
Abstract
NF-κB-inducing kinase (NIK) is well-known for its role in promoting p100/NF-κB2 processing into p52, a process defined as the alternative, or non-canonical, NF-κB pathway. Here we reveal an unexpected new role of NIK in TNFR1-mediated RIP1-dependent apoptosis, a consequence of TNFR1 activation observed in c-IAP1/2-depleted conditions. We show that NIK stabilization, obtained by activation of the non-death TNFRs Fn14 or LTβR, is required for TNFα-mediated apoptosis. These apoptotic stimuli trigger the depletion of c-IAP1/2, the phosphorylation of RIP1 and the RIP1 kinase-dependent assembly of the RIP1/FADD/caspase-8 complex. In the absence of NIK, the phosphorylation of RIP1 and the formation of RIP1/FADD/caspase-8 complex are compromised while c-IAP1/2 depletion is unaffected. In vitro kinase assays revealed that recombinant RIP1 is a bona fide substrate of NIK. In vivo, we demonstrated the requirement of NIK pro-death function, but not the processing of its substrate p100 into p52, in a mouse model of TNFR1/LTβR-induced thymus involution. In addition, we also highlight a role for NIK in hepatocyte apoptosis in a mouse model of virus-induced TNFR1/RIP1-dependent liver damage. We conclude that NIK not only contributes to lymphoid organogenesis, inflammation and cell survival but also to TNFR1/RIP1-dependent cell death independently of the alternative NF-κB pathway.
Similar content being viewed by others


Main
In mammals, cell death is not only required for the proper development of various organs and tissue remodeling but also occurs as a consequence of insults, such as viral infection, DNA damage or chronic inflammation. One form of cell death, called PCD (programmed cell death), appears in two varieties that are termed apoptosis and necroptosis, a caspase-dependent and -independent process, respectively. In the latter, caspase-8 has been shown to have an inhibitory role preventing spontaneous necroptosis induction.1 PCD has been described to take place downstream of death receptors of the tumor necrosis factor receptor (TNFR) family, such as TNFR 1 (TNFR1) or Fas (APO-1 or CD95).2 Beyond cell death, TNFR1 also triggers the activation of nuclear factor-kappa B (NF-κB) and the transcription of a plethora of pro-inflammatory cytokines and pro-survival factors. TNFR1-mediated NF-κB activation relies on a signaling platform called complex I.3, 4 The formation of this complex is initiated by the interaction of ligand-bound TNFR1 with TNF receptor-associated death domain (TRADD) and by the recruitment of the kinase receptor interacting protein kinase 1 (RIP1), the adaptor proteins TNF receptor-associated factor 2/5 and the ubiquitin ligase cellular inhibitor of apoptosis 1/2 (c-IAP1/2) and linear ubiquitin chain assembly complex (LUBAC).5, 6, 7, 8, 9 c-IAP1/2 and LUBAC target RIP1 for K11, K63 and linear polyubiquitination priming RIP1 for the recruitment of two kinase-containing complexes, namely, TAK1/TAK1-binding protein 1 (TAB1)/TAB2 and NEMO/inhibitor of NF-κB kinase α (IKKα)/IKKβ.5, 10, 11, 12, 13 Ultimately, activation of IKKβ leads to the phosphorylation and proteasomal degradation of IκBα and the nuclear translocation of NF-κB, a pathway defined as the classical, or the canonical, NF-κB pathway.14, 15, 16 Besides its role in the activation of NF-κB, the dissociation of complex I from TNFR1 triggers the recruitment of Fas-associated protein with death domain (FADD) and caspase-8 to form a cytosolic death complex, initially defined as complex II.3 TNFR1 induces apoptosis through two distinct caspase-8 activation pathways. Caspase-8 is either activated within a TRADD/FADD-containing complex (complex IIa) or in a RIP1/FADD-containing complex (complex IIb).17, 18 The former assembles when cells are deficient in p65 or pretreated with cycloheximide (CHX), whereas the latter forms in the absence of c-IAP1/2, in TAK1 kinase-inhibited conditions or in NEMO-depleted cells.19, 20 In addition, under caspase-repressed conditions RIP1 associates with RIP3 to mediate necroptosis, a form of programmed necrosis that takes place during viral infection or TNF alpha (TNFα)-induced systemic inflammatory response syndrome (SIRS).21, 22, 23, 24, 25
Synthetic small molecules mimicking Smac (Smac mimetics (SMs)) emerged as tools to induce c-IAP1/2 depletion and were consequently shown to sensitize particular cancer cell lines to TNFR1-mediated RIP1-dependent death.11, 26, 27, 28 Apart from these chemicals, a set of biological ligand of the TNF family, such as lymphotoxin LTα1β2, CD40L or Tweak, also appeared to target c-IAP1/2 toward the proteasomal and/or lysosomal degradative pathways.29, 30, 31, 32, 33, 34 c-IAP1/2 depletion leads to NIK (NF-κB-inducing kinase) stabilization and the phosphorylation of both IKKα and NF-κB2/p100 prior to the processing of p100 into p52, a pathway defined as the alternative, or non-canonical, NF-κB pathway.35, 36, 37
Genetic mouse models revealed that NIK is required for the proper development and/or maintenance of secondary lymphoid organs, for osteoclastogenesis, for T-cell activation as well for B-cell survival.35, 38, 39
In this paper, we highlight an unexpected role for NIK in TNFR1-induced cell death. We found that NIK diverges from the alternative NF-κB pathway to promote cell death. NIK is required for the activation of caspase-8 by promoting the assembly of the RIP1/FADD/caspase-8 death complex downstream of TNFR1. More importantly, we found that NIK activation, but not the processing of p100, was required for TNFR1-induced cortical thymic epithelia cell (cTEC) death and thymus involution, a function of NIK that is clearly distinct from its role in the differentiation of medullary thymic epithelial cells (mTECs) toward the induction of p100 processing. In addition, using a mouse model of viral infection we found a role for NIK in RIP1-dependent TNFR1-driven hepatocyte apoptosis.
