
Abstract
In order for cancer cells to survive during metastasis, they must overcome anoikis, a caspase-dependent cell death process triggered by extracellular matrix (ECM) detachment, and rectify detachment-induced metabolic defects that compromise cell survival. However, the precise signals used by cancer cells to facilitate their survival during metastasis remain poorly understood. We have discovered that oncogenic Ras facilitates the survival of ECM-detached cancer cells by using distinct effector pathways to regulate metabolism and block anoikis. Surprisingly, we find that while Ras-mediated phosphatidylinositol (3)-kinase signaling is critical for rectifying ECM-detachment-induced metabolic deficiencies, the critical downstream effector is serum and glucocorticoid-regulated kinase-1 (SGK-1) rather than Akt. Our data also indicate that oncogenic Ras blocks anoikis by diminishing expression of the phosphatase PHLPP1 (PH Domain and Leucine-Rich Repeat Protein Phosphatase 1), which promotes anoikis through the activation of p38 MAPK. Thus, our study represents a novel paradigm whereby oncogene-initiated signal transduction can promote the survival of ECM-detached cells through divergent downstream effectors.
Similar content being viewed by others


Main
Cancer metastasis, the spread of cancer cells to distant parts of the body, accounts for ~90% of cancer-related deaths and represents an inherently difficult clinical challenge.1, 2 It has become clear that for successful metastasis to occur, cells must overcome a caspase-dependent cell death mechanism, anoikis, which is triggered by detachment from the extracellular matrix (ECM).3 In addition to anoikis evasion, cancer cells must also contend with anoikis-independent cellular alterations that can compromise cellular viability.4 Chief among these alterations are metabolic deficiencies that are induced by ECM detachment.5, 6, 7 These metabolic alterations involve deficiencies in ATP generation, elevated levels of reactive oxygen species, and the induction of autophagy.6, 8, 9 Although recent studies have begun to unravel the strategies used by cancer cells to ameliorate metabolic deficiencies during ECM detachment,10 the signal-transduction cascades responsible for regulating metabolism during ECM detachment in cancer cells remain almost entirely unexplored.
The activation of oncogenic signaling pathways is critical to anchorage-independent growth and ultimately to the survival of a variety of distinct cancer cell types during ECM detachment.4, 11 Presumably, this oncogenic signaling is also necessary for resolving the aforementioned ECM-detachment-induced metabolic deficiencies. ErbB2 overexpression in mammary epithelial cells results in a stimulation of phosphatidylinositol (3)-kinase (PI(3)K)/Akt signaling to promote glucose uptake and ATP generation.6 These data raise the question as to how cancer cells that lack ErbB2 overexpression rectify metabolic deficiencies during ECM detachment. Does activation of other oncogenic signaling pathways facilitate ATP generation during ECM detachment, and are similar downstream effectors used? The design of novel chemotherapeutic approaches to eliminate ECM-detached cancer cells requires a better understanding of the signal-transduction cascades that regulate metabolism during ECM detachment.
The Ras oncogene is mutated in ~30% of all human cancers, typically resulting in constitutive activation via mutations in codon 12, 13, or 61.12, 13, 14 Given the frequency with which Ras mutations arise in cancers, it seems reasonable to speculate that Ras signaling may be involved in facilitating the survival of ECM-detached cells. Indeed, previous studies in intestinal epithelial cells suggest that Ras activation can promote anoikis evasion,15 although significant ambiguities exist with regard to the precise downstream signaling that is involved.16 Once activated, Ras is well known to activate ERK and Akt, which have well-documented roles in promoting cell survival in cancer cells.12 More specifically, the ability of Ras to modulate metabolism during ECM detachment would be consistent with its known capabilities to activate PI(3)K and the proficiency with which PI(3)K signaling promotes glucose uptake and ATP generation.17 Given this and the aforementioned studies demonstrating that ErbB2 promotes ATP generation during ECM detachment by activating PI(3)K and Akt,6 it seems reasonable to speculate that Ras promotes metabolic activity through a similar signal-transduction cascade.
Here, we uncover a surprising and novel signal-transduction pathway operating downstream of oncogenic Ras to promote the survival of ECM-detached cancer cells. Interestingly, rather than relying on PI(3)K/Akt to promote ATP generation, we have found that Ras overcomes ECM-detachment-induced ATP deficiencies through the activation of a distinct PI(3)K effector, serum and glucocorticoid-regulated kinase-1 (SGK-1). Furthermore, anoikis resistance in cells harboring Ras mutations relies on the downregulation of the PHLPP1 (PH Domain and Leucine-Rich Repeat Protein Phosphatase 1) phosphatase. Despite the well-known proclivity of PHLPP1 to dephosphorylate and deactivate Akt, we have discovered that PHLPP1 mediates anoikis through the activation of the p38 MAPK pathway. These data identify novel downstream targets that could be used for the development of chemotherapeutic approaches aimed at antagonizing the ability of Ras to eliminate deficits in ATP generation and block anoikis. Furthermore, our data substantially refine the current understanding of Ras signaling to suggest that Ras-mediated signal transduction can promote the survival of ECM-detached cells through divergent downstream effectors.
Results
Oncogenic Ras promotes ATP generation through a PI(3)K-dependent, Akt-independent signaling pathway in ECM-detached cells
To examine the ability of oncogenic Ras to promote ATP generation and cell viability during ECM detachment, we engineered MCF-10A cells to stably express constitutively active H-Ras (G12V, hereafter referred to as 10A HrasG12V) or K-Ras (G12V, hereafter referred to as 10A KrasG12V). Indeed, hyperactive Ras promotes ATP generation in ECM-detached (but not ECM-attached) cells (Figure 1a). This corresponds to an increase in the viability of ECM-detached cells (Figure 1b). Given that PI(3)K signaling is well known to operate downstream of Ras,6 we investigated if PI(3)K also operates to promote ATP generation. As expected, inhibition of PI(3)K signaling compromises ATP production in a dose-dependent manner in both 10A HrasG12V and 10A KrasG12V cells (Figure 1c). However, when examining phosphorylation of the PI(3)K effector Akt, we surprisingly observe minimal changes in Akt (S473) phosphorylation (Figure 1c). To confirm the efficacy of the inhibitor, we blocked PI(3)K signaling in MCF-10A cells overexpressing insulin growth factor receptor 1 (10A IGF-1R) and observed the expected loss of Akt phosphorylation (Figure 1d). These data therefore suggest that ATP generation in ECM-detached cells downstream of oncogenic Ras is occurring through a PI(3)K-dependent, but Akt-independent mechanism.

