human patients, the affected dogs lack procoagulant activity in activated platelets and carry loss-of-function mutations in the TMEM16F gene.95
Two mouse lines lacking TMEM16F in platelets have been established, by Yang et al.51 and by us.96 Yang et al. established conventional TMEM16F-knockout mice in a 129/B6-mixed background; the phenotype of these mice differs significantly from that of human or canine Scott syndrome. Although the PtdSer exposure in activated platelets depends significantly on TMEM16F, these mice produce tissue-factor-induced thrombin without TMEM16F; however, the tail-bleeding time is greatly increased in these mice. Yang et al. concluded that this phenotype was due to a defect in TMEM16F’s cation-channel activity. On the other hand, we were unable to detect ion-channel activity in TMEM16F, and our platelet-specific deletion of the TMEM16F gene in a C57/B6 background produced mice with phenotypes matching those in human and canine Scott syndrome. That is, the thrombin/collagen-induced PtdSer exposure in platelets is fully impaired in these mice, as is the tissue-factor-induced thrombin activation. However, similar to human and canine Scott syndrome, the tail-bleeding time is not affected in our platelet-specific TMEM16F-null mouse. The reason for the differences in these two TMEM16F-null mouse lines51, 96 is not clear.
A defect in Xkr8 causes the loss of PtdSer exposure in apoptotic cells, and the dead cells are not engulfed.66 Similarly, mutations in CED8, an Xkr8 ortholog in nematodes, causes the failure of PtdSer exposure in dying cells, and the inefficient or delayed engulfment of dead cells.71 Masking PtdSer or deleting molecules that recognize PtdSer, such as MFG-E8, Tim4 and TAM receptors, blocks the engulfment of apoptotic cells, and induces the development of systemic lupus erythematosus-type autoimmune diseases.97, 98, 99, 100, 101 Thus, it is likely that Xkr8-deficient mice develop similar autoimmune diseases, but this remains to be confirmed.
Perspectives
We have here described one flippase and two scramblase families (Ca2+-dependent and caspase-dependent), and have discussed how PtdSer is exposed on activated platelets and apoptotic cells. PtdSer and PtdEtn are exposed not only on activated platelets, but also on galectin-treated neutrophils,102 IgE-stimulated mast cells,103 activated CD4 T cells,104 capacitated sperm,105, 106 aged red blood cells,107 and cells activated through the P2X7 ATP receptor.104 PtdSer exposure on galectin-treated neutrophils,108 activated mast cells,109 activated lymphocytes and cells activated via the P2X7 receptor110 is mediated by (or at least accompanied by) increased intracellular calcium. It is tempting to speculate that PtdSer exposure in these processes involves TMEM16F or other TMEM16-family members (TMEM16C, 16D, 16F, 16G or 16J) with Ca2+-dependent scramblase activity.
Pyrenocytes (nucleus with a thin rim of cytoplasm surrounded by a plasma membrane) extruded from erythroblasts,111 milk-fat globules from mammary glands,112 exsosomes,113 enveloped viruses114 and microparticles released from activated platelets96 expose PtdSer. Pyrenocytes do not carry mitochondria, and quickly lose their ATP once separated from erythroblasts, causing a rapid Ca2+influx.111 A similar process may occur for milk-fat globules and enveloped viruses. Whether this loss of ATP and increased intracellular Ca2+ is responsible for activating or inactivating specific flippases or scramblases to expose PtdSer in these particles remains to be studied.
Tumor-associated endothelial cells expose PtdSer.115, 116 A monoclonal antibody against PtdSer was proposed as a treatment for cancer, to kill tumor-associated endothelial cells via antibody- or complement-mediated cytotoxicity.117 This treatment appears promising.118, 119, 120 Recent reports suggest that the PtdSer in the tumor-associated endothelial cells and the PtdSer exposed on tumor cells after treatment with anti-cancer drugs create an immune-suppressive tumor environment; the antibody against PtdSer reverses this effect, creating anti-tumor immunity.121 It will be interesting to study whether the flippase and scramblases discussed in this review are involved in the PtdSer exposure on tumor-associated endothelial cells.
