
Abstract
The preadipocyte-to-adipocyte differentiation (adipogenesis) is a key process in fat mass increase and thus it is regarded as a compelling target for preventing or treating obesity. Of adipogenic hormone receptors, peroxisome proliferator-activated receptor gamma (PPARγ) has crucial roles in adipogenesis and lipid accumulation within adipocytes. Here we demonstrate that the NEDD8 (neuronal precursor cell expressed, developmentally downregulated 8)-based post-translation modification (neddylation) of PPARγ is essential for adipogenesis. During adipogenesis, NEDD8 is robustly induced in preadipocytes and conjugates with PPARγ, leading to PPARγ stabilization. When the neddylation process was blocked by NEDD8-targeting siRNAs (or viral vectors) or an inhibitor MLN4924, adipocyte differentiation and fat tissue development were substantially impaired. We also demonstrate that MLN4924 effectively prevents the high-fat diet-induced obesity and glucose intolerance in mice. This study provides a better understanding of how the PPARγ signaling pathway starts and lasts during adipogenesis and a potential anti-obesity strategy that targets the neddylation of PPARγ.
Similar content being viewed by others

Main
Obesity is considered a chronic disease predisposing to various metabolic disorders, such as diabetes, cardiovascular diseases, and fatty liver.1 The preadipocyte-to-adipocyte differentiation (adipogenesis) is a key process in fat mass increase during childhood and adolescence. Even in adulthood, adipogenesis and adipocyte apoptosis counterbalance the volume of body fat in a dynamic mode. Indeed, approximately 10% of total adipocytes in adult body are renewed annually.2 Therefore, adipogensis is regarded as a compelling target for preventing or treating obesity.3
Adipogenesis is processed through multistep gene regulations that are mainly driven by nuclear hormone receptors, such as peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT/enhancer-binding proteins (C/EBP) α/β/δ.4 The heterodimeric complex of C/EBPβ and C/EBPδ initiates adipogenesis by expressing PPARγ and C/EBPα.5 Then, C/EBPα dimerizes with C/EBPβ or C/EBPδ, and the complex drives the late stage of adipogenesis.4, 6 In addition, PPARγ, which is induced during the middle stage of adipogenesis, leads to adipocyte differentiation and maturation.7 The expressions of PPARγ and C/EBPα are sustained in differentiated adipocytes to promote lipid accumulation and adipokine secretion.8
The ubiquitin-like peptide, NEDD8 (neuronal precursor cell expressed, developmentally downregulated 8) is a post-translational modifier that covalently binds to cellular proteins (so-called neddylation).9 In contrast with ubiquitination, which mainly induces the degradation of its substrates, neddylation has diverse actions on cell signaling by altering protein interactions and on gene regulation by modifying transcription factors. Interestingly, a DNA microarray study has demonstrated that NEDD8 was upregulated in white adipose tissues from obese mice whereas ubiquitin was downregulated.10 This finding encouraged us to explore a role of neddylation in obesity development. We here checked the possibility that the neddylation of adipogenic transcription factors is essential for adipogenesis. Furthermore, we evaluated the anti-obesity effect of a neddylation inhibitor MLN4924 in high-fat-dieted mice. Based on our results, we propose that neddylation could be a novel therapeutic target for obesity and obesity-associated diseases.
Results
NEDD8 has an essential role in adipogenesis and fat tissue development
NEDD8 was robustly induced in 3T3-L1 preadipocytes undergoing differentiation into fat-laden adipocytes. Temporally, C/EBPβ and C/EBPδ expression preceded the induction of NEDD8, PPARγ and C/EBPα (Figure 1a). Given that adipogenesis was blocked by knockingdown NEDD8 or the NEDD8-activating enzyme APPBP1 (amyloid beta precursor protein-binding protein 1; Figure 1b), the neddylation process might be required for adipogenesis. Adipogenesis is a multistep process involving a cascade of transcription factors. During adipogenesis, C/EBPβ and C/EBPδ are induced at early stage, followed by PPARγ and C/EBPα induction. Subsequently, PPARγ and C/EBPα induce each other in a positive feedback manner, leading to their sustained expression during adipogenesis.11 Our next concern was which adipogenic nuclear factors were regulated by NEDD8. PPARγ expression was substantially reduced by either NEDD8 or APPBP1 knockdown (Figures 1b and c). C/EBP isofroms including C/EBPα, C/EBPβ, and C/EBPδ were also downregulated by NEDD8 knockdown (Figure 1c). In addition, PPARγ and C/EBPα mRNAs were gradually induced during adipogenesis, which was substantially attenuated at all differentiation periods by NEDD8 knockdown (Figure 1d). C/EBPβ and C/EBPδ mRNAs were transiently induced only at the early phase of adipogenesis, but were not significantly reduced by NEDD8 knockdown (Figure 1d). These unexpected findings suggest that NEDD8 regulates both C/EBPβ and C/EBPδ at the post-transcriptional level. Lipid deposition-related factors, such as fatty acid synthase (FASN), fatty acid-binding protein 4 (FABP4), cluster of differentiation 36 (CD36), and adipokines, such as resistin and adiponectin, were also downregulated by NEDD8 knockdown (Figure 1d). Given the critical role of PPARγ in adipogenic gene regulation, we examined if PPARγ targets the adipogenic genes depending on NEDD8. Chromatin immunoprecipitation (ChIP) analyses showed that NEDD8 is required for the enrichment of PPARγ on the peroxisome proliferator response elements (PPREs) in the Fabp4 and Cebpa genes (Figures 1e and f). The suppression of PPARγ-PPRE binding by NEDD8 knockdown seems to be mainly attributed to the reduction of PPARγ protein amount (Figure 1g). Yet, whether neddylation affects the DNA affinity of PPARγ was not determined clearly. To investigate such a role of NEDD8 in vivo, NEDD8-deficient 3T3-L1 and 3T3-F442A cell lines were grafted into the subcutaneous tissue of mouse abdomens, and the histology and the expression of lipid droplet-associated perilipin in grafts were examined. Compared with the control cell lines, NEDD8-deficient preadipocytes barely developed into the adipose tissues (Figure 2). These results further support our notion that neddylation is essential for adipogenesis and adipose tissue development.