Results
Fn14- and lymphotoxin-beta receptor (LTβR)-mediated TNFR1-dependent cell death requires the activation of NIK
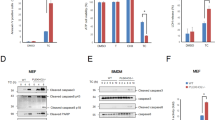
It was recently reported that Tweak-induced TNFR1-dependent cell death relies on the depletion of c-IAP1/2 proteins.33 Depletion of c-IAP1/2 leads to the stabilization of NIK, a kinase inducing the alternative NF-κB pathway, and involved in the development and homeostasis of lymphoid organs and cell survival of hematopoietic cell lineages.39 Thus, based on the latter function, we anticipated that inactivation of NIK would rather sensitize cells to TNFR1-induced death. To address this possibility, we first treated wild-type (NIK+/+), NIK knockout (NIK−/−) and NIK-deficient (aly/aly) MEFs with Tweak and/or human TNFα. We observed that co-stimulation of NIK+/+ MEFs with Tweak and TNFα led to substantial decrease in cell survival and an increase in AnnexinV/PI positivity, while these ligands had minor impact when used independently. Remarkably, this effect was drastically inhibited in the absence of NIK or in aly/aly MEFs (Figures 1a and b and Supplementary Figures S1a and b). Stimulation of NIK+/+ MEFs with increasing amount of Tweak and TNFα led to a substantial DEVDase activity overtime peaking at 4 h treatment (Figure 1c). The observed timing of caspase activation in NIK+/+ cells was consistent with the appearance of NIK expression and the cleaved forms of caspase-8 and -3 at 4 h posttreatment (Figure 1d). In contrast, Tweak/TNFα-stimulated NIK−/− MEFs displayed a severe defect in DEVDase activity and cleavage of caspase-8 and caspase-3. Importantly, complementation of NIK−/− cells with NIK WT, but not NIK kinase-dead, restored the ability of Tweak and TNFα to induce caspase-3 cleavage (Figure 1e). We next extended our analysis to LTβR, another TNF receptor that enables the activation of NIK and of the alternative NF-κB pathway.37, 40 Remarkably, we observed that co-stimulation of TNFR1 and LTβR induced an elevation of DEVDase activity and caspase-8 and -3 cleavage that was also dependent on the expression of NIK (Figures 1f and g). Together, these results revealed a pro-death activity for NIK downstream of TNFR1 but upstream of caspase-8.

Tweak- and Lymphotoxin-αβ-induced TNFR1-dependent cell death relies on NIK. (a) Cell survival of NIK+/+ and NIK−/− mouse embryonic fibroblasts (MEFs) was assessed by measuring the ATP levels using the Cell Titer-Glo Kit. Cells were treated for 24 h with Tweak or TNFα alone or both together. Data are represented as mean±S.Ds. of quadruplicates. Statistical analyses: ***P<0.001. (b) NIK+/+ and NIK−/− MEFs were treated for 6 h with Tweak or TNFα alone or both together, and Annexin V and/or PI positivity were measured by flow cytometry. (c) DEVDase activity of NIK+/+ and NIK−/− MEFs treated for various time with two doses of Tweak and a fixed concentration of TNFα. (d) NIK+/+ and NIK−/− MEFs were treated with Tweak and TNFα for different time periods. Levels of NIK, cleaved caspase-8, cleaved caspase-3 and actin were assessed by western blotting. (e) Western blotting for NIK, cleaved caspase-3 and actin expression in parental NIK−/− MEFs reconstituted with empty vector (EV), NIK WT or NIK kinase-dead (KD). (f and g) NIK+/+ and NIK−/− MEFs were treated with a LTβR agonist antibody and TNFα for different time periods. DEVDase activities and levels of NIK, cleaved caspase-8, cleaved caspase-3 and actin were assessed similar to panels (c and d)
NIK pro-death activity acts toward RIP1 cell death signaling
TNFR1 can trigger two independent apoptotic pathways depending on the cellular context and the stimuli. Indeed, as previously reported, RIP1-independent (TRADD-dependent) and RIP1-dependent apoptotic pathways can be activated when cells are stimulated with TNFα and CHX or TNFα and SMs, respectively.17 We found that NIK+/+ and NIK−/− MEFs equally died following exposure to TNFα/CHX, indicating that the RIP1-independent pro-apoptotic machinery was intact in NIK-deficient cells (Figure 2a). Conversely, similar to RIP1-deficient cells, NIK−/− MEFs were resistant to TNFα/SM (CmpA)-induced cell death (Figure 2b, Supplementary Figure S2a). The resistance of NIK−/− MEFs was not ascribed to a defect in TNF/SM-induced p-JNK or to c-FLIP expression levels (Supplementary Figures S2b–d). We recently showed that the RIP1 kinase inhibitor necrostatin-1 (Nec-1) prevented SM/TNFα-induced caspase activation.19 Similarly, we observed that Tweak/TNFα- or agonist LTβR /TNFα-induced caspase activation was equally inhibited in the presence of Nec-1 or Z-Val-Ala-DL-Asp-fluoromethylketone (z-VAD-FMK) (Figures 2c and d). We also found that Tweak/TNFα- or agonist LTβR/TNFα-induced RIP1/caspase-8 assembly was fully abrogated in the presence of Nec-1 (Figures 2e and f). These results are consistent with previous studies demonstrating a role for Nec-1 in the prevention of SM/TNFα-induced RIP1/caspase-8 assembly and apoptosis.19, 41 We further found that the DEVDase activity induced by Tweak/TNFα co-stimulation was strongly reduced in Bax/Bak−/− double KO MEFs when compared with Bax/Bak+/+ counterparts MEFs (Supplementary Figure S2e). Likewise, treatment of Bax/Bak−/− MEFs with Tweak and TNFα resulted in a slight alteration of caspase-8 processing but in a complete inhibition of caspase-3 cleavage when compared with Bax/Bak+/+ counterparts control MEFs, indicating that caspase-8 processing precedes the Bax/Bak-mediated activation of the executioner caspases (Supplementary Figure S2f).