Abbreviations
- PtdSer:
-
phosphatidylserine
- PtdEtn:
-
phosphatidylethanolamine
- PtdCho:
-
phosphatidylcholine
- SM:
-
sphingomyelin
- P4-ATPase:
-
type IV P-type ATPase
- TMEM:
-
transmembrane protein
- Xkr:
-
XK-related
- MEF:
-
mouse embryonic-fibroblast
- TNF:
-
tumor necrosis factor
References
van Meer G, Voelker D, Feigenson G . Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 2008; 9: 112–124.
Leventis PA, Grinstein S . The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys 2010; 39: 407–427.
Balasubramanian K, Schroit AJ . Aminophospholipid asymmetry: a matter of life and death. Annu Rev Physiol 2003; 65: 701–734.
Seigneuret M, Devaux P . ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc Natl Acad Sci USA 1984; 81: 3751–3755.
Zachowski A, Henry JP, Devaux PF . Control of transmembrane lipid asymmetry in chromaffin granules by an ATP-dependent protein. Nature 1989; 340: 75–76.
Moriyama Y, Nelson N . Purification and properties of a vanadate- and N-ethylmaleimide-sensitive ATPase from chromaffin granule membranes. J Biol Chem 1988; 263: 8521–8527.
Tang X, Halleck MS, Schlegel RA, Williamson P . A subfamily of P-type ATPases with aminophospholipid transporting activity. Science 1996; 272: 1495–1497.
Paulusma C, Oude Elferink R . The type 4 subfamily of P-type ATPases, putative aminophospholipid translocases with a role in human disease. Biochim Biophys Acta 2005; 1741: 11–24.
Folmer D, Elferink R, Paulusma C . P4 ATPases—lipid flippases and their role in disease. Biochim Biophys Acta 2009; 1791: 628–635.
Halleck MS, Pradhan D, Blackman C, Berkes C, Williamson P, Schlegel RA . Multiple members of a third subfamily of P-type ATPases identified by genomic sequences and ESTs. Genome Res 1998; 8: 354–361.
Tanaka K, Fujimura-Kamada K, Yamamoto T . Functions of phospholipid flippases. J Biochem 2011; 149: 131–143.
Panatala R, Hennrich H, Holthuis JC . Inner workings and biological impact of phospholipid flippases. J Cell Sci 2015; 128: 2021–2032.
Saito K, Fujimura-Kamada K, Furuta N, Kato U, Umeda M, Tanaka K . Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol Biol Cell 2004; 15: 3418–3432.
Lenoir G, Williamson P, Puts CF, Holthuis JCM . Cdc50p plays a vital role in the ATPase reaction cycle of the putative aminophospholipid transporter Drs2p. J Biol Chem 2009; 284: 17956–17967.
Hua Z, Fatheddin P, Graham TR . An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol Biol Cell 2002; 13: 3162–3177.
Lee S, Uchida Y, Wang J, Matsudaira T, Nakagawa T, Kishimoto T et al. Transport through recycling endosomes requires EHD1 recruitment by a phosphatidylserine translocase. EMBO J 2015; 34: 669–688.
Siegmund A, Grant A, Angeletti C, Malone L, Nichols JW, Rudolph HK . Loss of Drs2p does not abolish transfer of fluorescence-labeled phospholipids across the plasma membrane of Saccharomyces cerevisiae. J Biol Chem 1998; 273: 34399–34405.
Pomorski T, Lombardi R, Riezman H, Devaux PF, Van Meer G, Holthuis JCM . Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell 2003; 14: 1240–1254.
Stevens HC, Malone L, Nichols JW . The putative aminophospholipid translocases, DNF1 and DNF2, are not required for 7-nitrobenz-2-oxa-1,3-diazol-4-yl-phosphatidylserine flip across the plasma membrane of Saccharomyces cerevisiae. J Biol Chem 2008; 283: 35060–35069.
Darland-Ransom M, Wang X, Sun CL, Mapes J, Gengyo-Ando K, Mitani S et al. Role of C. elegans TAT-1 protein in maintaining plasma membrane phosphatidylserine asymmetry. Science 2008; 320: 528–531.