NEDD8 induction is essential for adipocyte differentiation. (a) NEDD8 and adipogenic transcription factors are induced in 3T3-L1 cells under differentiation. 3T3-L1 cells were stimulated with DMI for the indicated time, and protein expression was evaluated by western blotting. (b) Adipocyte differentiation is retarded by blocking neddylation. 3T3-L1 cells were transfected transiently with si-Control, si-NEDD8 or si-APPBP1 (10, 20 or 50 nM) and then stimulated with DMI for 8 days. Western blotting (upper panel) and Oil Red O staining (lower panel) were performed. (c) PPARγ and C/EBPs are downregulated by NEDD8 knockdown. 3T3-L1 cells were transfected with 50 nM of si-control or si-NEDD8, stabilized for 2 days, and then stimulated by DMI. C/EBPs and PPARγ protein levels were determined by immunoblot analysis in cell lysates and β-tubulin was used as a loading control. (d) NEDD8 is required for the expressions of adipogenic genes. 3T3-L1 cells were transfected with si-control (Si-Con) or si-NEDD8 (Si-N8) and then stimulated with DMI for the indicated lengths of time. Total RNA was purified from the cells, and RT-qPCR was performed to measure the expression levels of Pparg, Cebpa, Cebpb, Cebpd, Fasn, Fabp4, Cd36, adiponectin, resistin, and Nedd8. Expression levels are shown relative to those of 18S RNA (mean+S.D.). Data are presented as the means+S.D. (n=3); *P<0.05. (e and f) NEDD8 promotes PPARγ binding to the promoters of its target genes. Stable 3T3-L1 cell lines expressing non-targeting shRNA (pLKO.1-sh-Con), NEDD8-targeting shRNA (pLKO.1-sh-N8), pLVX-IRES, and pLVX-IRES-His-N8 vectors were stimulated with DMI, and chromatin was cross-linked and immunoprecipitated using an anti-PPARγ antibody. The precipitated Cebpa (f) and Fabp4 promoters (e) were amplified and quantified by qPCR. The results (mean+S.D., n=3) are expressed as percentages of the input level. PPRE, PPARγ response element; NS, non-specific element. (g) PPARγ protein is expressed depending on NEDD8 levels. Cell lysates stimulated with DMI in the stable 3T3-L1 cell lines expressing non-targeting shRNA (pLKO.1-sh-Con), NEDD8-targeting shRNA (pLKO.1-sh-N8), pLVX-IRES, and pLVX-IRES-His-N8 vectors were analyzed by immunoblot analysis. β-Tubulin was used as a loading control. The intensities of PPARγ protein bands were quantified using Image J. Data are presented as the means+S.D. (n=3); *P<0.05

NEDD8 is essential for adipose tissue differentiation of grafted preadipocytes. 3T3-L1 and 3T3-F442A preadipocytes, which stably expressed pLKO.1-sh-Control (Sh-Con) or pLKO.1-sh-NEDD8 (Sh-N8), were grafted into the subcutis of the abdomens of 9-week-old male BALB/c nude mice. Grafted fat pads were excised 5 weeks after grafting and then stained with hematoxylin and eosin (upper panel) or stained immunofluorescently with an anti-perilipin antibody (lower panel). Nuclei were stained with DAPI. Epididymal fat tissues were excised from the same mice and used as a positive control for well-differentiated fat tissues
Neddylation is involved in PPARγ stabilization
To search for the neddylated proteins, we isolated His-NEDD8-conjugated proteins using Ni2+ affinity beads, electrophoresed, identified them by liquid chromatography–mass spectrometry analysis (Supplementary Figure 1). PPARγ was identified as one of the neddylated proteins in differentiating 3T3-L1 cells. Of the major adipogenic factors examined, PPARγ alone was identified to be conjugated with NEDD8 (Figure 3a). Ectopically expressed PPARγ2 was also conjugated with NEDD8 but not with NEDD8ΔGG that is a conjugation-defective mutant due to the Gly-75/76 deletion. The NEDD8 conjugation of PPARγ2 was reversed by the sentrin-specific protease 8 (SENP8) deneddylase (Figure 3b). Immunoprecipitation analyses showed that the C-terminal region of PPARγ2 is strongly conjugated with NEDD8 while the middle region is done to a lesser extent (Figure 3c). In NEDD8-deficient 3T3-L1 cells, PPARγ was not induced even after DMI stimulation (Figure 3d). Conversely, ectopic PPARγ2 was upregulated by NEDD8 overexpression, but not by NEDD8ΔGG (Figure 3e). On the basis of the decay rate of PPARγ2 protein (Figure 3f), NEDD8 is likely to participate in PPARγ2 stabilization. As the ubiquitination of PPARγ2 at the C terminus leads to proteasomal degradation of PPARγ2,12 we checked whether neddylation interferes with this process. As expected, the polyubiquitinated forms of PPARγ2 was accumulated in the presence of a proteasome inhibitor MG132. The PPARγ2 ubiquitination was attenuated by NEDD8 overexpression but was facilitated by NEDD8 knockdown (Figure 3g). These results indicate that neddylation stabilizes PPARγ2 by competing with ubiquitination.

Neddylated PPARγ is stabilized by inhibition of its ubiquitination. (a) PPARγ is conjugated with NEDD8. 3T3-L1 cells stably expressing pLVX-IRES or pLVX-IRES-His-NEDD8 were differentiated with DMI and subjected to western blotting after pull-down purification using a Ni2+ column under denaturing conditions. (b) Ectopically expressed PPARγ2 is neddylated. HEK293 cells were cotransfected with pHA-PPARγ2, pHis-NEDE8, pHis-NEDD8ΔGG, and pMyc-SENP8 plasmids in the indicated combinations. Proteins isolated using Ni2+ were analyzed by western blotting. (c) Identification of neddylated domains of PPARγ. HEK293 cells were cotransfected with pHis-NEDE8 and one of four plasmids for HA-PPARγ2 fragments. PPARγ2 fragments were isolated using Ni2+ and then analyzed by western blotting. The amino acids of expressed peptides are indicated at the top panel. (d) NEDD8 is required for PPARγ expression. 3T3-L1 cells stably expressing sh-control (Sh-Con) or shNEDD8 (Sh-N8) were stimulated with DMI, and then cell lysates were subjected to western blotting. (e) NEDD8 promotes the expression of ectopic PPARγ. HEK293 cells were cotransfected with pHA- PPARγ2 and pHis-NEDD8 or pHis-NEDD8ΔGG, and then cell lysates were analyzed by western blotting using an anti-HA antibody. (f) PPARγ is stabilized by NEDD8. HEK293 cells were transfected with pHA-PPARγ2 and/or pHis-NEDE8, and then incubated with cycloheximide for the indicated time. Cell lysates were subjected to western blotting using anti-HA or anti-NEDD8 antibody (upper panel). Band intensities (mean±S.D., n=3) on blots were analyzed using ImageJ 1.36b and plotted (lower panel). (g) Neddylation competes with ubiquitination in PPARγ. HEK293 cells were cotransfected with the indicated plasmids or siRNAs. Then they were stabilized for 48 h. After incubated with 10 μM MG132 for 8 h, cells were subjected to immunoblot analyses. β-Tubulin was determined as a loading control
MDM2 binds and neddylates PPARγ
As a candidate for the E3 enzyme neddylating PPARγ2, we firstly considered murine double minute 2 (MDM2) because it has been known to be highly expressed in 3T3-L1 cells and to promote adipogenesis.13, 14 Ectopically expressed or endogenous MDM2 was identified to associate with PPARγ2 (Figures 4a and b). PPARγ2 neddylation by ectopic or endogenous MDM2 was verified using MDM2 siRNA and Nutlin-3 (an inhibitor of MDM2) (Figures 4c and d). It was also shown that MDM2 is required for PPARγ induction and lipid storage in differentiating 3T3-L1 cells (Figures 4e and f). Given that MDM2 is known to promote cell proliferation by antagonizing tumor suppressors,15 the adipogenesis inhibition by MDM2 knockdown could be attributed to growth arrest of preadipocytes. In particular, this possibility was tested in 3T3-L1 preadipocytes which undergo post-confluence mitosis (clonal expansion) at the early phase of adipogenesis.16 When 3T3-L1 cells were cultured at a lower confluence without DMI stimulation, MDM2 knockdown did not inhibit cell proliferation (Supplementary Figure 2), suggesting that MDM2 is not essential for cell growth in 3T3-L1 cells. When clonal expansion was induced with DMI, MDM2 siRNA-transfected cells were less proliferative than control siRNA-transfected cells. However, it should be noted that MDM2-deficient cells still showed a significant event of clonal expansion (Figure 4g). In cell cycle analyses, the S-phase population was substantially increased after DMI stimulation, which was somewhat repressed by MDM2 knockdown (Figure 4h). Considering that NEDD8 and PPARγ were induced after termination of clonal expansion, MDM2 knockdown is likely to attenuate adipogenesis by inhibiting either PPARγ neddylation or clonal expansion. However, the role of MDM2 in clonal expansion remains to be further investigated.