NIK pro-death function acts through the TNFR1/RIP1-dependent apoptotic axis. (a) NIK+/+ and NIK−/− mouse embryonic fibroblasts (MEFs) were stimulated with TNFα and CHX (5 μg/ml) for the indicated time periods, and cell survival was measured using the Cell Titer-Glo Kit. (b) NIK+/+ and NIK−/− MEFs were treated for 24 h with or without TNFα and the SMs CmpA, and cell survival was measured similar to panel (a). (c and d) Analysis of DEVDase activities in WT MEFs treated with Tweak and/or TNFα or LTβR Ago and/or TNFα in the absence or presence of the caspase inhibitor z-VAD-FMK and/or Nec-1. Statistical analyses: ***P<0.001. (e and f) MEFs were treated with Tweak/TNFα or LTβR Ago/TNFα in the absence or presence of Nec-1 for the indicated time periods prior to an immunoprecipitation with an anti-caspase-8 antibody and detection of both caspase-8 and RIP1 by western blotting
Altogether, our findings demonstrate that the apoptotic triggers that rely on the NIK pro-death function also depend on the kinase activity of RIP1.
NIK is a bona fide kinase of RIP1 in the TNFR1 cell death pathway
As we observed that NIK directs its pro-death activity toward RIP1 signaling, we sought to test the putative interaction of RIP1 with NIK. To do so, we ectopically expressed NIK (Flag-NIK-FL) with either full-length RIP1 (V5-RIP1-FL) or with mutated variants of RIP1 lacking the kinase domain (KD), the intermediate domain (ID) or the carboxyl-terminal region, including the death domain (DD), in HEK293T cells. Immunoprecipitations experiments not only confirmed the interaction between Flag-NIK-FL and RIP1-V5-FL but also showed that regions within the KD and ID domains, but not the DD domain, of RIP1 were required for the interaction with NIK (Figure 3a). Of note, the RHIM domain of RIP1, which is required for RIP3 binding and necroptosis induction, appeared dispensable for the interaction with NIK (Figure 3b and Supplementary Figure S3a).42 Importantly, the interaction between NIK and RIP1 was confirmed at the endogenous level in WT MEFs treated with Tweak/TNFα (Figure 3c).

NIK is a bona fide kinase of RIP1 in vivo and in vitro. (a) HEK293T cells were transiently transfected with expression vectors encoding full-length Flag-NIK-FL kinase dead together with either kinase dead RIP1-FL, truncated kinase domain (ΔKD) RIP1, truncated intermediate domain (ΔID) RIP1 or truncated death domain (ΔDD) RIP1. Flag immunoprecipitates were loaded on SDS-PAGE for detection of RIP1-V5. As control, anti-Flag immunoprecipitations were carried out in the presence of Flag peptide. (b) The same cells were transiently transfected with expression vectors encoding Flag-NIK-FL and FL kinase dead RIP1-V5 or ΔRHIM RIP1-V5 expression vectors. Flag-NIK-FL complexes were immunoprecipitated using an anti-Flag antibody, and RIP1 interactions were revealed by western blotting anti-RIP1. As control, anti-Flag immuniprecipitations were carry out in the presence of Flag peptide. (c) Mouse embryonic fibroblasts (MEFs) WT were treated or not with Tweak/TNFα and z-VAD (TTZ) and NIK-immunoprecipitates were analyzed by western blotting for the presence of endogenous RIP1. (d) In vitro kinase assays with recombinant NIK kinase domain (327–673) and active recombinant GST-RIP1 FL. (e) NIK+/+ and NIK−/− MEFs were left untreated or treated with a mix of TNFα and LTβR agonist antibody (Ago). The phosphorylation status of RIP1 was visualized by western blotting with cell extracts treated or not with the lambda phosphatase (λ phos.). (f) NIK+/+ and NIK−/− MEFs were treated with a mixture of TNFα and Tweak and analyzed similar to panel (e). (g) MEFs WT were treated with Tweak/TNFα in the absence or presence of Nec-1, and the protein extracts were analyzed by western blotting for RIP1 phosphorylation
We have found that NIK kinase activity contributes to its pro-apoptotic function (Figure 1e). We therefore investigated whether NIK could regulate RIP1-dependent apoptosis by directly phosphorylating RIP1. Using specific recombinant RIP1 and NIK proteins (Supplementary Figures S3b and c),43 we observed that the presence of NIK led to a significant increase in RIP1 phosphorylation level (Figure 3d). We also looked at RIP1 phosphorylation status in vivo. Interestingly, we found that agonist LTβR/TNFα stimulation triggered an upper shift of RIP1 that was fully abrogated in the presence lambda phosphatase in WT MEFs, indicative of RIP1 phosphorylation (Figure 3e). Remarkably, this phosphorylation was clearly deficient in NIK−/− cells treated with TNFα/LTβR Ago, TNFα/Tweak or TNFα/CmpA (Figures 3e and f and Supplementary Figure S3d). Importantly, NIK phosphorylation of RIP1 mainly increased RIP1 autophosphorylation activity. Indeed, preincubation of WT MEFs with Nec-1 altered TNFα/Tweak-induced RIP1 phosphorylation (Figures 2e and 3g). These results were also confirmed with in vitro kinase assays by combining Nec-1 with active recombinant RIP1 or using kinase-dead recombinant RIP1 (Supplementary Figures S3e and f).
Overall, we found that RIP1 is a NIK-interacting partner that is subjected to increased RIP1 autophosphorylation when cells are treated with apoptotic stimuli that induce NIK stabilization.