Chen B, Jiang Y, Zeng S, Yan J, Li X, Zhang Y et al. Endocytic sorting and recycling require membrane phosphatidylserine asymmetry maintained by TAT-1/CHAT-1. PLoS Genet 2010; 6: e1001235.
Segawa K, Kurata S, Yanagihashi Y, Brummelkamp T, Matsuda F, Nagata S . Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014; 344: 1164–1168.
Kotecki M, Reddy PS, Cochran BH . Isolation and characterization of a near-haploid human cell line. Exp Cell Res 1999; 252: 273–280.
Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat Biotechnol 2011; 29: 542–546.
Segawa K, Kurata S, Nagata S . Human type IV P-type ATPases that work as plasma membrane phospholipid flippases, and their regulation by caspase and calcium. J Biol Chem 2016; 291: 762–772.
Bitbol M, Fellmann P, Zachowski A, Devaux PF . Ion regulation of phosphatidylserine and phosphatidylethanolamine outside-inside translocation in human erythrocytes. Biochim Biophys Acta 1987; 904: 268–282.
Luthi AU, Martin SJ . The CASBAH: a searchable database of caspase substrates. Cell Death Differ 2007; 14: 641–650.
Martin SJ, Finucane DM, Amarante-Mendes GP, O'Brien GA, Green DR . Phosphatidylserine externalization during CD95-induced apoptosis of cells and cytoplasts requires ICE/CED-3 protease activity. J Biol Chem 1996; 271: 28753–28756.
Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM . Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem 1997; 272: 26159–26165.
Verhoven B, Schlegel RA, Williamson P . Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med 1995; 182: 1597–1601.
van den Eijnde SM, Boshart L, Baehrecke EH, De Zeeuw CI, Reutelingsperger CP, Vermeij-Keers C . Cell surface exposure of phosphatidylserine during apoptosis is phylogenetically conserved. Apoptosis 1998; 3: 9–16.
Venegas V, Zhou Z . Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol Biol Cell 2007; 18: 3180–3192.
Tung TT, Nagaosa K, Fujita Y, Kita A, Mori H, Okada R et al. Phosphatidylserine recognition and induction of apoptotic cell clearance by Drosophila engulfment receptor Draper. J Biochem 2013; 153: 483–491.
Bevers EM, Comfurius P, Zwaal RF . Changes in membrane phospholipid distribution during platelet activation. Biochim Biophys Acta 1983; 736: 57–66.
Bevers EM, Comfurius P, Zwaal RF . Platelet procoagulant activity: physiological significance and mechanisms of exposure. Blood Rev 1991; 5: 146–154.
Basse F, Stout JG, Sims PJ, Wiedmer T . Isolation of an erythrocyte membrane protein that mediates Ca2+-dependent transbilayer movement of phospholipid. J Biol Chem 1996; 271: 17205–17210.
Zhou Q, Zhao J, Stout J, Luhm R, Wiedmer T, Sims P . Molecular cloning of human plasma membrane phospholipid scramblase. A protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem 1997; 272: 18240–18244.
Bevers EM, Williamson PL . Phospholipid scramblase: an update. FEBS Lett 2010; 584: 2724–2730.
Sahu S, Gummadi S, Manoj N, Aradhyam G . Phospholipid scramblases: an overview. Arch Biochem Biophys 2007; 462: 103–114.
Suzuki J, Umeda M, Sims PJ, Nagata S . Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010; 468: 834–838.
Brunner JD, Lim NK, Schenck S, Duerst A, Dutzler R . X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 2014; 516: 207–212.
Suzuki T, Suzuki J, Nagata S . Functional swapping between transmembrane proteins TMEM16A and TMEM16F. J Biol Chem 2014; 289: 7438–7447.
Galietta L . The TMEM16 protein family: a new class of chloride channels? Biophys J 2009; 97: 3047–3053.
Yang Y, Cho H, Koo J, Tak M, Cho Y, Shim W et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008; 455: 1210–1215.
Schroeder B, Cheng T, Jan Y, Jan L . Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008; 134: 1019–1029.
Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008; 322: 590–594.