PPARγ neddylation is mediated by MDM2. (a) Ectopically expressed MDM2 interacts with PPARγ. HEK293 cells were cotransfected with pHA-PPARγ2 and pcMDM2. Proteins in cell lysates were immunoprecipitated by anti-PPARγ or anti-MDM2 antibody, and immunoprecipitates were analyzed by western blotting. (b) Endogenous MDM2 interacts with PPARγ. 3T3-L1 cells were differentiated by DMI for 2 days, and then cell lysates were subjected to immunoprecipitation with PPARγ antibody. Immunoprecipitates were analyzed by western blotting. (c) Ectopic MDM2 induces the neddylation of PPARγ. HEK293 cells were cotransfected with pHA-PPARγ2, pHis-NEDE8, pcMDM2, and/or si-MDM2. His-NEDD8-conjugated PPARγ was purified using a Ni2+ affinity column and analyzed by western blotting. Protein expressions (Input) were examined to verify transfection efficiency. (d) MDM2 neddylates PPARγ endogenously. 3T3-L1 cells were cotransfected with the NEDD8 plasmid and si-Control or si-MDM2. Transfected cells were treated with DMI to stimulate adipogenesis, and then lysed in a denaturing condition. Neddylated PPARγ was pulled-down with Ni2+ affinity resin, and then analyzed by immunoblotting. β-Tubulin was used as an input control. (e) MDM2 is required for PPARγ expression. 3T3-L1 cells were transfected with si-Control or si-MDM2, and then cell lysates were subjected to western blotting for checking MDM2 or PPARγ expression. (f) MDM2 is required for lipid storage in adipocytes. 3T3-L1 cells, which had been transfected with si-control or si-MDM2 at the indicated concentrations, were differentiated with DMI for 8 days and subjected to Oil Red O staining. (g) Knockdown of MDM2 partially blocks clonal expansion in 3T3-L1 preadipocytes. 3T3-L1 cells, which had been transfected with non-targeting or MDM2-targeting siRNAs, were cultured to reach confluence, and the cells were further cultured for two days. Cells were treated with DMI and incubated for the indicated times. Cell numbers were counted using a hemocytometer and presented relatively to that on day 0. Each bar represents the mean+S.D. from three independent experiments. *denotes P<0.05. (h) Two-day post-confluent 3T3-L1 preadipocytes, which had been transfected with siRNAs, were incubated with DMI. On the indicated times, cells were stained with propidium iodide and subjected to flow cytometry to analyze cell cycle. The data are presented as the mean values from three independent experiments
MLN4924 inhibits adipogenesis and lipid storage by deregulating PPARγ
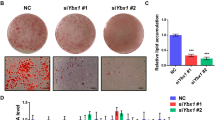
NEDD8 is activated by the NEDD8-activating (E1) enzyme complex composed of APPBP1 and UBA3 (ubiquitin-like modifier activating enzyme 3) during the first step of the neddylation process.17 MLN4924 is a synthetic compound that inhibits this process and has been under investigation as an anticancer drug in phase 2 clinical studies.18 Given that adipogenesis does not occur without NEDD8 or APPBP1, we were motivated to evaluate the anti-obesity effect of MLN4924. MLN4924 downregulated PPARγ by destabilizing the protein (Figures 5a and b) and by doing so blocked adipogenesis in 3T3-L1 cells and human adipose tissue-derived mesenchymal stem cells (H-ADSCs) isolated from four obese patients (Figure 5c). As shown in NEDD8 knockdown 3T3-L1 cells (Figure 1c), MLN4924 blocked the induction of both PPARγ and C/EBPα during adipogenesis but not that of C/EBPβ and C/EBPδ (Figure 5d). MLN4924 also reduced the mRNA levels of other adipogenic factors (Figure 5d). To examine the effect of MLN4924 on lipid storage, we treated 3T3-L1 and H-ADSC cells with DMI. After the 5 days span of differentiation, we treated the cells with MLN4924. Consequently, the number and size of lipid droplets in 3T3-L1 and H-ADSC cells decreased (Figure 6a). The effect of MLN4924 against lipid droplet formation was confirmed by immunoblotting perilipin (Figure 6b). MLN4924 was also found in both cell lines to repress the mRNA levels of the genes responsible for lipid storage (Figure 6c). Even after 3T3-L1 cells already differentiated into adipocytes, MLN4924 was able to inhibit the lipid accumulation in adipocytes (Figure 6d) and the expression of lipid storage-related genes (Figure 6e). From the 4′,6-diamidino-2-phenylindole (DAPI) staining, we found out that there was a slight reduction in the number of MLN4924-treated cells (Figure 6a). It is possible that long term treatment of MLN4924 may has another effect on the cell proliferation or cell cycle neddylation independently, but this phenomenon is still an open question at present. These results encouraged us to explore the anti-obesity action of MLN4924 in vivo.