Oncogenic Ras promotes ATP generation through a PI(3)K-dependent, Akt-independent signaling axis. (a) ATP levels were measured using CellTiter Glo in the indicated cells 24 h after plating in either attachment or detachment (poly-HEMA). (b) Cell viability was measured after 24 h in detached conditions in the indicated cells using AlamarBlue. (c) ATP levels were measured using the ATP Determination Kit in the indicated cells 24 h after plating in detachment. Detached cells were treated with the indicated dose of LY294002 (PI(3)K inhibitor) for 24 h. Lysates were prepared and immunoblotted for p-Akt and GAPDH. (d and e) Detached cells were treated with the indicated dose of LY294002 for 24 h. Lysates were prepared and then underwent immunoblotting for IGF-1R, p-Akt, p-Sek1, GAPDH, or β-tubulin. (f) The indicated (ECM-detached) cells were immunoblotted for Ras, p-Sek1, p-Akt, GAPDH, FLAG-tag (on K-Ras G12V), α-tubulin, or β-actin. All data are presented as mean±S.D. and statistical significance was calculated using a two-tailed t-test. Fold change is calculated as a ratio compared with empty vector (EV). a.u., Arbitrary units
SGK-1 activation is sufficient to promote ATP production and cell survival during ECM detachment
Given that Akt does not appear to be a relevant mediator of ATP generation downstream of PI(3)K (in the presence of oncogenic Ras) during ECM detachment, we assessed the contribution of SGK-1, another AGC family kinase known to operate downstream of PI(3)K.6 SGK-1 shares significant sequence identity with Akt and has previously been linked to the regulation of glucose transporter localization.6, 18, 19, 20 In our 10A HrasG12V and 10A KrasG12V cells, we see a decrease in SGK-1 activation upon PI(3)K inhibition as measured by phosphorylation status of Sek1 at serine 80 (Figure 1e), a canonical downstream SGK-1 target.21 In addition, we observe an increase in the phosphorylation of Sek1 in the presence of oncogenic Ras mutations (Figure 1f). This increase in Sek1 phosphorylation downstream of oncogenic Ras occurs in both ECM-detached and -attached cells with similar kinetics (Supplementary Figures S1A and S1B).
To examine if SGK-1 is sufficient to promote ATP generation and survival during ECM detachment, we generated MCF-10A cells that express constitutively active SGK-1 (S422D, hereafter referred to as 10A S422D). Overexpression and constitutive activation of SGK-1 was confirmed via immunoblotting for SGK-1 and phospho (p)-Sek1 (Figure 2a). When grown in ECM-detached conditions, we observe an increase in glucose uptake (Figure 2b), ATP production (Figure 2c), and cellular viability (Figure 2d) in 10A S422D cells. This increase in productive metabolism and ATP generation in ECM-detached 10A S422D cells does not seem to be a result of alterations in SGK-1 localization (Supplementary Figure S2A) and occurs in both glucose-rich and -limiting conditions (Supplementary Figure S2B). Furthermore, using a 3D cell culture model of mammary acini where the hollow lumen is normally generated by ECM-detachment-induced cell death,6 we find that constitutive activation of SGK-1 can promote luminal filling and cell survival (Figure 2e). Interestingly, constitutively active SGK-1 does not influence caspase activation in either ECM-attached or -detached cells (Figure 2f), suggesting that the effects of SGK-1 activation on ATP generation and viability are independent of anoikis.

SGK-1 kinase signaling is sufficient to promote metabolic maintenance and survival in ECM-detached cells independently of modulating caspase-3/7 activity. (a) The indicated (ECM-detached) cells underwent immunoblotting for SGK-1, p-Sek1 or GAPDH. (b) Glucose uptake levels were measured in detached MCF-10A and 10A S422D cells after 24 h using the Amplex Red Glucose Assay Kit. (c) ATP levels were measured in detached MCF-10A cells and 10A S422D cells after 24 h using the ATP Determination Kit. (d) Cell viability was measured via AlamarBlue after 24 h in detached MCF-10A cells and 10A S422D cells. (e) MCF-10A and 10A S422D cells were plated in Matrigel and allowed to form acini for 7 days. Acini were stained with DAPI (blue) and anti-Laminin 5 (red). Acini were scored as clear, mostly clear, mostly filled, or filled. Representative images of these categories are shown above at × 40. (f) Caspase-3/7 activity was measured in MCF-10A and 10A S422D cells in attachment (left) and detachment (right) after 24 h using the CaspaseGlo 3/7 assay. 10A S422D cells were treated with 1 μM STS at the time of plating as a control for cell death. (g) The indicated (ECM-detached) cells underwent immunoblotting for SGK-1, p-Sek1, and GAPDH. (h) ATP levels were measured using the ATP Determination Kit following 24 h of ECM detachment in the indicated cells. (i) Cell viability was measured via AlamarBlue after in the indicated cells following 24 h of detachment. All data are presented as mean±S.D. and statistical significance was calculated using a two-tailed t-test. Fold change is calculated as a ratio compared with empty vector (EV). a.u., Arbitrary units
To ascertain the importance of SGK-1 kinase activity for the observed changes in ATP generation and cell viability during ECM detachment, we expressed kinase-dead SGK-1 (K127M) in MCF-10A cells (hereafter referred to as 10A K127M). Immunoblotting for p-Sek1 confirmed the lack of SGK-1 kinase activity in these cells (Figure 2g). In contrast to constitutively active SGK-1 (S422D), the expression of SGK-1 (K127M) does not promote ATP production or cell viability in ECM-detached cells (Figures 2h and i). Thus, our data suggest that the SGK-1-mediated increase in ATP generation and cell viability is a direct consequence of SGK-1 kinase activity.
SGK-1 is required for the promotion of ATP generation downstream of oncogenic Ras during ECM detachment
Given that SGK-1 kinase activity is sufficient to promote ATP generation during ECM detachment, we next investigated the role of SGK-1 downstream of oncogenic Ras during ECM detachment. Indeed, 10A HrasG12V and 10A KrasG12V cells have elevated SGK-1 activity (Figure 1f), ATP production (Figures 1a and c), glucose uptake (Figure 3a), and cellular viability (Figure 1b). To more directly examine the role of SGK-1 in Ras-mediated regulation of ATP generation, we eliminated SGK-1 expression (and concomitant activity; Figure 3b) in 10A HrasG12V cells using short hairpin RNA (shRNA) (10A HrasG12V shSGK-1). Elimination of SGK-1 in these cells results in a loss of glucose uptake (Figure 3c), diminished ATP generation (Figure 3d), and a loss of cell viability (Figure 3e). Interestingly, the loss of SGK-1 signaling is only detrimental to ECM-detached cells, as SGK-1 knockdown does not alter ATP production in ECM-attached cells (Figure 3d). As we observed in our 10A S422D cells, the loss of ATP production in 10A HrasG12V shSGK-1 cells occurs in both glucose-rich and -limiting conditions (Supplementary Figure S3A).