Tweak and Lymphotoxin-αβ signaling require NIK to induce TNFα-mediated RIP1/caspase-8 death complex IIb assembly
RIP1 is a key kinase at the crossroad of two antagonist pathways downstream of TNFR1. TNFα stimulation induces RIP1 recruitment to TNFR1 to form the so-called complex I in which RIP1 is ubiquitinated by c-IAP1/2 to mediate the pro-survival response.3, 4 However, in conditions of c-IAP1/2 depletion, non-ubiquitinated RIP1 dissociates from TNFR1 and recruits FADD and caspase-8 to form the cytosolic pro-death complex IIb required for caspase-8 activation and apoptosis.11, 17, 18 In order to decipher how NIK promotes TNFR1-mediated RIP1-dependent apoptosis, we next analyzed the assembly of complex I and complex IIb in NIK+/+ and NIK−/− MEFs. Complex I was isolated by immunoprecipitating TNFR1 following 5 min of stimulation with Flag-TNFα. No significant differences in recruitment of ubiquitinated RIP1 and IKKα to TNFR1 or in TNFα-mediated IκBα degradation were observed between NIK+/+ and NIK−/− MEFs (Figures 4a and b). We further showed that prestimulation of NIK+/+ and NIK−/− MEFs with Tweak prior to TNFα stimulation did not affect the recruitment of de-ubiquitinated RIP1 to TNFR1 (Figure 4c).

NIK is required for the assembly of complex IIb downstream of TNFR1. (a) NIK+/+ and NIK−/− mouse embryonic fibroblasts (MEFs) were stimulated 5 min with Flag-TNFα (1 μg/ml). TNFR1-associated proteins were pulled-down by immunoprecipitating Flag-TNFα-bound TNFR1. Presence of TNFR1, ubiquitinated RIP1 (Ub-RIP1) and IKKα in the immunoprecipitates was detected by western blotting. (b) Responsiveness of NIK+/+ and NIK−/− MEFs to TNFα-mediated IκBα degradation. Cells were stimulated with TNFα for the indicated time. (c) NIK+/+ and NIK−/− MEFs were pretreated for 2 h with Tweak followed by a stimulation with Flag-TNFα for 5 min TNFR1-associated proteins were pulled-down by immunoprecipitation of Flag-TNFα-bound TNFR1. Presence of TNFR1 and RIP1 in the immunoprecipitates was detected by western blotting. (d) NIK+/+ and NIK−/− MEFs were treated with Tweak and/or TNFα for the indicated time periods prior to an immunoprecipitation with an anti-caspase-8 antibody and detection of both caspase-8 and RIP1 by western blotting. (e) NIK+/+ and NIK−/− MEFs were treated with a LTβR agonist antibody (Ago) and TNFα, and association of caspase-8 and RIP1 was analyzed similar to panel (d). (f and g) Level of c-IAP1/2 depletion in NIK+/+ and NIK−/− MEFs treated with Tweak or a LTβR Ago. (h) z-VAD-FMK-pretreated NIK+/+ and NIK−/− MEFs were stimulated for the indicated time period with Tweak and/or TNFα, and stabilization of NIK was analyzed by western blotting. (i) Cell extracts prepared in panel (h) were subjected to an immunoprecipitation with anti-NIK prior to an in vitro caspase-8 activity assay. (j) Measurement of in vitro caspase-8 activity associated with FADD-containing complexes. NIK+/+ and NIK−/− MEFs were co-stimulated or not with Tweak and TNFα for 1 h prior to immunoprecipitation of FADD. NS represents non-specific signals, and Ig represents a cross-reactivity with heavy chain immunoglobulins. Statistical analyses: **P<0.01; ***P<0.001
Next, we evaluated whether NIK deficiency could affect complex IIb formation. In NIK+/+ MEFs, we detected a robust association of caspase-8 with RIP1 as early as 90 min of co-stimulation with Tweak/TNFα, which persisted up to 180 min posttreatment (Figure 4d). Remarkably, the level of RIP1/caspase-8 association drastically decreased in the absence of NIK, despite an equal amount of caspase-8 immunoprecipitated in NIK+/+ and NIK−/− MEFs. We observed similar results when NIK+/+ and NIK−/− MEFs were either treated with agonist LTβR/TNFα or CmpA/TNFα (Figure 4e and Supplementary Figure S4a). The lack of RIP1/caspase-8 assembly could not be ascribed to a defect in the expression of RIP1, FADD or caspase-8 (Supplementary Figure S4b). As previously reported, degradation of c-IAP-1/2 is critical for Tweak/TNFα- or SM/TNFα-induced cell death.26, 28, 33 Yet, we observed that the level of Tweak-, agonist LTβR- or CmpA-induced c-IAP1/2 degradation was similar in NIK+/+ and NIK−/− MEFs, indicating that c-IAP1/2 depletion is not compromised in the absence of NIK (Figures 4f and g and Supplementary Figure S4c). Together, these results indicate that NIK acts downstream of complex I formation but upstream or at the level of RIP1/caspase-8 assembly. We have previously shown that NIK can associate with RIP1 upon Tweak/TNFα stimulation (Figure 3c). To further investigate whether active caspase-8 could also co-immunoprecipitate with NIK, NIK+/+ and NIK−/− MEFs were stimulated for 60 or 90 min with TNFα and Tweak or with Tweak alone as control (Figure 4h). Lysates from these cells were then subjected to control or anti-NIK immunoprecipitation prior to an in vitro caspase-8 assay (Figure 4i and Supplementary Figure S4d). We observed that treatment of NIK+/+ MEFs with Tweak/TNFα led to a time-dependent increase in caspase-8 activity. In contrast, similar treatment of NIK−/− MEFs did not reveal any caspase-8 activity (Figure 4i). Of note, stimulation of NIK+/+ MEFs with Tweak alone did not induce caspase-8 activation besides a marked stabilization of NIK (Figures 4h and i). Consistent with these findings, caspase-8 activity associated with immunoprecipitated FADD in response to Tweak and TNFα was greatly inhibited in the absence of NIK (Figure 4j). Altogether, our results demonstrate that biological ligands of TNFR family members or synthetic chemicals that trigger c-IAP1/2 depletion by various means require NIK for the assembly of the RIP1/caspase-8 complex IIb.