MLN4924 blocks adipocyte differentiation by destabilizing PPARγ. (a) MLN4924 downregulates PPARγ. 3T3-L1 cells or human adipose tissue-derived stem cells were treated with 0.1 or 0.5 μM MLN4924 for 2 days, and then stimulated with DMI for 8 days. Cell lysates were subjected to western blotting. (b) MLN4924 destabilizes PPARγ protein. 3T3-L1 cells were treated by DMI to stimulate differentiation and treated with MLN4924 for 24 h, and then cells were further treated with cycloheximide for the indicated times. PPARγ protein levels were determined by immunoblot analysis (upper). Band intensities were quantified using ImageJ. Half-lives (t1/2) were calculated from the slopes of first-order decay curves (lower). (c) MLN4924 blocks adipogenesis. 3T3-L1 cells and four human adipose tissue-derived stem cells were treated with 0.1 or 0.5 μM MLN4924 for 2 days, and then stimulated with DMI for 8 days. Cells were subjected to Oil Red O staining. (d) MLN4924 inhibits the expression of adipogenic genes. 3T3-L1 cells were differentiated by DMI with vehicle or 0.5 μM MLN4924 for the indicated time, and then RT-qPCRs were performed to evaluate the expression of Pparg, Cebpa, Cebpb, Cebpd, Fabp4, Cd36, adiponectin, resistin, Fasn, and Scd1 mRNAs. The mRNA levels (mean+S.D., n=3) are shown as relative values to 18S RNA levels. *P<0.05



MLN4924 blocks lipid storage in adipocytes. (a) MLN4924 reduces lipid accumulation in mid-differentiated adipocytes. After 3T3-L1 and H-ADSC cells were stimulated with DMI for 5 days, mid-differentiated cells were treated with MLN4924 and further differentiated until day 21 (3T3-L1 cells) or day 30 (H-ADSCs). On the last day, cell morphology was analyzed by phase contrast microscopy, and cells were then subjected to immunofluorescence staining with anti-perilipin antibody. Nuclei were stained with DAPI. (b) Cell lysates from 3T3-L1 and H-ADSCs were subjected to western blotting using anti-perilipin antibody. (c) MLN4924 reduces the expressions of lipid-storing genes. 3T3-L1 and H-ADSC cells were stimulated with DMI, and then MLN4924 (0.5 μM) were administered into culture media from differentiation day 3 (d3) or day 6 (d6). After differentiation of 20-days, RNAs were prepared from the adipocytes, and the expressions of Perilipin, Acs, Cd36, Lpl, Scd1 and Fabp4 were quantified by RT-qPCR. The mRNA levels (mean+S.D., n=3) are presented as relative values to 18S RNA levels. *P<0.05. (d) MLN4924 reduces lipid accumulation in differentiated adipocytes. After 3T3-L1 cells were differentiated by DMI treatment for 8 days, the adipocytes were treated with 0.1 or 0.5 μM MLN4924 for 8 more days. On the last day, cells were subjected to immunofluorescence analysis using anti-perilipin antibody. (e) RNAs were prepared from the adipocytes, and the cellular levels of Perilipin, Lpl, Acs, Scd1, Cd36 and Fabp4 mRNAs were quantified by RT-qPCR. The mRNA levels (mean+S.D., n=3) are presented as relative values to 18S RNA levels. *P<0.05
MLN4924 ameliorates obesity and glucose intolerance in young mice
Obesity is developed through two different programs: adipocyte hyperplasia due to increased adipogenesis and adipocyte hypertrophy due to lipid accumulation.3 In childhood obesity, adipocyte hyperplasia largely contributes to the increase of fat volume.19 As the early stage of adipogenesis is blocked by the inhibition of neddylation, MLN4924 was expected to prevent childhood obesity rather than adult obesity. For this reason, we induced obesity in young mice. C57BL/6 male mice were fed with either normal control diet (NCD) or high-fat diet (HFD) for 12 weeks and were also injected intraperitoneally with vehicle or MLN4924 (30 mg/kg) twice a week. Amazingly, MLN4924 prevented HFD-induced overweight (Figure 7a). Moreover, MLN4924 did not reduce body weight in the NCD group, indicating that the dose used is not toxic. Given that MLN4924 did not reduce food intake (Figure 7b), the weight reduction by MLN4924 in the HFD group might not result from loss of appetite. After MLN4924 treatment was stopped, body weight in the HFD group increased again, suggesting that the action of MLN4924 is reversible. To determine how obese HFD-fed mice were, the abdominal fat of the mice was analyzed by computed tomography (CT; Figure 7c; Supplementary Figure 3). This CT-based volumetric analysis revealed that the volume of abdominal fat is substantially reduced by MLN4924 (Figure 7c), suggesting that MLN4924 has the anti-obesity effect. Naturally, HFD increased visceral (epididymal and perirenal) and subcutaneous fat levels and the size of adipocytes; these changes were prevented by MLN4924 (Figure 7d; Supplementary Figures 4–6). Interestingly, MLN4924 increased the expression of uncoupling protein 1, which is a marker protein of browning of white adipocyte, in epididymal adipose tissue of both NCD and HCD group (Supplementary Figure 7). In contrast, MLN4924 did not reduce the fat level and cell volume in NCD-fed mice. As obesity is a leading cause of type 2 diabetes,20 we investigated whether MLN4924 could ameliorate diabetic symptoms in HFD-fed young mice. As expected, the fasting serum glucose level was increased substantially in HFD-fed mice, and HFD-induced hyperglycemia was inhibited significantly by MLN4924 injection (Figure 7e, left panel). Likewise, the fasting insulin level was increased by >400% in the HFD group compared with the NCD group, and HFD-induced insulin overproduction was corrected by MLN4924 (Figure 7e, right panel). To further characterize HFD-induced hyperglycemia, we performed the glucose tolerance test and found that MLN4924 alleviates HFD-induced glucose intolerance (Figure 7f). In addition to its anti-obesity action, MLN4924 may have an ancillary effect to prevent obesity-associated metabolic disorders. We further characterized serum lipid profiles by analyzing free fatty acid, triglycerides (TG), high-density lipoproteins and low-density lipoproteins (LDL). The LDL level was higher in HFD group than in NCD group, whereas the TG level was shown in a reverse pattern. However, there were no significant differences in lipid profiles between the non-treated groups and MLN4924-treated groups (Supplementary Figure 7). Therefore, MLN4924 may not affect the lipid metabolism very much. To examine if MLN4924 inhibits adipogenesis in vivo, the expressions of PPARγ and lipid storage-related genes were measured in epididymal fat tissues. As expected, MLN4924 inhibited the PPARγ induced in HFD-fed mice but did not affect the basal level of PPARγ in NCD-fed mice (Figure 8a). Furthermore, the expression of lipid-depositing genes increased in epididymal fat of HFD-fed mice; this response was also blocked by MLN4924 (Figure 8b). The in vivo results suggest that MLN4924 prevents obesity in young mice by blocking HFD-induced adipogenesis and fat accumulation. Nonetheless, MLN4924 did not disturb the development of normal fat tissues. The obesity-specific effect of MLN4924 might be a stepping stone in the development of a selective anti-obesity drug.

MLN4924 prevents obesity and glucose intolerance induced by high-fat diet. (a) MLN4924 prevents excess gaining of weight in mice fed with HFD. Mice were divided into five groups (n=8 per group): vehicle treatment in NCD-fed mice and HFD-fed mice, MLN4924 treatment (30 mg/kg) in NCD-fed mice and HFD-fed mice, and MLN4924 withdrawal after 6 weeks treatment in HFD-fed mice. All mice were weighed every week and the body weights (mean±S.E.M.) are plotted as a function of time (left panel). Mice were photographed on the 12th week (right panel). (b) Weekly food intakes of the mice were monitored and the amounts (mean+S.E.M., n=8) are plotted. (c) MLN4924 reduces the volume of abdominal fat tissues. Representative CT images from the optimal single slice with both renal hila are presented (left panel). Mice underwent a CT scan before and after HFD feeding with vehicle or MLN4924 treatment. The volumes of abdominal fat tissues (mean+S.E.M., n=8) were calculated based on three-dimensional CT images (right panel). *P<0.05. (d) MLN4924 prevents HFD-induced overgrowth of fat tissues in mice. Gross appearance (left panel) and hematoxylin and eosin-stained microscopic pictures (right panel) of mouse epididymal, subcutaneous, and perirectal fat tissues and staining of these fat tissues are presented. (e) MLN4924 reduces the burden of insulin in HFD-fed mice. After 12 h of fasting, glucose and insulin levels were measured in serum. All data are presented as the means+S.E.M. (n=8). *P<0.05. (f) MLN4924 ameliorates HFD-induced glucose intolerance. Glucose was administered intraperitoneally after 12 h fasting, and then serum glucose levels (mean±S.E.M., n=8) were measured at the indicated time

MLN4924 inhibits the expressions of PPARγ and lipid-storing genes in fat tissues. (a) MLN4924 downregulates PPARγ in fat tissues. Protein expression of PPARγ in epididymal fat tissues was analyzed by Western blotting (top panel). The band intensities (mean+S.D., n=3) were calculated using ImageJ and plotted in the bottom panel. (b) MLN4924 represses lipid-storing genes in fat tissues. RNAs prepared from epididymal fat tissues were subjected to RT-qPCR to check the expressions of Pparg, Fabp4, Cd36, Perilipin, Fasn, Acs, and Lpl mRNAs. The mRNA levels (mean+S.D., n=3) are shown as relative values to 18S RNA levels. *P<0.05
Discussion
NEDD8 is induced in preadipocytes undergoing differentiation and has an essential role in adipogenesis and fat accumulation. MDM2-dependent neddylation participates in PPARγ stabilization though the post-translational level by inhibiting poly-ubiquitination, which facilitates the transcription of the genes essential for adipogenesis and lipid storage. Given previous reports, PPARγ expression and activity are distinctly regulated by post-translational modifications, such as acetylation at K268/K293, phosphorylation at S112/273, and sumoylation at K107/K395.20, 21, 22, 23, 24 As far as we know, this study is the first to identify the PPARγ regulation by neddylation. We also found that MLN4924 inhibits either adipogenesis or fat accumulation and prevents obesity and glucose intolerance induced by HFD. These results suggest that the neddylation of PPARγ is a potential target for treating obesity.