Hartzell HC, Yu K, Xiao Q, Chien LT, Qu Z . Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J Physiol 2009; 587: 2127–2139.
Flores CA, Cid LP, Sepúlveda FV, Niemeyer MI . TMEM16 proteins: the long awaited calcium-activated chloride channels? Braz J Med Biol Res 2009; 42: 993–1001.
Schreiber R, Uliyakina I, Kongsuphol P, Warth R, Mirza M, Martins J et al. Expression and function of epithelial anoctamins. J Biol Chem 2010; 285: 7838–7845.
Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, Okada Y . TMEM16F is a component of a Ca2+-activated Cl- channel but not a volume-sensitive outwardly rectifying Cl- channel. Am J Physiol Cell Physiol 2013; 304: C748–C759.
Yang H, Kim A, David T, Palmer D, Jin T, Tien J et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012; 151: 111–122.
Kunzelmann K, Nilius B, Owsianik G, Schreiber R, Ousingsawat J, Sirianant L et al. Molecular functions of anoctamin 6 (TMEM16F): a chloride channel, cation channel, or phospholipid scramblase? Pflug Arch Eur J Phy 2014; 466: 407–414.
Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S . Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem 2013; 288: 13305–13316.
Yu K, Whitlock JM, Lee K, Ortlund EA, Yuan Cui Y, Hartzell HC . Identification of a lipid scrambling domain in ANO6/TMEM16F. eLife 2015; 4: e06901.
Scudieri P, Caci E, Venturini A, Sondo E, Pianigiani G, Marchetti C et al. Ion channel and lipid scramblase activity associated with expression of tmem16F/ANO6 isoforms. J Physiol 2015; 593: 3829–3848.
Wang Y, Alam T, Hill-Harfe K, Lopez AJ, Leung CK, Iribarne D et al. Phylogenetic, expression, and functional analyses of anoctamin homologs in Caenorhabditis elegans. Am J Physiol Regul Integr Comp Physiol 2013; 305: R1376–R1389.
Milenkovic VM, Brockmann M, Stöhr H, Weber BH, Strauss O . Evolution and functional divergence of the anoctamin family of membrane proteins. BMC Evol Biol 2010; 10: 319.
Li Z, Venegas V, Nagaoka Y, Morino E, Raghavan P, Audhya A et al. Necrotic cells actively attract phagocytes through the collaborative action of two distinct PS-exposure mechanisms. PLoS Genet 2015; 11: e1005285.
Yu K, Duran C, Qu Z, Cui Y-y, Hartzell HC . Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res 2012; 110: 990–999.
Gu X . A simple statistical method for estimating type-II (cluster-specific) functional divergence of protein sequences. Mol Biol Evol 2006; 23: 1937–1945.
Carafoli E . Calcium pump of the plasma membrane. Physiol Rev 1991; 71: 129–153.
Hussain JF, Mahaut-Smith MP . Reversible and irreversible intracellular Ca2+ spiking in single isolated human platelets. J Physiol 1999; 514: 713–718.
Marsault R, Murgia M, Pozzan T, Rizzuto R . Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J 1997; 16: 1575–1581.
Williamson P, Christie A, Kohlin T, Schlegel RA, Comfurius P, Harmsma M et al. Phospholipid scramblase activation pathways in lymphocytes. Biochemistry 2001; 40: 8065–8072.
Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Yao Y, Mason KD et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 2009; 114: 663–666.
Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S . Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013; 341: 403–406.
Fadeel B, Gleiss B, Högstrand K, Chandra J, Wiedmer T, Sims PJ et al. Phosphatidylserine exposure during apoptosis is a cell-type-specific event and does not correlate with plasma membrane phospholipid scramblase expression. Biochem Biophys Res Commun 1999; 266: 504–511.
Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL . Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J Biol Chem 2001; 276: 1071–1077.
Calenda G, Peng J, Redman CM, Sha Q, Wu X, Lee S . Identification of two new members, XPLAC and XTES, of the XK family. Gene 2006; 370: 6–16.
Suzuki J, Imanishi E, Nagata S . Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J Biol Chem 2014; 289: 30257–30267.