SGK-1 is necessary for the promotion of ATP generation in ECM-detached cells harboring oncogenic Ras. (a) Glucose uptake levels were measured at 24 h in the indicated detached cells using the Amplex Red Glucose Assay Kit. (b) Immunoblotting against SGK-1, p-Sek1 and GAPDH was used to confirm the SGK-1 knockdown (shSGK-1) in detached 10A HrasG12V cells. (c) Glucose uptake levels were measured using the Amplex Red Glucose Assay Kit at 24 h in detached 10A HrasG12V and shSGK-1 cells. (d) ATP levels were measured in 10A HrasG12V and shSGK-1 cells after 24 h in either attachment (left) or detachment (right) using CellTiter Glo. (e) Cell viability was measured in detached 10A HrasG12V and shSGK-1 cells at 24 h using AlamarBlue. (f) Immunoblotting against p-Sek1 and β-tubulin was used following treatment of detached cells for 24 h with 25 μM of SGK-1 kinase inhibitor EMD638683 to confirm inhibitor efficacy. (g) ATP levels were measured using the ATP Determination Kit in ECM-detached 10A HrasG12V cells following 24 h of treatment with 25 μM EMD638683. (h) Cell viability was measured using AlamarBlue in ECM-detached 10A HrasG12V cells after being treated with 25 μM EMD638683. All data are presented as mean±S.D. and statistical significance was calculated using a two-tailed t-test. Fold change is calculated as a ratio compared with empty vector (EV, shEV) or dimethyl sulfoxide (DMSO) (vehicle) treatment. a.u., Arbitrary units
To further validate the shRNA data, we used a pharmacological inhibitor of SGK-1 kinase activity, EMD638683, to assess the contribution of SGK-1 kinase activity to ATP generation and viability in 10A HrasG12V cells. Successful inhibition of SGK-1 in these cells was confirmed via immunoblotting for p-Sek1 (Figure 3f). As expected, pharmacological inhibition of SGK-1 kinase activity resulted in a decrease in ATP production (Figure 3g) and viability (Figure 3h) in ECM-detached cells. Furthermore, neither the shRNA-mediated reduction in SGK-1 nor the inhibition of SGK-1 kinase activity altered caspase activation (Supplementary Figures S3B and C). These data suggest that, analogous to the results observed in 10A S422D cells, loss of ATP generation and viability in ECM-detached cells with constitutively active Ras mutations occurs independently of anoikis induction.
To extend our studies into cancer cell lines with endogenous-activating Ras mutations, we used a colorectal cancer cell line (HCT116), which contains a G13D mutation in K-Ras.22 HCT116 cells were engineered to have reduced SGK-1 (hereafter HCT116 shSGK-1 cells) and knockdown was confirmed via immunoblotting for SGK-1 and p-Sek1 (Figure 4a). In line with our data in MCF-10A cells, HCT116 shSGK-1 cells have diminished glucose uptake (Figure 4b), lower ATP generation (Figure 4c), and reduced viability during ECM detachment (Figure 4d). Furthermore, the loss of ATP generation in HCT116 shSGK-1 cells occurs in both glucose-rich and -limiting conditions. (Supplementary Figure S4A). The loss of SGK-1 signaling also leads to a significant reduction in anchorage-independent growth in HCT116 cells (Figure 4e). Building on these findings, we treated HCT116 cells with the SGK-1 kinase inhibitor (EMD638683) (Figure 4f) and discovered a decrease in ATP generation (Figure 4g) and viability (Figure 4h) in ECM-detached cells. In line with our data in MCF-10A cells, the loss of ATP generation and viability observed during ECM detachment in HCT116 shSGK-1 cells and HCT116 cells treated with the SGK-1 inhibitor occurs independently of alterations in caspase activation (Supplementary Figures S4B and S4C). These data suggest that the ability of SGK-1 kinase activity to promote the survival during ECM detachment is also present in cancer cells with endogenous-activating mutations in Ras.

SGK-1 is required for ATP maintenance and survival of ECM-detached colon cancer cells. (a) Immunoblotting against SGK-1, p-Sek1 and β -tubulin was used to confirm the SGK-1 knockdown (shSGK-1) in detached HCT116 cells. (b) Glucose uptake levels were measured using the Amplex Red Glucose Assay Kit in HCT116 and shSGK-1 cells after 24 h in detachment. (c) ATP levels were measured using CellTiter Glo after 24 h in either attachment (left) or detachment (right) in HCT116 and shSGK-1 cells. (d) Cell viability for HCT116 and shSGK-1 cells was measured via AlamarBlue following 24 h in detachment. (e) HCT116 and shSGK-1 cells were plated in soft agar. After 5 days, images were taken following iodonitrotetrazolium chloride (INT-violet) staining. Colony count was determined using ImageJ. (f) Immunoblotting against p-Sek1 and β-tubulin was used following treatment of detached cells for 24 h with 25 μM EMD638683 to confirm the efficacy of the inhibitor. (g) ATP levels were measured using the ATP Determination Kit in ECM-detached HCT116 cells after 24 h of treatment with 25 μM EMD638683. (h) Cell viability was measured in ECM-detached HCT116 cells using AlamarBlue following 24 h of treatment with 25 μM EMD638683. All data are presented as mean±S.D. and statistical significance was calculated using a two-tailed t-test. Fold change is calculated as a ratio compared with empty vector (shEV) or DMSO (vehicle) treatment. a.u., Arbitrary units
Oncogenic Ras downregulates PHLPP1 to evade anoikis
The survival of cancer cells during ECM detachment requires rectifying ECM-detachment-induced loss of ATP generation (accomplished in this case by Ras-mediated SGK-1 activation) and blocking caspase activation (anoikis).4 Given that we did not discern a role for Ras-mediated SGK-1 signaling in anoikis regulation (Figure 2f and Supplementary Figures S3B, S3C, S4B and S4C), we hypothesized that oncogenic Ras may use a distinct signaling pathway to block anoikis. Indeed, oncogenic Ras (either H-Ras or K-Ras) can effectively inhibit caspase-3/7 activation (Figure 5a) and promote cell viability (Figure 5b) during ECM detachment. These changes in caspase activation are specific to ECM-detached cells, as the introduction of oncogenic Ras does not inherently alter caspase activation in ECM-attached cells (Figure 5a). While contemplating Ras-mediated signaling pathways that may be responsible for this anoikis inhibition, we revisited our surprising results from Figure 1c: Akt phosphorylation in cells with oncogenic Ras is only marginally affected when PI(3)K is inhibited. We reasoned that this might be due to diminished phosphatase activity and hence the resulting phosphorylated (and activated) Akt may have an important role in anoikis inhibition. The phosphatase PHLPP1 is known to negatively regulate Akt by dephosphorylating the hydrophobic motif (S473).23, 24 To examine if PHLPP1 is negatively regulated by Ras, we assessed PHLPP1 levels in the presence and absence of oncogenic Ras. Indeed, the expression of oncogenic Ras is sufficient to lower the quantities of PHLPP1 in MCF-10A cells (Figure 5c). To examine if changes in PHLPP1 have the capacity to regulate anoikis, we engineered MCF-10A cells to be deficient in PHLPP1 (10A shPHLPP1) and confirmed that PHLPP1 reduction did lead to an increase in Akt phosphorylation (Figure 5d). Reduction of PHLPP1 in MCF-10A cells is sufficient to inhibit caspase-3/7 activation during ECM detachment (Figure 5e) and to promote the viability of ECM-detached cells (Figure 5f). Furthermore, acinar structures derived from MCF-10A cells deficient in PHLPP1 have increased luminal filling, which is indicative of enhanced survival during ECM detachment (Figure 5g).