PPARγ is activated by endogenous ligands like free fatty acids or their derivatives, and then stores excess lipids by expressing FASN, FABP4, and CD36.25 In addition, PPARγ powerfully drives the differentiation of preadipocytes into adipocyte.26 Under some circumstances, PPARγ has been known to convert even non-adipose cells to adipocyte-like cells.27 The Pparg gene in mammalian cells is transcribed as several splicing variants including two major variants PPARγ1 and PPARγ2.28 PPARγ2 is exclusively expressed in adipose cells, whereas PPARγ1 is ubiquitously expressed in many types of cells. For this reason, we focused on the neddylation of PPARγ2 in this study. In a view-point of protein structure, PPARγ2 is composed of four compartments: an N-terminal ligand-independent activation domain (aa 1–136), a DNA-binding domain (aa 137–210), a hinge domain, and a C-terminal ligand-binding domain (aa 319–505).29 As shown in Figure 3c, the C-terminal fragment of PPARγ2 is profoundly neddylated, whereas other fragments are less neddylated or not done at all. The C-terminal domain is responsible for the ligand-dependent PPARγ2 binding to the promoters of target genes, and also provides the docking site for retinoid X receptor (RXR).30 In addition, the C terminus has been reported to induce the proteasomal degradation of PPARγ2 by ubiquitination.12 Therefore, it is plausible that the neddylation at the C terminus stabilizes PPARγ2 by competing with ubiquitination and thereby activating the PPARγ2-driven adipogenesis. Then, the neddylated PPARγ2 starts to run the positive feedback regulation between C/EBPα and PPARγ2, where PPARγ2 expresses C/EBPα, which in turn expresses PPARγ.11 In effect, PPARγ2 and C/EBPα both were suppressed together due to the disruption of such a reciprocal regulation in the NEDD8-deficient cells. The effect of neddylation on PPARγ2 stabilization is not surprising because neddylation has been reported to stabilize other proteins, such as hypoxia-inducible factor 1α, regulator of calcineurin 1 and Hu antigen R.31, 32, 33 Although NEDD8 resembles ubiquitin structurally, it might counteract ubiquitin functionally. The PPARγ2 protein level is regulated by the counterbalance between NEDD8 and ubiquitin.
Given the crucial roles of PPARγ in adipogenesis and lipid storage, PPARγ might be a most compelling target for treating obesity. However, only a few PPARγ antagonists are currently available. Bisphenol A diglycidyl ether (BADGE) has been reported as a PPARγ antagonist that inhibits adipogenesis in 3T3-L1 and 3T3-F442A cells.34 Dimethyl a-(dimethoxyphophinyl)-p-chlorobenzyl phosphate (SR-202) has been also reported to improve insulin sensitivity in ob/ob mice by inhibiting the PPARγ-driven transcription.35 In the present study, MLN4924 is introduced as a PPARγ inhibitor that prevents obesity and glucose intolerance in young mice. However, since numerous anti-obesity drugs have been withdrawn from the market due to their adverse effects, the toxicity of MLN4924 should be carefully checked the preclinical stage during the drug development. MLN4924 makes a MLN4924-NEDD8 adduct by covalently binding to NEDD8, and the adduct inhibits the enzymatic action of NAE. Indeed, MLN4924 has successfully passed through preclinical tests and is under evaluation for clinical stages.36, 37 A phase 1 study has been done on patients with acute myeloid leukemia and myelodysplastic syndromes. The patients were treated with a 60-min intravenous infusion of MLN4924 according to two different administration schedules, which were repeated every 21 days. Consequently, the majority of adverse effects were mild or moderate symptoms, such as pyrexia, chill, diarrhea, fatigue, and others. However, some dose-limiting toxicities, including neutropenia, thrombocytopenia and elevated aspartate transaminase, were also observed in both cohorts. Based on the patients’ responses, the maximum tolerated dose (MTD) of MLN4924 was finally determined as either 59 or 83 mg/m2 in two cohorts.37 However, as MLN4924 has been tested as an anticancer drug, the tolerable upper limit of toxicity may be higher than that of developing anti-obesity drugs. This is why we treated mice with MLN4924 at a lower dose. Previously, MLN4924 was injected either daily at doses of 30 and 60 mg/kg or twice a week at a dose of 60 mg/kg in preclinical studies evaluating its anticancer effect.17 In this study, we reduced the dose of MLN4924 approximately by half because the drug was administered twice a week at a dose of 30 mg/kg. Even after being injected with MLN4924 for 12 weeks, all mice appeared healthy and active without weight loss in the NCD group. Also, there were no pathologic findings in the abdominal viscera (data not shown). Practically, we cannot simply compare the toxic doses of drugs between humans and mice because generally mice have much higher xenobiotic rates than humans. Therefore, according to the standard protocol for toxicological study, the toxicity of MLN4924 should be further evaluated in the future.
A better understanding of the molecular mechanisms underlying obesity will provide an innovative platform for discovering more effective and tolerable anti-obesity drugs. The mode-of-actions of currently used anti-obesity drugs can be roughly classified into reduced appetite, increased energy expenditure, and reduced fat absorption.38 However, considering that the overgrowth of fat tissue is the prime event in obesity, it is plausible that adipogenesis is regarded as one of ideal targets for obesity control. We here identified that PPARγ neddylation is essential for both adipogenesis and fat accumulation and it also showed the possibility that obesity is pharmacologically controlled through inhibition of the neddylation. This study provides a scientific basis for the development of next-generation anti-obesity drugs.