NIK displays a pro-death function in vivo in a mouse model of TNFR1/LTβR-mediated thymus involution independently of the alternative NF-κB pathway
TNFR1 is a key receptor that contributes to inflammation, cell death and, in some circumstances, to tissue destruction. To address the in vivo pro-death function of NIK in a context of TNFR1 activation, we decided to use a mouse model in which TNFR1-induced cell death and tissue destruction relies on a co-stimulatory inflammatory signal that activate NIK and the alternative NF-κB pathway. Tg Lck-LTαβ mice develop a fulminant thymus involution that is dependent on LTα3/TNFR1 and LTα1β2/LTβR signaling within the cTEC recapitulating virus infection-related thymic involution by activated LTαβ-overexpressing T cells.44, 45 Therefore, to assess the role of NIK in cell death of cTEC, we performed bone marrow (BM) reconstitution assays using WT or Tg Lck-LTαβ mice as donors and WT or aly/aly NIK-deficient recipient mice.46 For phenotypic analyses, 8 weeks postreconstitution mice were killed and thymi were imaged and quantitatively and qualitatively analyzed. As previously reported, reconstitution of Lck-LTαβ BM into WT recipient mice led to a drastic reduction in thymus size compared with the reconstitution of WT BM into WT recipient mice (Figure 5a).44 Reciprocal reconstitution experiments using Lck-LT αβ BM into aly/aly recipient mice greatly protected from LTαβ-induced thymus involution. Indeed, thymi were comparable in size to those obtained from reconstitution experiments of WT BM into WT recipient mice or aly/aly BM into aly/aly recipient mice. To further quantify the severity of thymus involution, the thymi were dissected and analyzed for relative body weight and absolute cell numbers. Reconstitution of Lck-LTαβ BM into WT mice led to a marked reduction of the relative thymus weight (P<0.001) and absolute thymocyte numbers (P<0.001) when compared with WT recipient mice that received WT BM (Figures 5b and c). Importantly, reciprocal reconstitution experiments using Lck-LTαβ BM and aly/aly recipient mice showed no statistically significant changes in relative thymic weight (P<0.5) and absolute thymocyte numbers (P<0.5) when compared with WT and aly/aly recipient mice that received WT BM or aly/aly BM, respectively.

TNFR1/LTβR-induced thymus involution and cell death require the kinase NIK. (a) Macroscopic images of thymi indicated by dashed white line from postnatal (8 weeks) BM reconstitution experiments. Lethally irradiated WT recipient mice were either reconstituted with BM from WT donor mice (n=7) or from Tg Lck-LTαβ mice (n=6). In addition, aly/aly (NIK-defective) recipient mice were either reconstituted with BM from Tg Lck-LTαβ mice (n=6) or from aly/aly mice (n=3); scale bar, 5 mm. (b and c) Quantitative analyses of relative thymus weight and absolute cell number of the reconstitution experiments achieved in panel (a). (d) Analysis of thymic architecture (H&E staining) of the indicated postreconstitution (5–6 weeks) experiments. Medulla areas are surrounded by asterisks; scale bar, 250 μm. (e and f) TUNEL staining (scale bar, 75 μm) and flow cytometry analysis of Annexin V/7-AAD double-positive cells of thymi from the indicated postreconstitution (5–6 weeks) experiments. Statistical analyses: **P<0.01; ***P<0.001, NS, not statistically significant. See also Supplementary Figure S5
Histological analyses revealed that LTαβ-mediated reduction of cortical thymic area along with enlargement of the medulla were abrogated in aly/aly recipient mice reconstituted with Lck-LTαβ BM (Figure 5d). In addition, TUNEL staining were performed on cryosections of thymi postreconstitution and clearly indicated that aly/aly mice were protected from LTαβ-induced thymic epithelial cell apoptosis (Figure 5e). These results were confirmed by monitoring the percentage of Annexin V/7-AAD double-positive thymic cells (P<0.01) by flow cytometry (Figure 5f). Importantly, NIK knockout recipient mice were similarly fully protected from LTαβ-induced thymic involution (Supplementary Figures S5a–c). Altogether, our data demonstrate that TNFR1/ LTβR-induced thymus involution and thymic epithelial cell death relies on the pro-death activity of NIK in vivo.
We previously showed that LTβR induces two NF-κB pathways, one of which is the alternative NF-κB pathway that relies on the activation of the kinase NIK and the processing of NF-κB2/p100.40 Thus, to address whether the in vivo pro-death activity NIK originates from an activation of the alternative NF-κB pathway, we analyzed the nfkb2−/− mice in the LTαβ-induced thymus involution model. First, we performed control experiments using WT and nfkb2−/− (p100/p52-deficient) recipient mice reconstituted with BM derived from WT mice and did not observe any difference in thymus reconstitution, relative thymic weight and absolute thymocyte numbers (Figures 6a–c). Second, we performed reconstitution of WT and nfkb2−/− recipient mice with Lck-LTαβ BM. Interestingly, we observed in both cases a significant reduction in relative thymic weight (P<0.001) and absolute thymocyte numbers (P<0.001) compared with control experiments (Figures 6a–c). Moreover, the extent of thymic epithelial cell death (TUNEL+ cells) was comparable between WT and nfkb2−/− recipient mice that received Lck-LTαβ BM (Figure 6d), indicating that the absence of p100 did not rescue LTαβ-mediated thymus involution and thymic epithelial cell death. We also confirmed in vitro that p100 was dispensable to trigger cell death with other TNFR family members (Supplementary Figure S6).

TNFR1/LTβR-induced thymus involution and cell death does not rely on the activation of the alternative NF-κB pathway. (a) Macroscopic images of thymi indicated by dashed white line from postnatal (8 weeks) reconstituted experiments. Lethally irradiated WT recipient mice were either reconstituted with BM from WT donor mice (n=4) or from Tg Lck-LTαβ mice (n=4). In addition, nfkb2−/− (p100-deficient) recipient mice were either reconstituted with BM from Tg Lck-LTαβ mice (n=5) or from WT mice (n=3), scale bar, 5 mm. (b and c) Lethally irradiated WT-recipient mice were either reconstituted with BM from WT donor mice (n=4) or from Tg Lck-LTαβ mice (n=4) and nfkb2−/− (p100-deficient) recipient mice were either reconstituted with BM from Tg Lck-LTαβ mice (n=5) or from WT mice (n=3). Quantitative analyses of relative thymus weight and absolute cell number are depicted, respectively. Data are represented as mean±S.E.M. (NS, not statistically significant). (d) TUNEL staining of thymi from the indicated postreconstitution (5–6 weeks) experiments, scale bar, 75 μm. Statistical analyses: ***P<0.001, NS, not statistically significant
Altogether, our data demonstrate that the in vivo ability of NIK to induce TNFR1/LTβR-dependent thymus involution and thymic epithelial cell death does not require a transcriptional program under the control of p52-containing NF-κB complexes.