Loss of PHLPP1 leads to anoikis evasion and enhanced viability in ECM-detached cells. (a) Caspase-3/7 activity was measured using the CaspaseGlo 3/7 assay in the indicated cells following 48 h in either attachment (left) or detachment (right). (b) Cell viability was measured at 48 h in detachment in the indicated cells using AlamarBlue. (c) The indicated (ECM-detached) cells underwent immunoblotting for PHLPP1 and GAPDH. (d) Detached MCF-10A and 10A shPHLPP1 (shPHLPP1) cells underwent immunoblotting for PHLPP1 and β-actin. p-Akt levels were measured as a functional control. (e) Caspase-3/7 activity was measured in MCF-10A and shPHLPP1 cells after 48 h in either attachment (left) or detachment (right) using the CaspaseGlo 3/7 assay. (f) Cell viability was measured using AlamarBlue in MCF-10A and shPHLPP1 cells following 48 h in detachment. (g) MCF-10A and shPHLPP1 cells were plated in Matrigel and allowed to form acini for 10 days. Acini were stained with DAPI (blue) and anti-Laminin 5 (red), and scored as clear, mostly clear, mostly filled, or filled. Representative images are shown above at × 40. All data are presented as mean±S.D. and statistical significance was calculated using a two-tailed t-test. Fold change is calculated as a ratio compared with empty vector (EV, shEV). a.u., Arbitrary units
To examine if the Ras-mediated elimination of PHLPP1 is necessary for anoikis inhibition, we rescued PHLPP1 expression in MCF-10A derivatives with activating Ras mutations and HCT116 cells, and confirmed PHLPP1 expression in each cell line (Figure 6a). Indeed, when PHLPP1 expression is rescued, there is a substantive increase in caspase-3/7 activation (Figure 6b). Furthermore, in HCT116 cells with PHLPP1 overexpression, there is a marked decrease in anchorage-independent growth in soft agar (Figure 6c). Treatment with the pancaspase inhibitor z-VAD-fmk rescued anchorage-independent growth in HCT116 cells engineered to express PHLPP1 (Figure 6c), but had a negligible effect on HCT116 EV cells. These data suggest that PHLPP1-mediated caspase activation is responsible for the loss of colony formation in soft agar. Interestingly, the increase in anoikis induction as a result of PHLPP1 expression occurs independently of the regulation of ATP generation (Supplementary Figures S5A, S5B and S5C).

PHLPP1 downregulation is necessary for Ras-induced anoikis evasion. (a) The indicated cells were immunoblotted for PHLPP1, α-tubulin, or β-actin. (b) Caspase-3/7 levels were measured using the CaspaseGlo 3/7 assay in the indicated cells after 48 h in detachment. (c) HCT116 EV and HCT116 PHLPP1 cells treated with 20 μM of caspase inhibitor z-VAD-fmk (or DMSO vehicle) were plated in soft agar and images were taken after 5 days following staining with INT-violet. z-VAD-fmk or DMSO was added at plating every day as the cells were fed with new media. Colony count was determined using ImageJ. All data are presented as mean±S.D. and statistical significance was calculated using a two-tailed t-test. Fold change is calculated as a ratio compared with empty vector (EV). a.u., Arbitrary units
PHLPP1-mediated rescue of anoikis in cells with Ras mutations is dependent on p38 MAPK
Much to our surprise, when we examined the phosphorylation of Akt following PHLPP1 expression in either 10A HRasG12V or HCT116 cells, we did not observe an appreciable loss of p-Akt (Figure 7a). This suggested that the ability of PHLPP1 to promote anoikis might be independent of its ability to dephosphorylate Akt. In support of this possibility, we found that the levels of FKBP5, which has been shown to function as a scaffolding protein to target PHLPP1 to Akt,20 are substantially reduced in the presence of oncogenic Ras (Figure 7b). This loss of FKBP5 would render PHLPP1 obsolete in its ability to target and dephosphorylate Akt, suggesting that PHLPP1-mediated anoikis is unlikely to be due to Akt regulation. Another common target of PHLPP1 is the ERK/MAPK pathway,25, 26 but we observe no change in p-ERK when PHLPP1 is overexpressed (Figure 7a).

PHLPP1 mediates anoikis through a p38-dependent signaling axis. (a) The indicated (ECM-detached) cells underwent immunoblotting for p-Akt, p-ERK, GAPDH, α-tubulin, or β-actin. (b) Immunoblotting for FKBP5 and β-actin was performed on the indicated (ECM-detached) cells. (c) The indicated (ECM-detached) cells were treated with SB203580 (p38 inhibitor) at 10 μM (HCT116 PHLPP1) or 20 μM (10A HrasG12V PHLPP1) concentrations. Caspase-3/7 activity levels were measured in the indicated cells using the CaspaseGlo 3/7 assay after 24 h (10A HrasG12V) or 48 h (HCT116) in detachment. Immunoblotting for p-MAPKAPK2 and GAPDH was performed to confirm the efficacy of the inhibitor. (d) Immunoblotting for PHLPP1, β-tubulin and p-Sek1 was performed on the indicated (ECM-detached) cells. The indicated cells were plated in soft agar and images were taken after 5 days following staining with INT-violet. Colony count was calculated using ImageJ. (e) Cell viability was measured using AlamarBlue in the indicated attached cells following treatment for 24 h with DMSO, 10 μM LY294002 (PI(3)K inhibitor), or 10 μM SB203580 (p38 inhibitor). (f) Cell viability was measured by AlamarBlue in ECM-detached 10A EV and 10A S422D cells treated with the indicated inhibitor (DMSO, 10 μM LY294002, or 10 μM SB203580) for 24 h. (g) RPPA data for colorectal adenocarcinoma patients was analyzed. (Top) p-p27/Kip1 (T157) versus oncogenic K-Ras (Pearson’s coefficient=0.47; P<0.01). (Bottom) p-p38 MAPK (T180) versus oncogenic K-Ras (Pearson’s coefficient=−0.37; P<0.05). All data are presented as mean±S.D. and statistical significance was calculated using a two-tailed t-test. Fold change is calculated as a ratio compared to empty vector (EV, shEV). a.u., Arbitrary units
Given that neither Akt nor ERK seem to be the relevant target of PHLPP1 responsible for anoikis induction, we next investigated the role of the p38 MAPK pathway in PHLPP1-mediated anoikis. Recently, Mst-1 has been identified as a substrate of PHLPP1. Desphosphorylation activates Mst-1 and ultimately leads to stimulation of p38 MAPK activity.25 Indeed, in 10A HrasG12V and HCT116 cells, the expression of PHLPP1 results in an increase in p38 MAPK activation (that can be blocked by the p38 MAPK inhibitor SB203580) as measured by phosphorylation of the downstream target MAPKAPK227 (Figure 7c). Furthermore, SB203580 treatment leads to a significant reduction in PHLPP1-mediated caspase-3/7 activation (Figure 7c). Taken together, these data demonstrate that the rescue of anoikis by PHLPP1 in cells harboring mutations in oncogenic Ras is dependent on activation of the p38 MAPK pathway.