Materials and Methods
Antibodies and reagents
Antibodies against PPARγ, C/EBPα, C/EBPβ, C/EBP-δ, β-tubulin, MDM2, and Ub were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against NEDD8 and perilipin were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-HA was obtained from Roche (Basel, Switzerland). The antibody against APPBP1 was obtained from NOVUS (St. Charles, MO, USA). 3-Isobutyl-1-methylxanthine, dexamethasone, insulin, cycloheximide, HA affinity beads, human insulin, Oil Red O and EZview Red anti-HA affinity gel were obtained from Sigma-Aldrich (St. Louis, MO, USA). Nutlin-3 was obtained from Cayman Chemical Company (Ann Arbor, MI, USA). MG132 was obtained from ENZO Life Sciences (Farmingdale, NY, USA). Ni-NTA agarose beads were obtained from Qiagen (Hilden, Germany). MLN4924 was synthesized, as previously described.39 Bovine serum, fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium (DMEM) were obtained from Thermo Fisher Scientific (Waltham, MA, USA).
Cell culture and adipocyte differentiation
HEK293 (human embryonic kidney) cells were obtained from the American Type Culture Collection (Manassas, VA, USA). 3T3-L1 and 3T3-F442A preadipocytes were kindly given by Dr. Jae-Woo Kim (Yonsei University, Seoul, Korea). Cells were cultured in DMEM supplemented with 10% FBS. Differentiation of 3T3-L1 cells was induced with DMI (1 μM dexamethasone, 500 μM 3-isobutyl-a-methylxanthine and 5 μg/ml of insulin). From 2–8 days after DMI treatment, cells were maintained in DMEM containing 10% FBS and 1 μg/ml of insulin and the medium was changed every other day. Human adipose tissue was obtained from patients who provided informed consent, and the institutional review board of Asan Hospital (Seoul, Korea) approved the use of the clinical samples for this research (approve #, 2012-0283). Human adipose tissue-derived stem cells (hADSCs) were isolated from 35 to 55 years old women undergoing liposuction and passage 3–5 cells were used in this study. The cells were isolated using ADSC markers under flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). More than 95% of hADSCs were identified to express CD105, CD90, CD73, and HLA-abc. Adipocyte differentiation of hADSCs was induced by DMI and 200 μM indomethacin in α-MEM supplemented with 10% FBS. Cells were incubated in 5% CO2 and 20% O2 at 37 °C.
Animals
This animal experiment was approved by the Seoul National University Animal Experiments Ethics Committee (approve #, 130318-1). C57BL/6J mice (Central Lab. Animal Inc., Seoul, Korea) were housed in a pathogen-free facility under a 12-h light/12-h dark cycle. Young mice (4 weeks old) were fed with NCD or HFD with 60% of calories from fat (Research Diets Inc., New Brunswick, NJ, USA) for 12 weeks. A vehicle or MLN4924 (30 mg/kg) was injected intraperitoneally into mice twice a week. Food consumption was measured daily for individually housed mice, and body weight was measured once a week. Blood samples were collected by inserting a capillary tube into the peri-orbital plexus. Fat tissues were excised under anesthesia, weighed, and cut into two parts, which were fixed with 4% formalin or frozen in liquid nitrogen.
Plasmids, siRNAs, shRNAs, and transfection
The plasmid of HA-tagged mouse PPARγ was constructed, as previously described.40 NEDD8 cDNA obtained by reverse transcription-PCR (RT-PCR) was cloned into His6-tagged pcDNA3. The NEDD8-ΔGG mutant was generated by site-directed mutagenesis.41 Ubiquitin, PPARγ-NT (aa 1–140), PPARγ-M (aa 141–281), or PPARγ-CT (aa 282–505) amplified by PCR was inserted into HA-tagged pcDNA3 (Clontech Laboratories, Mountain View, CA, USA). PCR-amplified cDNAs of human MDM2 and SENP8 were inserted into pcDNA, Myc-tagged pcDNA, or SFB-pIRES2-EGFP. siRNAs were synthesized by Integrated DNA Technologies (Coralville, IA, USA) and their sequences are listed in Supplementary Table S2. The plasmids and siRNAs were transiently transfected into cells using Lipofectamine (Invitrogen, Calsbad, CA, USA). shRNAs, such as shNEDD8-I, shNEDD8-II, shAPPBP1-I, shAPPBP1-II, and shControl, were inserted into the pLKO.1-puro vector (Sigma-Aldrich). The nucleotide sequences of shRNAs are listed in Supplementary Table S3. His-NEDD8 and HA-PPAR-γ plasmids were subcloned into the NotI and SpeI sites of the lentiviral shuttle vector pLVX-IRES-puro (Clontech). 3T3-L1 cells and 3T3-F442A cells were infected with lentiviral vectors (pLKO.1-puro and pLKO.1-puro-shNEDD8-I). Then, cells stably expressing viral vectors were selected using 5 μg/ml of puromycin. HEK293 cells were transfected with pLVX-IRES-puro, pLVX-IRES-puro-His-NEDD8, and HA-PPARγ.
Grafting of 3T3-L1 and 3T3-F442A preadipocytes
This animal experiment was approved by the Seoul National University Animal Experiments Ethics Committee (approve #, 110718-1). 3T3-L1 and 3T3-F442A cells stably expressing pLKO.1-puro-shControl or pLKO.1-puro-shNEDD8 were trypsinized and suspended in DMEM supplemented with 10% bovine serum. The cells (3 × 107 cells) were injected into the subcutis of the abdomens of 9-week-old male BALB/c nude mice (Orient Bio. Inc., Gyeonggi-do, Korea). After 5 weeks, fat pads produced from implanted preadpocytes were fixed with formalin and stained with hematoxylin and eosin.42
Identification of NEDD8 conjugation
We identified NEDD8 conjugation, as previously described.43 After transfected with the plasmid for His6-NEDD8 or His6-NEDD8ΔGG mutant, cells were sub-cultured in two dishes. The cells in one dish were lysed for western blotting to confirm the expression level of input proteins, while those in the other dish were lysed in a denaturing buffer (6 M guanidine hydrochloride, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-Cl (pH 8), 10 mM imidazole, and 10 mM β-mercaptoethanol). The lysates were incubated with Ni2+-NTA-agarose beads (Qiagen) at room temperature for 4 h. To remove non-covalently bound proteins, the beads were washed in a denaturing buffer (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-Cl (pH 8), 20 mM imidazole, 10 mM β-mercaptoethanol, and 0.2% Triton X-100) at pH 8.0 for 5 min and done again in the buffer at pH 6.3 for 5 min. Then, His-tagged proteins bound to Ni2+ were eluted in a 2 × SDS denaturing buffer and subjected to western blotting.
RNA preparation and quantitative RT-PCR
Total RNA was isolated from cultured cells using TRIzol (Invitrogen). The EasyScript cDNA Synthesis Kit (Applied Biological Materials Inc., Richmond, Canada) was used to synthesize cDNAs. The mouse cDNAs, such as Pparg, Cebpa, Cebpb, Cebpd, Adiponectin, Resistin, Cd36, Fabp4, Fas, Scd1, Perilipin, Lpl, Acs, Fasn, Nedd8, Tnfa, Il6, Mcp1, F40/80, C11b, C11c, and 18S rRNA, were amplified with EvaGreen qPCR master mix reagent (Applied Biological Materials) in StepOne Real-time PCR System (Applied Biosystems, Foster, CA, USA). The 18S rRNA level was used to quantify the level of cDNAs for each corresponding sample. The sequences of qPCR primers are listed in Supplementary Table S1.