Chen Y-Z, Mapes J, Lee E-S, Skeen-Gaar RR, Xue D . Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat Commun 2013; 4: 1–9.
Rich T, Watson CJ, Wyllie A . Apoptosis: the germs of death. Nat Cell Biol 1999; 1: E69–E71.
Berghe TV, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P . Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014; 15: 135–147.
Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell 2004; 15: 1089–1100.
Krysko DV, Denecker G, Festjens N, Gabriels S, Parthoens E, D'Herde K et al. Macrophages use different internalization mechanisms to clear apoptotic and necrotic cells. Cell Death Differ 2006; 13: 2011–2022.
Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T . Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int Immunol 2013; 25: 363–372.
Nagata S, Hanayama R, Kawane K . Autoimmunity and the clearance of dead cells. Cell 2010; 140: 619–630.
Segawa K, Suzuki J, Nagata S . Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci USA 2011; 108: 19246–19251.
Segawa K, Nagata S . An apoptotic 'eat me' signal: phosphatidylserine exposure. Trends Cell Biol 2015; 25: 649–650.
Kornberg RD, McConnell HM . Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry 1971; 10: 1111–1120.
Nakano M, Fukuda M, Kudo T, Matsuzaki N, Azuma T, Sekine K et al. Flip-Flop of phospholipids in vesicles: kinetic analysis with time-resolved small-angle neutron scattering. J Physical Chem B 2009; 113: 6745–6748.
Driscoll M, Chalfie M . The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 1991; 349: 588–593.
Bianchi L, Gerstbrein B, Frøkjær-Jensen C, Royal DC, Mukherjee G, Royal MA et al. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci 2004; 7: 1337–1344.
Cai Z, Jitkaew S, Zhao J, Chiang H-C, Choksi S, Liu J et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014; 16: 55–65.
Bergsbaken T, Fink SL, den Hartigh AB, Loomis WP, Cookson BT . Coordinated host responses during pyroptosis: caspase-1-dependent lysosome exocytosis and inflammatory cytokine Maturation. J Immunol 2011; 187: 2748–2754.
Siggs OM, Schnabl B, Webb B, Beutler B . X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc Nat Acad Sci USA 2011; 108: 7890–7895.
Siggs OM, Arnold CN, Huber C, Pirie E, Xia Y, Lin P et al. The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol 2011; 12: 434–440.
Yabas M, Teh CE, Frankenreiter S, Lal D, Roots CM, Whittle B et al. ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat Immunol 2011; 12: 441–449.
Yabas M, Coupland LA, Cromer D, Winterberg M, Teoh NC, D'Rozario J et al. Mice deficient in the putative phospholipid flippase ATP11C exhibit altered erythrocyte shape, anemia, and reduced erythrocyte life span. J Biol Chem 2014; 289: 19531–19537.
Manno S, Takakuwa Y, Mohandas N . Identification of a functional role for lipid asymmetry in biological membranes: phosphatidylserine-skeletal protein interactions modulate membrane stability. Proc Nat Acad Sci USA 2002; 99: 1943–1948.
Matsuzaka Y, Hayashi H, Kusuhara H . Impaired hepatic uptake by organic anion-transporting polypeptides is associated with hyperbilirubinemia and hypercholanemia in Atp11c mutant mice. Mol Pharmacol 2015; 88: 1085–1092.
Wielders S, Broers J, ten Cate H, Collins P, Bevers E, Lindhout T . Absence of platelet-dependent fibrin formation in a patient with Scott syndrome. Thromb Haemost 2009; 102: 76–82.
Castoldi E, Collins PW, Williamson PL, Bevers EM . Compound heterozygosity for 2 novel TMEM16F mutations in a patient with Scott syndrome. Blood 2011; 117: 4399–4400.
Brooks M, Catalfamo J, Brown H, Ivanova P, Lovaglio J . A hereditary bleeding disorder of dogs caused by a lack of platelet procoagulant activity. Blood 2002; 99: 2434–2441.
Brooks MB, Catalfamo JL, MacNguyen R, Tim D, Fancher S, McCardle JA . A TMEM16F point mutation causes an absence of canine platelet TMEM16F and ineffective activation and death-induced phospholipid scrambling. J Thromb Haemost 2015; 13: 2240–2252.