SGK-1 and PHLPP1 cooperatively function to promote the survival of ECM-detached cancer cells
Given that our data support a model by which Ras facilitates cell survival during ECM detachment through distinct downstream signaling pathways, we sought to investigate if there is any synergy between SGK-1 and PHLPP1/p38 MAPK in promoting anchorage-independent growth. To examine this possibility, we engineered HCT116 shSGK-1 cells to express PHLPP1 (Figure 7d). Indeed, in HCT116 shSGK-1 cells we see a decrease in anchorage-independent growth that is further diminished by the addition of PHLPP1 (Figure 7d). These data suggest that the simultaneous modulation of both SGK-1 and PHLPP1/p38 MAPK signaling during ECM detachment can function cooperatively to influence anchorage-independent growth.
Given the aforementioned data, it is reasonable to surmise that simultaneous activation of SGK-1 and inhibition of p38 MAPK may be able to enhance the viability of cells treated with PI(3)K inhibitors. Indeed, overexpression of constitutively active SGK-1 (S422D) and inhibition of p38 MAPK (with SB203580) rescues cell death induced by PI(3)K inhibition (Figure 7e). We next sought to further assess the individual contributions of the SGK-1 and p38 MAPK pathway. Interestingly, while p38 MAPK inhibition alone is not sufficient to promote viability of ECM-detached cells upon PI(3)K inhibition, p38 MAPK inhibition did cooperate with constitutive activation of SGK-1 to elevate viability (Figure 7f). Taken together, these data suggest that simultaneous activation of SGK-1 and inhibition of p38 MAPK can promote the viability of cells treated with a PI(3)K inhibitor.
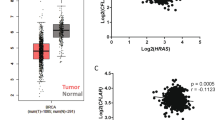
We next sought to ascertain if patients afflicted with colorectal adenocarcinomas containing Ras mutations harbored evidence of concomitant regulation of SGK-1 and PHLPP1. Indeed, in these patients, there is a statistically significant negative correlation between levels of oncogenic K-Ras and phosphorylation of p38 MAPK at threonine 180 (Figure 7g). Furthermore, in these same patients, there exists a statistically significant positive correlation between levels of oncogenic K-Ras and phosphorylation of p27/KIP1 at threonine 157, a site well known to be phosphorylated by SGK-128 (Figure 7g). In aggregate, these data suggest that differential and concurrent regulation of ATP production and caspase activation (anoikis) downstream of oncogenic K-Ras through SGK-1 and PHLPP1/p38 MAPK is occurring in human colorectal adenocarcinoma.
Discussion
Given that it is now appreciated that cancer cells must rectify metabolic defects (e.g. ATP generation) in addition to blocking caspase activation (anoikis) to survive during ECM detachment,4 it is critically important to understand the precise molecular mechanisms involved in mediating both anoikis and metabolism. The data described here outline a unique, multifaceted signaling pathway operating downstream of oncogenic Ras to permit anoikis inhibition and stimulate ATP production (Figure 8). Our data place Ras signaling in contrast to ErbB2 with regard to metabolic regulation during ECM detachment, as previous studies have shown that ErbB2 promotes ATP generation through PI(3)K-mediated activation of Akt (rather than SGK-1).6 These data suggest that the strategies used by cancer cells to alleviate deleterious metabolic changes (and block anoikis) may vary considerably across tumor types and contexts.11, 29, 30 Additional studies examining the relationship between oncogenic signaling and metabolism in ECM-detached cancer cells are warranted to better ascertain the best strategies to facilitate their elimination.

Model for oncogenic Ras-mediated cell survival during ECM detachment
The identification of SGK-1 as a primary mediator of ATP generation downstream of oncogenic Ras adds to the growing body of literature, suggesting that molecules other than Akt can function downstream of PI(3)K to modulate distinct cellular functions in cancer cells.18, 31 Our studies suggest that ECM-detached cells with oncogenic Ras mutations could be compromised by SGK-1 inhibition. Indeed, the development of SGK-1 antagonists has already commenced in other cancers,32, 33 and our results suggest that similar inhibitors may be efficacious in eliminating ECM-detached cells with Ras mutations. Interestingly, our findings are also consistent with data suggesting that SGK-1 activity can mediate the sensitivity of cancer cells to targeted therapies.34 It is noteworthy that other investigators have discovered that SGK-1 can interact with proteins at the mitochondria and can inhibit the induction of necrotic cell death.19, 35 Our data implicating SGK-1 in ATP generation may provide additional insight into these connections between SGK-1 and mitochondrial integrity.
Much like SGK-1, interest in understanding PHLPP1 regulation during tumorigenesis has intensified over the past few years. Studies from a number of labs have now implicated loss of PHLPP1 activity as an important facet of cancer cell survival.23, 36, 37, 38 That being said, despite the fact that PHLPP1 has a predicted Ras-associated domain,39 our study is the first (to our knowledge) to directly link downregulation of PHLPP1 to oncogenic Ras signaling. In addition, the majority of studies aimed at understanding PHLPP1 in cancer cells have focused on PHLPP1’s ability to dephosphorylate Akt. Our data suggest that PHLPP1-mediated anoikis is not reliant on Akt dephosphorylation, but is instead dependent on activation of the p38 MAPK pathway. Our data provide an important and novel physiological setting by which PHLPP1-mediated activation of p38 MAPK could be important in the context of tumor progression.
In fact, our data suggest that the most effective way to eliminate ECM-detached cells with Ras mutations would be by stimulating p38 MAPK (and inducing anoikis) while simultaneously antagonizing SGK-1 (and blocking ATP generation). Indeed, our analysis of publically available reverse phase protein array (RPPA) data from colorectal cancer patients (Figure 7g) demonstrates a correlation between mutant K-Ras levels, activation of SGK-1, and inactivation of p38 MAPK. However, while these data are consistent with the notion that simultaneous activation of p38 MAPK and inhibition of SGK-1 might be an effective therapeutic strategy, future experiments aimed at assessing the efficacy of such a regimen will be critical to the development of novel chemotherapies designed to eradicate ECM-detached cancer cells.