Western blotting and immunoprecipitation
Proteins in cell lysates were electrophoresed on SDS-polyacrylamide gels and transferred to Immobilon-P membranes (Millipore, Billerica, MA, USA). Membranes were preincubated with 5% skim milk in TTBS (tris-buffered saline containing 0.1% Tween 20) for 30 min and incubated overnight with primary antibody diluted 1 : 500 to 1 : 3000 in the TTBS. The membranes were further incubated with a horseradish peroxidase-conjugated secondary antibody for 1 h and visualized using the ECL Plus kit (Thermo Fisher Scientific, Waltham, MA, USA). To precipitate HA-tagged proteins, transfected cells were lysed in a buffer containing 5 mM EDTA, 50 mM Tris-Cl, 100 mM NaCl, 0.1% NP-40, and the protease inhibitor cocktail (Sigma-Aldrich). Cell lysates (1 mg proteins) were incubated with EZview anti-HA affinity beads sequentially incubated with a primary antibody (10 μg/ml) and protein A/G-Sepharose beads (GE Healthcare, Uppsala, Sweden) at 4 °C for 4 h. Proteins bound to the beads were eluted in a 2 × SDS denaturing buffer and subjected to western blotting.
Immunofluorescence
Fat tissues were fixed with formalin and cut into 4-μm slices. The sections were deparaffinized and hydrated, and antigens were then retrieved by heating the specimens in 10 mM sodium citrate buffer (pH 6.0) using microwave for 20 min. After blocking, the specimens were incubated overnight with anti-perilipin (1 : 500) and then with Alexa Fluor 488 anti-IgG antibody (Invitrogen) for visualization. Nuclei were stained with DAPI (1 μg/ml) in PBS.
Chromatin immunoprecipitation
Cell lysates from 1 × 107 cells were cross-linked with 1% formaldehyde for 10 min, and glycine (finally 125 mM) was added to quench the cross-linking reaction. Cell extracts were subjected to sonication until the resulting DNA fragments were <500 bp. Chromatin complexes were incubated with an antibody overnight at 4 °C and precipitated with Protein A/G-Sepharose beads (10 μl) for 2 h. After washing with saline, DNA–protein complexes were eluted with 1% SDS and incubated overnight at 65 °C to reverse the cross-linking. The eluted DNAs were purified using a MEGAquick-spin DNA purification kit (Intron Biotech., Seoul, Korea), and quantified by the ABI StepOne Real-Time PCR System using EvaGreen qPCR master mix. The sequences (5′ to 3′) of primers used for ChIP-PCR are CTGAGCTACACCCTCGGCTC and TCCCCACCGGAGGGCATGAG for the PPRE of mouse Cebpa; TTTGCTTCCACTTAATTCCT and GCTCAGGGTGTGCAAGCAGG for the PPRE of mouse Fabp4.
CT and measurement of intra-abdominal adipose tissue
A 128-slice multi-detector computed tomography (MDCT) was performed (Ingenuity CT; Philips Healthcare, Cleveland, OH, USA). The tube voltage was set at 120 kV, with a constant current of 175 mA. The slice thickness was 0.7 mm, and the field of view was set at 50 × 50 mm. Reconstruction in the axial, sagittal and coronal planes were done for all CT scans. Before CT scanning, mice were anesthetized with a mixture of Zoletil (Virbac, Carros, France) (50 mg/kg) and xylazine (10 mg/kg), administered by intraperitoneal injection. The mice were placed in a prone position in the appropriate holders. First, a whole-body scannogram was generated to ensure proper placement in the holder and to set the scan area. Second, whole-body scans of the entire mice were performed. To avoid artificially including leg fat in the abdominal area, the hind limbs of the mice were extended. We selected the optimal single slice in which both renal hila were well observed around L3–L5. Cross-sectional abdominal contours were estimated by delineating the skin manually with a graph pen through the muscular structures and vertebral corpora. Hounsfield unit (HU) cut-off values of −250 to −50 were assigned for adipose tissue in the slice. The area between −250 HU and −50 HU pixels was calculated automatically by the post-processing CT software (Rapidia 2.8, INFINITT, Seoul, Korea). Abdominal fat was assessed by CT: sex difference in distribution.44
Blood chemistry, glucose tolerance test, and Oil red O staining
Sera were isolated from mice that had fasted for 12 h. Serum glucose content was measured using a one-touch glucometer Accu-Chek Active system (Roche). Serum levels of insulin and leptin were measured using ELISA kits provided by ALPCP Diagnostics (Salem, NH, USA) according to the manufacturer’s instructions. For glucose tolerance test, mice were fasted overnight and then were injected intraperitoneally with d-glucose at a dose of 2 g/kg body weight. The plasma level of glucose was measured 0, 15, 30, 60, 90, and 120 min after glucose treatment using the glucometer. Adipocytes were fixed with formalin and lipids were stained with 0.3% Oil red O solution (Sigma-Aldrich) dissolved in isopropanol.
Cell counting and cell cycle analysis
Cells were harvested in PBS containing 0.05% trypsin and 0.02% EDTA. An aliquot of cell suspension was applied to a hemocytometer plate to count cells. Cells were twice washed by centrifugation and resuspension with PBS, fixed with 75% cold ethanol overnight at –20 °C, and washed again with PBS. Cells were incubated with 10 μg/ml of propidium iodide and 100 μg/ml of RNase A for 20 min, and then analyzed using FACSCalibur flow cytometer (BD Bioscience, San Jose, CA, USA).
Statistical analysis
Means, S.D., and S.E.M. were calculated using Microsoft Excel 2010. The means of two groups were compared using two-tailed, unpaired Student’s t-test, and the difference was considered statistically significant when P<0.05.
Abbreviations
- APPBP1:
-
amyloid beta precursor protein-binding protein 1
- CD36:
-
cluster of differentiation 36
- C/EBP:
-
CCAAT/enhancer-binding protein
- ChIP:
-
chromatin immunoprecipitation
- CT:
-
computed tomography
- DAPI:
-
4′,6-diamidino-2-phenylindole
- FABP4:
-
fatty acid-binding protein 4
- FASN:
-
fatty acid synthase
- H-ADSCs:
-
human adipose tissue-derived mesenchymal stem cells
- HFD:
-
high-fat diet
- LDL:
-
low-density lipoproteins
- MDM2:
-
murine double minute 2
- NCD:
-
normal control diet
- NEDD8:
-
neuronal precursor cell expressed developmentally downregulated 8
- Neddylation:
-
NEDD8-based post-translation modification
- PPARγ:
-
peroxisome proliferator-activated receptor gamma
- PPREs:
-
peroxisome proliferator response elements
- SENP8:
-
sentrin-specific protease 8
- TG:
-
triglycerides
References
Kopelman PG . Obesity as a medical problem. Nature 2000; 404: 635–643.
Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O et al. Dynamics of fat cell turnover in humans. Nature 2008; 453: 783–787.
Camp HS, Ren D, Leff T . Adipogenesis and fat-cell function in obesity and diabetes. Trends Mol Med 2002; 9: 442–447.
Cao Z, Umek RM, McKnight SL . Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev 1991; 5: 1538–1552.
Steger DJ, Grant GR, Schupp M, Tomaru T, Lefterova MI, Schug J et al. Propagation of adipogenic signals though an epigenomic transition state. Genes Dev 2010; 24: 1035–1044.