Fujii T, Sakata A, Nishimura S, Koji Eto, Nagata S . TMEM16F is required for phosphatidylserine exposure and microvesicle release in activated mouse platelets. Proc Nat Acad Sci USA 2015; 112: 12800–12805.
Miyanishi M, Segawa K, Nagata S . Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. Int Immunol 2012; 24: 551–559.
Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 2004; 304: 1147–1150.
Asano K, Miwa M, Miwa K, Hanayama R, Nagase H, Nagata S et al. Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med 2004; 200: 459–467.
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001; 411: 207–211.
Behrens EM, Gadue P, Gong SY, Garrett S, Stein PL, Cohen PL . The mer receptor tyrosine kinase: expression and function suggest a role in innate immunity. Eur J Immunol 2003; 33: 2160–2167.
Stowell SR, Karmakar S, Stowell CJ, Dias-Baruffi M, McEver RP, Cummings RD . Human galectin-1, -2, and -4 induce surface exposure of phosphatidylserine in activated human neutrophils but not in activated T cells. Blood 2007; 109: 219–227.
Martin S, Pombo I, Poncet P, David B, Arock M, Blank U . Immunologic stimulation of mast cells leads to the reversible exposure of phosphatidylserine in the absence of apoptosis. Int Arch Aller Imm 2000; 123: 249–258.
Elliott JI, Surprenant A, Marelli-Berg FM, Cooper JC, Cassady-Cain RL, Wooding C et al. Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat Cell Biol 2005; 7: 808–816.
de Vries KJ, Wiedmer T, Sims PJ, Gadella BM . Caspase-independent exposure of aminophospholipids and tyrosine phosphorylation in bicarbonate responsive human sperm cells. Biol Reprod 2003; 68: 2122–2134.
Gadella BM, Harrison RA . Capacitation induces cyclic adenosine 3',5'-monophosphate-dependent, but apoptosis-unrelated, exposure of aminophospholipids at the apical head plasma membrane of boar sperm cells. Biol Reprod 2002; 67: 340–350.
Lutz HU, Bogdanova A . Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front Physiol 2013; 4: 387.
Karmakar S, Cummings RD, McEver RP . Contributions of Ca2+ to galectin-1-induced exposure of phosphatidylserine on activated neutrophils. J Biol Chem 2005; 280: 28623–28631.
Smrz D, Draberova L, Draber P . Non-apoptotic phosphatidylserine externalization induced by engagement of glycosylphosphatidylinositol-anchored proteins. J Biol Chem 2007; 282: 10487–10497.
Taylor SRJ, Gonzalez-Begne M, Dewhurst S, Chimini G, Higgins CF, Melvin JE et al. Sequential shrinkage and swelling underlie P2X7-stimulated lymphocyte phosphatidylserine exposure and death. J Immunol 2008; 180: 300–308.
Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S . Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 2005; 437: 754–758.
Hanayama R, Nagata S . Impaired involution of mammary glands in the absence of milk fat globule EGF factor 8. Proc Natl Acad Sci USA 2005; 102: 16886–16891.
Théry C, Ostrowski M, Segura E . Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009; 9: 581–593.
Amara A, Mercer J . Viral apoptotic mimicry. Nat Rev Microbiol 2015; 13: 461–469.
Stafford JH, Thorpe PE . Increased exposure of phosphatidylethanolamine on the surface of tumor vascular endothelium. Neoplasia 2011; 13: 299–308.
Ran S, Downes A, Thorpe PE . Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res 2002; 62: 6132–6140.
Huang X, Bennett M, Thorpe PE . A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumors in mice. Cancer Res 2005; 65: 4408–4416.
Zörnig M, Grzeschiczek A, Kowalski M-B, Hartmann KU, Möröy T . Loss of Fas/Apo-1 receptor accelerates lymphogenesis in Em L-MYC transgenic mice but not in animals infected with MoMuLV. Oncogene 1995; 10: 2397–2401.