Materials and Methods
ATP assays
To measure ATP levels in ECM-detached cells, 400 000 cells were plated in 6-well poly-(2-hydroxyethyl methacrylate)- (poly-HEMA; Sigma-Aldrich, St. Louis, MO, USA) coated plates for the amount of time indicated in the figure legends. The ATP Determination Kit (Invitrogen, Carlsbad, CA, USA) was used following normalization of total protein concentration according to the manufacturer’s instructions. In LY294002 (Calbiochem, Billerica, MA, USA) and EMD638683 (ApexBio, Houston, TX, USA) experiments, the indicated concentration of inhibitor was added at the time of plating. All data presented are representative images from at least three independent replicates.
Caspase-3/7 assays
Cells were plated at a density of 13 000 cells per well on 96-well poly-HEMA-coated plates. Caspase activation was measured using the CaspaseGlo 3/7 Assay Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. In p38 (SB203580; Cell Signaling Technologies, Danvers, MA, USA), SGK-1 kinase (EMD638683), and staurosporine (STS; Sigma-Aldrich) experiments, the indicated concentration of the inhibitor was added at the time of plating. For SB203580 experiments, the inhibitor was added every 24 h (12 h for 10A HrasG12V PHLPP1) throughout the duration of the experiment. All data presented are representative images from at least three independent replicates.
Cell culture
MCF-10A cells (ATCC, Manassas, VA, USA) and derivatives were cultured in Dulbecco’s modified Eagle's medium/F12 (Gibco, Waltham, MA, USA) supplemented with 5% horse serum (Invitrogen), 20ng/ml epidermal growth factor (EGF), 10 μg/ml insulin, 500 μg/ml hydrocortisone, 100 ng/ml cholera toxin, and 1% penicillin/streptomycin. HCT116 cells (ATCC) and derivatives were cultured in McCoy’s media (Gibco) plus 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin.
CellTiter Glo assays
Cells were plated at a density of 13 000 cells per well on either a 96-well plate (attached) or a 96-well poly-HEMA-coated plate (detached) for the indicated time. ATP levels were measured using the CellTiter Glo Assay (Promega) according to the manufacturer’s instructions. All data presented are representative images from at least three independent replicates.
Cell viability assays
Cells were plated at a density of 13 000 cells per well on 96-well poly-HEMA-coated plates. AlamarBlue reagent (Invitrogen) was added to each well according to the manufacturer’s instructions. EMD638683, LY294002, and SB203580 were added at the indicated concentrations at the time of plating. All data presented are representative images from at least three independent replicates.
Glucose-limiting assays
The indicated cells were plated at 400 000 cells per well in 6-well poly-HEMA-coated plates for 24 h. MCF-10A derivatives were plated in standard media (described above) or glucose-free Dulbecco’s modified Eagle's medium/F12 (US Biologicals, Salem, MA, USA) supplemented with 5% horse serum (Invitrogen) 20 ng/ml EGF, 10 μg/ml insulin, 500 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 1% penicillin/streptomycin, and 0.4 mM glucose solution (Gibco). HCT116 derivatives were plated in either standard media (described above) or glucose-free Dulbecco’s modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Invitrogen), 1% penicillin/streptomycin, and 0.4 mM glucose solution (Gibco).
Glucose uptake assays
Glucose uptake was measured in ECM-detached cells using the Amplex Red Glucose Assay Kit (Invitrogen) according to the manufacturer’s instructions. Cells were plated at a density of 13 000 cells per well, and baseline glucose measurements were taken from a media-only control plated at the same time as the cells. All data presented are representative images from at least three independent replicates.
Immunoblotting
ECM-detached cells were harvested, washed once with cold PBS, and lysed in 1% Nonidet P-40 supplemented with protease inhibitors leupeptin (5 μg/ml), aprotinin (1 μg/ml), and PMSF (1 mM) and the Halt Phosphatase Inhibitor Mixture (Thermo Scientific, Waltham, MA, USA). Lysates were collected after spinning for 30 min at 4 °C at 14 000 r.p.m. and normalized by BCA Assay (Pierce Biotechnology, Waltham, MA, USA). Normalized lysates underwent SDS-PAGE and transfer/blotting was performed as described previously.10 The following antibodies were used for western blotting: IGF-1Rβ (Santa Cruz Biotechnology, Dallas, TX, USA; no. sc-81167), SGK-1 (EMD Millipore, Billerica, MA, USA; no. 07-315), p-Akt (Cell Signaling Technology; no. 4060), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technologies; no. 5174), Ras (Cell Signaling Technology; no. 3965), p-Sek1 (Abcam, Cambridge, MA, USA; ab39403), DYKDDDDK (FLAG) tag (Cell Signaling Technology; no. 2368), α-tubulin (Cell Signaling Technology; no. 2144), β-tubulin (Cell Signaling Technology; no. 2146), β-actin (Sigma-Aldrich; no. A1978), PHLPP (Bethyl Laboratories, Montgomery, TX, USA; no. A304-029A), p-Erk1/2 (Invitrogen; no. 368800), FKBP5 (Cell Signaling Technology; no. 12210), and p-MAPKAPK2 (Cell Signaling Technology; no. 3007).
Immunofluorescence
Cells were plated at a density of 50 000 cells per well in a 6-well plate (attached), or a 6-well poly-HEMA-coated plate (detached) for 24 h. The cells were then harvested, washed two times with cold PBS, and deposited onto slides with a Shandon Cytospin3 (Thermo Scientific) at 800 r.p.m. for 5 min. Cells were then fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 in PBS. Cells were washed with 100 mM glycine in PBS three times and blocked with immunofluorescence (IF) buffer containing 130 mM NaCl, 7 mM Na2HPO4, 3.5 mM NaH2PO4, 7.7 mM NaH3, 0.1% BSA (Millipore), 1.2% Triton X-100, and 0.5% Tween-20, supplemented with 10% goat serum (Invitrogen). Slides were stained with SGK-1 (EMD Millipore, no. 07-315) at a concentration of 1 : 200 in IF buffer. For secondary visualization, AlexaFluor 568 (Invitrogen; no. A11031) was used at 1 : 200 in IF buffer. Nuclei were stained with 5 μg/ml 4,6-diamidino-2-phenylindone (DAPI; Invitrogen) and mounted with ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA, USA). Imaging was completed using the Applied Precision DeltaVision OMX fluorescent microscope (Applied Precision; GE Healthcare, Issaquah, WA, USA). Images shown are representative images at × 60 magnification.