Yeh WC, Cao Z, Classon M, McKnight SL . Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev 1991; 9: 168–181.
Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM . mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 1994; 8: 1224–1234.
Tontonoz P, Hu E, Spiegelman BM . Stimulation of adipogenesis in fibroblasts by PPARγ, a lipid-activated transcription factor. Cell 1994; 79: 1147–1156.
Osaka F, Kawasaki H, Aida N, Saeki M, Chiba T, Kawashima S et al. A new NEDD8-ligating system for cullin-4A. Genes Dev 1998; 12: 2263–2268.
Nadler ST, Stoehr JP, Schueler KL, Tanimoto G, Yandell BS, Attie AD . The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc Natl Acad Sci USA 2000; 97: 11371–11376.
Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE et al. Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell 1999; 3: 151–158.
Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM . Degradation of the peroxisome proliferator-activated receptor gamma is linked to ligand-dependent activation. J Biol Chem 2000; 24: 18527–18533.
Berberich SJ, Litteral V, Mayo LD, Tabesh D, Morris D . Mdm-2 gene amplification in 3T3-L1 preadipocytes. Differentiation 1999; 64: 205–212.
Hallenborg P, Feddersen S, Francoz S, Murano I, Sundekilde U, Petersen RK et al. Mdm2 controls CREB-dependent transactivation and initiation of adipocyte differentiation. Cell Death Differ 2012; 19: 1381–1389.
Wade M, Li YC, Wahl GM . MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013; 13: 83–96.
Tang QQ, Otto TC, Lane MD . Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci USA 2003; 100: 44–49.
Rabut G, Peter M . Function and regulation of protein neddylation. 'Protein modifications: beyond the usual suspects' review series. EMBO Rep 2008; 9: 969–976.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009; 458: 732–736.
Knittle JL, Timmers K, Ginsberg-Fellner E, Brown RE, Katz DP . The growth of adipose tissue in children and adolescents. Cross-sectional and longitudinal studies of adipose cell-number and size. J Clin Invest 1979; 63: 239–246.
Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012; 150: 620–632.
Camp HS, Tafuri SR . Regulation of peroxisome proliferator-activated receptor gamma activity by mitogen-activated protein kinase. J Biol Chem 1997; 272: 10811–10816.
Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010; 466: 451–456.
Ohshima T, Koga H, Shimotohno K . Transcriptional activity of peroxisome proliferator-activated receptor gamma is modulated by SUMO-1 modification. J Biol Chem 2004; 279: 29551–29557.
Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 2005; 437: 759–763.
Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med 2013; 19: 557–566.
Tontonoz P, Hu E, Spiegelman BM . Regulation of adipocyte gene expression and differentiation by peroxisome proliferator activated receptor gamma. Curr Opin Genet Dev 1995; 5: 571–576.
Tontonoz P, Hu E, Spiegelman BM . Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994; 79: 1147–1156.
Tontonoz P, Spiegelman BM . Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem 2008; 77: 289–312.
Kilroy GE, Zhang X, Floyd ZE . PPAR-gamma AF-2 domain functions as a component of an ubiquitin-dependent degradation signal. Obesity 2009; 17: 665–673.
Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP et al. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 2008; 456: 350–356.
Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS . Hypoxia-inducible factor α subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem 2011; 28: 6963–6970.
Noh EH, Hwang HS, Hwang HS, Min B, Im E, Chung KC . Covalent NEDD8 conjugation increases RCAN1 protein stability and potentiates its inhibitory action on calcineurin. PLoS One 2012; 7: e48315.
Embade N, Fernández-Ramos D, Varela-Rey M, Beraza N, Sini M, Gutiérrez de Juan V et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology 2012; 55: 1237–1248.
Wright HM, Clish CB, Mikami T, Hauser S, Yanagi K, Hiramatsu R et al. A synthetic antagonist for the peroxisome proliferator-activated receptor-g inhibits adipocyte differentiation. J Biol Chem 2000; 3: 1873–1877.
Rieusset J, Touri F, Michalik L, Escher P, Desvergne B, Niesor E et al. A new selective peroxisome proliferator-activated receptor-g antagonist with antiobesity and antidiabetic acitivity. Mol Endocrinol 2002; 11: 2628–2644.
Smith MA, Maris JM, Gorlick R, Kolb EA, Lock R, Carol H et al. Initial testing of the investigational NEDD8-activating enzyme inhibitor MLN4924 by the pediatric preclinical testing program. Pediatr Blood Cancer 2012; 2: 246–253.
Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M et al. Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol 2015; 169: 534–543.
Hainer V, Hainerová IA . Do we need anti-obesity drugs? Diabetes Metab Res Rev 2012; 28: 8–20.
Lee HW, Nam SK, Choi WJ, Kim HO, Jeong LS . Stereoselective synthesis of MLN4924, an inhibitor of NEDD8-activating enzyme. J Org Chem 2011; 76: 3557–3561.
Chung SS, Ahn BY, Kim M, Kho JH, Jung HS, Park KS . SUMO modification selectively regulates transcriptional activity of peroxisome-proliferator-activated receptor γ in C2C12 myotubes. Biochem J 2011; 433: 155–161.
Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS . Hypoxia-inducible factor α subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem 2011; 286: 6963–6970.
Mandrup S, Loftus TM, MacDougald OA, Kuhajda FP, Lane MD . Obese gene expression at in vivo levels by fat pads derived from s.c. implanted 3T3-F442A preadipocytes. Proc Natl Acad Sci USA 1997; 94: 4300–4305.
Jaffray EG, Hay RT . Detection of modification by ubiquitin-like proteins. Methods 2006; 38: 35–38.
Dixon AK . Abdominal fat assessed by computed tomography: sex difference in distribution. Clin Radiol 1983; 34: 189–191.
Acknowledgements
This work was supported by a Korean Health Technology R&D grant (A121106, A120476, HI15C2695), National Research Foundation grants of Korea government (2012R1A5A2A44671346, 2009-0090188). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
YSC designed experiments. HSP, UIJ, JYS, DHS and SWK performed experiments and interpreted results. JWP, JYC, LSJ, SYK, JY, HWL and YSC provided critical materials. HSP, JWP, JBK, KSP and YSC interpreted results and analyzed statistics. HSP, UIJ, JWP and YSC wrote the manuscript
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by N Bazan
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Park, HS., Ju, UI., Park, JW. et al. PPARγ neddylation essential for adipogenesis is a potential target for treating obesity. Cell Death Differ 23, 1296–1311 (2016). https://doi.org/10.1038/cdd.2016.6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.6
This article is cited by
-
Neddylation of insulin receptor substrate acts as a bona fide regulator of insulin signaling and its implications for cancer cell migration
Cancer Gene Therapy (2024)
-
Targeting protein modifications in metabolic diseases: molecular mechanisms and targeted therapies
Signal Transduction and Targeted Therapy (2023)
-
Neddylation inhibition induces glutamine uptake and metabolism by targeting CRL3SPOP E3 ligase in cancer cells
Nature Communications (2022)
-
Latexin deficiency attenuates adipocyte differentiation and protects mice against obesity and metabolic disorders induced by high-fat diet
Cell Death & Disease (2022)
-
Neddylation regulation of mitochondrial structure and functions
Cell & Bioscience (2021)