NIK/RIP1-dependent cell death in mouse models of liver damage
TNFα is a key cytokine that contributes to disease-mediated liver damage in human patients. To address the role of NIK in hepatocyte apoptosis, we used two different mouse models of TNFα-induced liver damage. First, we used TNFα in association with D-galactosamine in aly/aly mice and littermate controls aly/wt mice. We did not observe differences for cleaved caspase-3 staining nor in the level of the aminotransferase sALT and sAST in the serum (Figure 7a and Supplementary Figures S7a and b). To investigate whether this TNF-driven apoptotic model of liver damage was dependent on RIP1 kinase activity, we pretreated WT mice with DMSO or Nec-1 for 30 min prior to D-Gal-N/TNFα treatment for 4 h. We observed no significant differences in the level of sALT and sAST or in the number of cleaved caspase-3-positive hepatocytes (Figure 7b and Supplementary Figures S7c and d).

RIP1 and NIK control TNFα-induced hepatocytes apoptosis. (a) Cleaved caspase-3 staining of liver sections from control aly/wt and aly/aly mice treated with D-Gal-N in combination with TNFα. Scale bars, 100 μm. (b) Cleaved caspase-3 staining of liver sections from wild-type mice treated with D-Gal-N in combination with DMSO/TNFα or Nec-1/TNFα. Scale bars, 100 μm. (c and d) Cleaved caspase-3 staining of liver sections from Ad-GFP-infected aly/wt (n=6) and aly/aly (n=8) mice treated with TNFα. Representative pictures are shown, and a quantification of cleaved caspase-3+ hepatocytes for each genotype. Scale bars, 100 μm. Statistical analyses: **P<0.01. (e) Western blotting analysis of NIK, actin and GFP proteins of the livers from uninfected or adenovirus-infected mice. (f) Representative pictures of immunostaining for NIK-positive hepatocytes (black arrows) derived from liver sections analyzed in panel (e). Scale bars, 100 μm. Statistical analyses: **P<0.01. (g) Quantification of NIK-positive heaptocytes/mm2 in uninfected (n=3) and Ad-GFP-infected (n=3) mice. (h and i) Cleaved caspase-3 staining of liver sections from Ad-GFP-infected wild-type mice treated with DMSO/TNFα or Nec-1/TNFα. Representative pictures are shown and a quantification of cleaved caspase-3+ hepatocytes for DMSO control (n=8) and Nec-1-injected mice (n=8). Scale bars, 100 μm. Statistical analyses: **P<0.01
Next, we used a mouse model known to predispose to TNFα-induced hepatocyte cell death by cytotoxic T lymphocytes (CTLs).47 Intravenous injection of TNFα subsequent to adenoviral infection (Ad-GFP) recapitulated this CTL-derived, TNFα-mediated capacity of hepatocyte cell killing. We observed that aly/wt mice were sensitive to adenovirus/TNFα-induced hepatocyte damage while aly/aly mice showed a strong reduction in the number of cleaved caspase-3+ hepatocytes (Figures 7c and d). Moreover, we monitored the capacity of adenovirus infection to activate NIK. We analyzed NIK expression levels in the livers of uninfected or adenovirus-infected WT mice and observed a strong increase of hepatocyte NIK protein level from adenovirus-infected mice (Figure 7e). These data were also strengthened by immunohistochemical analyses, which revealed that the numbers of NIK-expressing hepatocytes were higher in adenovirus-infected mice compared with control mice (Figures 7f and g).
Remarkably, we found that the inhibition of RIP1 kinase activity sufficed to significantly reduce the amount of cleaved caspase-3 positive hepatocytes in Ad-GFP-infected mice treated with TNFα (Figures 7h and i). In conclusion, we have tested the in vivo role of NIK in two different models of TNF-driven apoptotic liver damage, one relying on RIP1 kinase activity and the other one not relying on this activity. We found that NIK only contributed to the TNF-induced RIP1-dependent apoptotic model. Our data highlight, in line with our in vitro and in vivo data described above, another mouse model in which viral-infected hepatocyte rely on NIK and RIP1 kinase activity to trigger TNFα-induced apoptosis.
Discussion
In this study, we found that inducers of the alternative NF-κB pathway such as Tweak, LTβR agonist or SMs required NIK to sensitize to TNFα/TNFR1-induced apoptotic death. Previous studies reported that, at least in certain cell types, Tweak- and SM-induced cell death relied on the NIK-dependent neosynthesis of endogenous TNFα via induction of the classical and/or the alternative NF-κB pathway.11, 26, 27, 33 We can rule out this possibility in our in vitro and in vivo genetic models. Indeed, we showed that Tweak-, LTβR agonist- or SM-induced NIK stabilization in MEFs is by itself not sufficient to induce cell death and that NIK deficiency protected the MEFs from TNFR1-mediated death induced by exogenous TNFα. In our in vivo models, TNFR1 was either stimulated by injection of recombinant TNFα or by ectopically expressed transgenic LTα, strongly suggesting that NIK pro-death activity rather impinges downstream of TNFR1.
Our results demonstrated that NIK was associated with and required for the formation of the RIP1/FADD/caspase-8 death complex IIb. Indeed, we showed that (1) Tweak/TNFα or agonist LTβR/TNFα stimulation led to caspase-8 and RIP1 interaction; (2) RIP1 kinase inhibition by Nec-1 inhibits cell death and blocks caspase-8 and RIP1 association; (3) cell death and RIP1/caspase-8 assembly are severely impaired in NIK-deficient MEFs stimulated with Tweak/TNFα or agonist LTβR/TNFα; and (4) Tweak/TNFα-induced association of FADD with active caspase-8 is impaired in NIK-deficient MEFs.