Retroviral generation of stable cell lines
The pBABE-Puro-based retroviral vectors encoding constitutively active H-Ras, constitutively active K-Ras, and PHLPP1, the pBABE-Neo-based retroviral vectors encoding constitutive active H-Ras and constitutive active K-Ras, and the pLPCX-Puro-based retroviral vectors encoding constitutive active SGK-1 and kinase-dead SGK-1 were used to generate stable cell lines. HEK293T cells were transfected with 0.75 μg target DNA along with the packaging vector pCLAmpho (0.75 μg) with Lipofectamine 2000 (Life Technologies). Virus was collected at 48 and 72 h post-transfection, filtered through a 0.45 μm filter (EMD Millipore), and used for transduction of MCF-10A and HCT116 cells in the presence of 8 μg/ml polybrene. Stable populations of puromycin-resistant cells were obtained using 2 μg/ml puromycin (Invivogen, San Diego, CA, USA). Stable populations of neomycin-based cells were obtained using 300 μg/ml G418 (Nalgene, Waltham, MA, USA).
shRNA generation of stable cell lines
MISSION shRNA constructs against SGK-1 (NM_005627; TRCN0000094957, TRCN0000040175) and PHLPP1 (NM_194449; TRCN0000082796) in the puromycin-resistant pLKO.4 vector along with an empty vector control were purchased from Sigma-Aldrich. HEK293T cells were transfected with 0.5 μg target DNA along with the packaging vectors pCMV-D8.9 (0.5 μg) and pCMV-VSV-G (60 ng) using Lipofectamine 2000 and PLUS reagent (Life Technologies). Virus was collected 24 and 48 h post-transfection and filtered through a 0.45 μm filter (EMD Millipore), and used for transduction of MCF-10A and HCT116 cells in the presence of 8 μg/ml polybrene. Stable populations of cells were selected using 2 μg/ml puromycin (Invivogen).
Soft agar assays
Cells were plated at densities of 20 000–30 000 cells per well in 1.5 ml of growth media plus 0.4% low-melt agarose (Sigma-Aldrich) and layered onto a 3 ml bed of growth media with 0.5% low-melt agarose. Cells were fed daily with 1 ml of growth media, plus the indicated concentration of z-VAD-fmk when indicated. At the indicated time, growth media was removed and viable colonies were stained using INT-violet (Sigma-Aldrich). Colony number was determined using ImageJ (Bethesda, MD, USA). All data presented are representative images of at least three independent replicates.
Statistics
Statistical significance was determined using a two-tailed t-test. Error bars represent S.D.
Transient transfection
The indicated cells were transfected with 1.5 μg target DNA for the pcDNA3-based vectors including an empty vector control and PHLPP1 (Addgene, Cambridge, MA, USA; plasmid no. 22404) using Lipofectamine 2000 and PLUS reagent (Life Technologies) according to the manufacturer’s instructions. Cells were plated for soft agar assays and immunoblotting was completed 48 h post-transfection.
3D cell culture
To generate acini, 3000–4000 cells were plated per well in an 8-well chamber slide on a Matrigel (Lot. no. 9044) bed and allowed to form acinar structures as described previously40, 41 and according to the protocol at http://brugge.med.harvard.edu/. Overlay media (Dulbecco’s modified Eagle's Medium/F12 supplemented with 2% horse serum, 5 ng/ml EGF, 10 μg/ml insulin, 500 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 2% Matrigel, and 1% penicillin/streptomycin) was added at days 0 and 4. Overlay media, consisting of the above without the 2% Matrigel, was added every 4 subsequent days until fixing. At the indicated time, acinar structures were harvested with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 in PBS. Fixed structures then underwent three glycine (100 mM) washes in IF buffer (described above). Laminin 5 (Millipore; no. MAB88198) primary antibody was used at 1 : 150 in IF buffer plus 10% goat serum. For secondary visualization, AlexaFluor 568 (Invitrogen; no. A11031) was used at 1 : 200 in IF buffer plus 10% goat serum. Nuclei were counterstained with 5 μg/ml DAPI and slides were mounted using ProLong Gold Antifade Reagent (Waltham, MA, USA). Luminal filling was examined using a Nikon A1R-MP microscope (Nikon, Melville, NY, USA) and scored as clear (~0–10% filled), most clear (~10–50% filled), mostly filled (~50–90% filled), or filled (~90–100% filled). All data are representative of at least three independent replicates and images are shown at × 40.
Analysis of RPPA data
R studio version 3.1.3 was used to mine colorectal adenocarcinoma (TCGA, Provisional) for RPPA clinical data from MD Anderson Cancer Center (Houston, TX, USA). K-Ras mutations and RPPA data for K-Ras, and p27 phosphorylated at threonine 157 (p-p27/KIP1 T157) and p38/MAPK phosphorylated at threonine 180 (phospho-p38/MAPK T180) data were collected. The data were parsed into a data set that contained patients with highly expressed oncogenic K-Ras (samples with positive values of relative K-Ras levels). These patient data were then used to generate scatter plots in Excel separating K-Ras on the X axis and either p27_pT157 or p38_pT180 on the Y axis and a best fit line was drawn for visualization of the overall trend of relative protein levels. R studio was used to calculate the Pearson's coefficients (r-values) and P-values based on Fisher's Z-scores (*95% confidence interval, P<0.05 and **99% confidence interval, P<0.001).
Abbreviations
- ECM:
-
extracellular matrix
- PI(3)K:
-
phosphatidylinositol (3)-kinase
- SGK-1:
-
serum and glucocorticoid-regulated kinase-1
- PHLPP1:
-
PH domain and leucine rich repeat protein phosphatase
- poly-HEMA:
-
poly-(2-hydroxyethyl methacrylate)
- EGF:
-
epidermal growth factor
- DAPI:
-
4,6-diamidino-2-phenylindone
- RPPA:
-
reverse phase protein array
- INT-violet:
-
iodonitrotetrazolium chloride
- STS:
-
staurosporine
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- EV:
-
empty vector
References
Nguyen DX, Massague J . Genetic determinants of cancer metastasis. Nat Rev Genet 2007; 8: 341–352.
Li X, Stevens PD, Yang H, Gulhati P, Wang W, Evers BM et al. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene 2013; 32: 471–478.
Frisch SM, Francis H . Disruption of epithelial cell–matrix interactions induces apoptosis. J Cell Biol 1994; 124: 619–626.
Buchheit CL, Weigel KJ, Schafer ZT . Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer 2014; 14: 632–641.
Buchheit CL, Rayavarapu RR, Schafer ZT . The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev Biol 2012; 23: 402–411.
Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009; 461: 109–113.
Davison CA, Schafer ZT . Keeping a breast of recent developments in cancer metabolism. Curr Drug Targets 2010; 11: 1112–1120.
Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol 2011; 31: 3616–3629.
Fung C, Lock R, Gao S, Salas E, Debnath J . Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19: 797–806.