Digumarti R, Bapsy PP, Suresh AV, Bhattacharyya GS, Dasappa L, Shan JS et al. Bavituximab plus paclitaxel and carboplatin for the treatment of advanced non-small-cell lung cancer. Lung Cancer 2014; 86: 231–236.
Chalasani P, Marron M, Roe D, Clarke K, Iannone M, Livingston RB et al. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med 2015; 4: 1051–1059.
Yin Y, Huang X, Lynn KD, Thorpe PE . Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res 2013; 1: 256–268.
Gong EY, Park E, Lee HJ, Lee K . Expression of Atp8b3 in murine testis and its characterization as a testis specific P-type ATPase. Reproduction 2009; 137: 345–351.
Naito T, Takatsu H, Miyano R, Takada N, Nakayama K, Shin HW . Phospholipid flippase ATP10A translocates phosphatidylcholine and is involved in plasma membrane dynamics. J Biol Chem 2015; 290: 15004–15017.
Takatsu H, Tanaka G, Segawa K, Suzuki J, Nagata S, Nakayama K et al. Phospholipid flippase activities and substrate specificities of human type IV P-type ATPases localized to the plasma membrane. J Biol Chem 2014; 289: 33543–33556.
Paulusma C, Folmer D, Ho-Mok K, de Waart D, Hilarius P, Verhoeven A et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 2008; 47: 268–278.
Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 2010; 16: 1120–1127.
Munchau A, Valente EM, Davis MB, Stinton V, Wood NW, Quinn NP et al. A Yorkshire family with adult-onset cranio-cervical primary torsion dystonia. Mov Disord 2000; 15: 954–959.
Charlesworth G, Plagnol V, Holmström KM, Bras J, Sheerin U-M, Preza E et al. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am J Hum Genet 2012; 91: 1041–1050.
Tsutsumi S, Kamata N, Vokes T, Maruoka Y, Nakakuki K, Enomoto S et al. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD). Am J Hum Genet 2004; 74: 1255–1261.
Savarese M, Di Fruscio G, Tasca G, Ruggiero L, Janssens S, De Bleecker J et al. Next generation sequencing on patients with LGMD and nonspecific myopathies: findings associated with ANO5 mutations. Neuromuscular Disord 2015; 25: 533–541.
Vermeer S, Hoischen A, Meijer RPP, Gilissen C, Neveling K, Wieskamp N et al. Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am J Hum Genet 2010; 87: 813–819.
Balreira A, Boczonadi V, Barca E, Pyle A, Bansagi B, Appleton M et al. ANO10 mutations cause ataxia and coenzyme Q(1)(0) deficiency. J Neurol 2014; 261: 2192–2198.
Danek A, Rubio JP, Rampoldi L, Ho M, Dobson-Stone C, Tison F et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol 2001; 50: 755–764.
Acknowledgements
We thank all of the members of our laboratory for discussions and M. Fujii for secretarial assistance. Experiments in our laboratory were supported in part by grants-in-aid from the Ministry of Education, Science, Sports and Culture in Japan, and by Core Research for Evolutional Science and Technology from Japan Science Technology Corporation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Kroemer
Rights and permissions
About this article

Cite this article
Nagata, S., Suzuki, J., Segawa, K. et al. Exposure of phosphatidylserine on the cell surface. Cell Death Differ 23, 952–961 (2016). https://doi.org/10.1038/cdd.2016.7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.7
This article is cited by
-
Apoptosis-mediated ADAM10 activation removes a mucin barrier promoting T cell efferocytosis
Nature Communications (2024)
-
Metabolic regulation of microglial phagocytosis: Implications for Alzheimer's disease therapeutics
Translational Neurodegeneration (2023)
-
Targeting Xkr8 via nanoparticle-mediated in situ co-delivery of siRNA and chemotherapy drugs for cancer immunochemotherapy
Nature Nanotechnology (2023)
-
All-trans retinoic acid and dexamethasone regulate phagocytosis-related gene expression and enhance dead cell uptake in C2C12 myoblast cells
Scientific Reports (2023)
-
High efficiency preparation of monodisperse plasma membrane derived extracellular vesicles for therapeutic applications
Communications Biology (2023)