A 2-MDa death-inducing complex containing the core components RIP1/FADD/caspase-8, referred as ripoptosome, was reported to form in response to genotoxic stress or to TLR3 activation in some cancer cell lines.48, 49 In both the studies, the authors found that the assembly of the ripoptosome occurred independently of TNFR1 signaling. However, in our experimental settings, a robust activation of NIK in the absence of TNFR1 stimulation could neither promote a NIK-associated caspase-8 activity nor an interaction between RIP1 and caspase-8. Therefore, as opposed to the ripoptosome, NIK-induced complex IIb formation is strictly dependent on TNFR1 signaling.
In any case, spontaneous ripoptosome formation upon c-IAP1/2 depletion was reported to occur in response to DNA damage or TLR3 activation.48, 49 Yet, we found that despite the efficiency of Tweak-induced c-IAP1/2 depletion in NIK-deficient MEFs, RIP1 and caspase-8 failed to assemble, indicating that TNFα-induced complex IIb formation is a highly regulated process that is primed by c-IAP1/2 depletion but requires the kinase NIK.
So far, the differentiation of mTECs requires the integration of signaling from CD40, RANK and LTβR toward NIK, IKKα and p52/Bcl-3 for the establishment of self-tolerance.50, 51, 52, 53, 54, 55, 56, 57 As opposed to mTECs, cTEC number is not affected in mice with defect in the alternative NF-κB pathway.58 However, in our study, we highlight a new role for NIK downstream of LTβR in induction of TNFR1-driven cTEC death and thymus involution. Notably, we showed that NIK pro-death function in cTEC was independent of the processing of p100 into p52, which is in contrast to the requirement of p52 for mTEC differentiation. Such biological functions of NIK independent of the processing of p100 have been also described in other settings and highlight a role for NIK beyond the activation of the alternative NF-κB pathway.59, 60, 61
We also found that NIK pro-death function has a role in a model of viral infection associated with TNFα-induced hepatocytes apoptosis. Indeed, we have observed that adenovirus-infected NIK-deficient hepatocytes were resistant to TNFα-mediated cell death. Similarly, inhibition of RIP1 kinase activity lowered caspase-3 activation in adenovirus-infected hepatocytes. These results are also consistent with the fact that hepatocyte-specific FADD-deficient mice and QVD-Oph pretreated WT mice appeared to resist to adenovirus/TNFα-induced hepatocytes damage, suggesting a link between RIP1, FADD and caspase-8 in that model.47 Therefore, the NIK/RIP1 pathway could be crucial for the clearance of infected cells through the TNFα/TNFR1 death signaling axis. Overall, we have identified a new function of NIK in cell death that extends beyond its role already described in development, inflammation and cell survival.
Materials and Methods
Mouse strains, thymic and liver analyses
Mice deficient for NIK (aly/aly) and NIK KO mice were provided by B Becher, (University of Zurich, Zurich, Switzerland) and p100/p52 (nfkb2−/−) mice were provided by J Caamano (University of Birmingham, Birmingham, UK), respectively, and Lck-LTαβ mice were previously described.44 All mice were on a C57BL/6 genetic background and were bred and maintained under specific pathogen-free conditions and within protocols approved by the Institute of Virology, Technische Universität München (TUM)/Helmholtz Zentrum München, Munich, Germany and the MRC Centre of Immune Regulation, University of Birmingham, Birmingham, UK. For control of lethal irradiation (1100 rad) and reconstitution efficiency, reconstitution into WT recipient mice was performed using BM derived from β-Actin Green Fluorescent Protein (β-Act GFP) transgenic mice and gave a reconstitution efficiency of 89–97% (data not shown). Relative thymus weight, histology analysis, TUNEL staining and Annexin V/7-AAD staining were performed as previously described.44 Pictures were taken with an Olympus BX53 microscope. Cell counting were performed with the ‘CellSens Dimension’ software from Olympus (Hamburg, Germany). Adenoviral infection, hTNF injection and sALT and sAST measurement were peformed as previously described.47 For the D-galactosamine/TNFα experiments, D-Gal-N (800 mg/kg) was injected peritonally 30 min prior to human TNFα (20 μg/kg) injection and mice were killed 4 h latter. To address the role of RIP1 kinase activity in both models, DMSO or Nec-1 (5 mg/kg) was injected 30 min prior to TNFα.
Immunohistochemistry
Thymic and liver tissues were fixed with 4% paraformaldehyde and embedded in paraffin.
Paraffin sections (2 μm) were either stained with haematoxylin/eosin or automated immunohistochemistry staining were performed using rabbit-anti-cleaved caspase-3 (Cell Signaling, Danvers, MA, USA, no. 9661), rabbit anti-NIK (Abcam, Cambridge, UK, ab7204) and (Hamburg, Germany)an anti-rabbit-poly HRP (Leica Bond Polymer Refine Detection, Nussloch, Germany, DS9800). For pretreatment of paraffin sections, the bond max H2 (20) program (Leica, Wetzlar, Germany) was applied. Image acquisition was performed on the DotSlide BX51 (Olympus), SCN400 (Leica), Axio Z1 (Zeiss, Göttingen, Germany) or BX53 (Olympus) microscope. For quantification of NIK-positive hepatocytes, eight different areas (four around vessels and four in random fields) were counted for each mouse.
Statistical analysis
Statistical analysis was performed with the GraphPad Prism software (GraphPad Software, La Jolla, CA, USA). All data are presented as mean±S.D. and were analyzed by ANOVA with the post hoc Bonferroni multiple comparison test. Analysis of two samples was performed with Student’s t test.