Davison CA, Durbin SM, Thau MR, Zellmer VR, Chapman SE, Diener J et al. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res 2013; 73: 3704–3715.
Buchheit CL, Angarola BL, Steiner A, Weigel KJ, Schafer ZT . Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ 2015; 22: 1275–1286.
Downward J . Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3: 11–22.
Prior IA, Lewis PD, Mattos C . A comprehensive survey of Ras mutations in cancer. Cancer Res 2012; 72: 2457–2467.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D . RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011; 11: 761–774.
McFall A, Ulku A, Lambert QT, Kusa A, Rogers-Graham K, Der CJ . Oncogenic Ras blocks anoikis by activation of a novel effector pathway independent of phosphatidylinositol 3-kinase. Mol Cell Biol 2001; 21: 5488–5499.
Eckert LB, Repasky GA, Ulku AS, McFall A, Zhou H, Sartor CI et al. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res 2004; 64: 4585–4592.
Bryant NJ, Govers R, James DE . Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 2002; 3: 267–277.
Bruhn MA, Pearson RB, Hannan RD, Sheppard KE . Second AKT: the rise of SGK in cancer signalling. Growth Factors 2010; 28: 394–408.
Brickley DR, Agyeman AS, Kopp RF, Hall BA, Harbeck MC, Belova L et al. Serum- and glucocorticoid-induced protein kinase 1 (SGK1) is regulated by store-operated Ca2+ entry and mediates cytoprotection against necrotic cell death. J Biol Chem 2013; 288: 32708–32719.
Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA . Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J 1999; 18: 3024–3033.
Kim MJ, Chae JS, Kim KJ, Hwang SG, Yoon KW, Kim EK et al. Negative regulation of SEK1 signaling by serum- and glucocorticoid-inducible protein kinase 1. EMBO J 2007; 26: 3075–3085.
Brink M, de Goeij AF, Weijenberg MP, Roemen GM, Lentjes MH, Pachen MM et al. K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 2003; 24: 703–710.
Newton AC, Trotman LC . Turning off AKT: PHLPP as a drug target. Annu Rev Pharmacol Toxicol 2014; 54: 537–558.
Gao T, Furnari F, Newton AC . PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 2005; 18: 13–24.
Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W et al. Mst1 is an interacting protein that mediates PHLPPs' induced apoptosis. Mol Cell 2010; 38: 512–523.
Suljagic M, Laurenti L, Tarnani M, Alam M, Malek SN, Efremov DG . Reduced expression of the tumor suppressor PHLPP1 enhances the antiapoptotic B-cell receptor signal in chronic lymphocytic leukemia B-cells. Leukemia 2010; 24: 2063–2071.
Xu L, Chen S, Bergan RC . MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 2006; 25: 2987–2998.
Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM . mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell 2008; 30: 701–711.
Rayavarapu RR, Heiden B, Pagani N, Shaw MM, Shuff S, Zhang S et al. The role of multicellular aggregation in the survival of ErbB2-positive breast cancer cells during extracellular matrix detachment. J Biol Chem 2015; 290: 8722–8733.
Weigel KJ, Jakimenko A, Conti BA, Chapman SE, Kaliney WJ, Leevy WM et al. CAF-secreted IGFBPs regulate breast cancer cell anoikis. Mol Cancer Res 2014; 12: 855–866.
Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A . SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol Cell 2014; 56: 595–607.
Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res 2008; 68: 7475–7483.
Skor MN, Wonder EL, Kocherginsky M, Goyal A, Hall BA, Cai Y et al. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res 2013; 19: 6163–6172.
Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR . Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J 2013; 452: 499–508.
O'Keeffe BA, Cilia S, Maiyar AC, Vaysberg M, Firestone GL . The serum- and glucocorticoid-induced protein kinase-1 (Sgk-1) mitochondria connection: identification of the IF-1 inhibitor of the F(1)F(0)-ATPase as a mitochondria-specific binding target and the stress-induced mitochondrial localization of endogenous Sgk-1. Biochimie 2013; 95: 1258–1265.
Wang P, Zhou Z, Hu A, Ponte de Albuquerque C, Zhou Y, Hong L et al. Both decreased and increased SRPK1 levels promote cancer by interfering with PHLPP-mediated dephosphorylation of Akt. Mol Cell 2014; 54: 378–391.
Wen YA, Stevens PD, Gasser ML, Andrei R, Gao T . Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol Cell Biol 2013; 33: 4594–4605.
Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y et al. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res 2013; 73: 5402–5415.
Warfel NA, Newton AC . Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP): a new player in cell signaling. J Biol Chem 2012; 287: 3610–3616.
Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS . BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell 2007; 12: 221–234.
Debnath J, Muthuswamy SK, Brugge JS . Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30: 256–268.
Acknowledgements
We thank Raju Rayavarapu, Kelsey Weigel, Mark Hawk, Meaghan Boyd, Kimberly Hagel, Veronica Schafer, Patricia Champion, Athanasia Panopoulos, Crislyn D’Souza-Schorey, and the entire Schafer Lab for their discussions, comments, and experimental assistance. We also express gratitude to Chris Counter (Duke University) for the KrasG12V plasmid, Suzanne Conzen (University of Chicago) for the constitutively active and kinase-dead SGK-1 plasmids, Tianyan Gao (University of Kentucky) for the PHLPP1 plasmid, and Alexandra Newton (UC San Diego) for the pcDNA3-PHLPP1 plasmid. This work was supported by a Lee National Denim Day Research Scholar Grant (RSG-14-145-01-CSM) from the American Cancer Society (to ZTS), a research grant from the Phi Beta Psi National Project (to ZTS), and a Pathway to Independence Award (5R00CA158066) from the National Cancer Institute (to SZ). ZTS is also the recipient of a Career Catalyst Research Grant (CCR14302768) from Susan G Komen.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by S Fulda
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Mason, J., Davison-Versagli, C., Leliaert, A. et al. Oncogenic Ras differentially regulates metabolism and anoikis in extracellular matrix-detached cells. Cell Death Differ 23, 1271–1282 (2016). https://doi.org/10.1038/cdd.2016.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.15
This article is cited by
-
Chronic stress in solid tumor development: from mechanisms to interventions
Journal of Biomedical Science (2023)
-
EGFR/MET promotes hepatocellular carcinoma metastasis by stabilizing tumor cells and resisting to RTKs inhibitors in circulating tumor microemboli
Cell Death & Disease (2022)
-
Small leucine zipper protein functions as a modulator for metabolic reprogramming of colorectal cancer cells by inducing nutrient stress-mediated autophagy
Cellular and Molecular Life Sciences (2022)
-
Oncogenic signaling inhibits c-FLIPL expression and its non-apoptotic function during ECM-detachment
Scientific Reports (2021)
-
RIPK1-mediated induction of mitophagy compromises the viability of extracellular-matrix-detached cells
Nature Cell Biology (2018)