Antibodies, chemicals and cytokines
Mouse monoclonal anti-RIP1 (610459) was from BD Biosciences, San Jose, CA, USA. Rabbit polyclonal anti-RIP1 (sc-7881), anti-TNFR1 (sc-1070) and anti-FADD and anti-TRADD were purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA. Mouse monoclonal anti-caspase-8 (ALX-804-447) was obtained from Alexis (San Diego, CA, USA). Monoclonal anti-Flag M2 antibody (F3165), anti-Flag M2 conjugated beads (F2426) were purchased from Sigma (St. Louis, MO, USA). Rabbit monoclonal cleaved-caspase-8 XP (8592), anti-cleaved caspase-3 (17 kDa) (9661) and anti-IKKα (2682) were from Cell Signaling Technology. Rat anti-cFLIP (Dave-2) antibody was purchased from Adipogen (Liestal, Switzerland). The rabbit polyclonal anti-cIAP1/2 was provided by Robert Korneluk (University of Ottawa, Ottawa, ON, Canada). Rabbit polyclonal antiserum against recombinant murine caspase-8 was prepared at the Centre d'Economie Rurale (Laboratoire d'Hormonologie Animale, Marloie, Belgium), purified by Protein A-Sepharose 4B affinity chromatography (Invitrogen, Life Technologies Europe, Gent, Belgium) and showed specificity for the p20 fragment of recombinant caspases by western blotting.62 Anti-GFP, anti-NIK and anti-actin were previously described.29
z-VAD-FMK and Nec-1 were purchased from Promega (Madison, WI, USA), SM Biochemicals LLC (Anaheim, CA, USA) and BD Biosciences, respectively. Compound A was a gift of Srinivas Chunduru (TetraLogic Pharmaceuticals, Malvern, PA, USA). The NIK kinase inhibitor Cmp1 was a gift from Genentech (South San Francisco, CA, USA) and was previously described.43 Recombinant Fc-Tweak and Flag-TNFα were provided by Pascal Schneider (University of Lausanne, Lausanne, Switzerland) and Peter Vandenabeele (VIB, Ghent, Belgium), respectively. Recombinant human TNFα was purchased from Roche (Mannheim, Germany).
Recombinant protein and kinase assays
Recombinant active GST-RIP1-FL was purchased from Sigma (SRP5370). Active human NIK (327–673) recombinant protein was obtained from Sarah Hymowitz (Genentech/Roche, Pleasanton, CA, USA). Recombinant inactive GST-RIP1-FL was obtained from Abnova (H00008737) (Heidelberg, Germany). For the kinase assays with recombinant NIK (500 ng) and GST-RIP1 (200 ng), the proteins were incubated 30’ at 30 °C in the absence or presence of Nec-1 (50 μM) or Cmp1 (20 nM) in a kinase buffer containing 20 mM Hepes, pH 7,5, 10 mM MgCl2, 10 mM MnCl2, 2 mM DTT supplemented with protease and phosphatase inhibitors, 100 μM ATP and 1 μl ATP γ-32 P (Perkin Elmer (Waltham, MA, USA), BLU002A2). For the in vivo kinase assays, NIK+/+ and NIK−/− MEFs were treated 2 h with TNFα/Tweak or TNFα/CmpA or 4 h with TNFα/LTβR Ago, and cells were lysed in NP-40-containing buffer supplemented with phosphatase inhibitors. Then, cell extracts were further incubated at 30 °C for 1 h in the lambda phosphatase buffer (Bioké, Leiden, The Netherlands, P0753S) in the absence or presence of 400 units of lambda phosphatase.
Cell survival and caspase activation assays
Cell survival analysis was performed using the Cell Titer-Glo Luminescent Cell Viability Assay kit (Promega). In brief, cells were plated in 96-well plates to reach 50–60% confluence before induction of cell death with Fc-Tweak (200 ng/ml), LTβR agonist Ab (2 μg/ml) or CmpA (250 nM) and/or human TNFα (50 or 200 U/ml). Cells were then incubated with 100 μl of Cell Titer-Glo reagent at room temperature for 5 min prior to luminescent monitoring with a Victor2 reader (Perkin Elmer).
For caspase 3/7 activity measurement, cells were plated in 96-well plates. After cell death stimulation, cells were lysed for 30 min at 4 °C in lysis buffer (50 mM HEPES pH 8, 150 mM NaCl, 20 mM EDTA, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 0.2% Triton X-100) and lysates were cleared at 10 000 × g for 15 min at 4 °C. Each assay was performed in duplicate with 10 μg of protein prepared from control or stimulated cells. Briefly, cellular extracts were then incubated in a 96-well plate, with 0.2 mM of Ac-DEVD-AMC (7-amino-4-methylcoumarin) and 5 mM DTT for various times at 37 °C. Caspase activity was measured following emission at 460 nm in the presence or absence of 10 μM of Ac-DEVD-CHO. Enzyme activities were determined as initial velocities expressed as relative intensity per min and per mg of protein. Alternatively, caspase 3/7 activity was monitored using the Caspase-Glo 3/7 Assay Kit (Promega) following the manufacturer’s instructions.
For measurement of caspase-8 activity associated with FADD or NIK-containing complex, cells were pretreated 30 min with z-VAD-FMK as previously described,41 prior to Tweak and/or TNFα stimulation, and then were harvested in lysis buffer containing 20 mM Tris HCl (pH 7.5), 150 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, and complete EDTA-free protease Inhibitor (Roche). Cells were left on ice for 20 min and centrifuged at 10 000 × g for 15 min. One milligram of protein extract was used with 2 μg of antibody for immunoprecipitation. The immunoprecipitates were washed four times with the lysis buffer and two times with cold PBS. Beads were resuspended in 100 μl PBS, and 100 μl of Caspase-8 Glo reagent was added for 1-h incubation at room temperature.
Immunoprecipitation and western blotting
MEF cell extracts were made in NP-40-containing buffer as previously described.11 Cell extracts were either used directly for western blotting or for co-immunoprecipitation experiments. For caspase-8/RIP1 complex analysis, 20 μl of protein G-Agarose beads (Santa Cruz) were incubated O/N with 2 μg of anti-murine caspase-8 antibody at 4 °C. Cell lysates were added and incubated overnight at 4 °C. The following day, the beads were washed four times with the lysis buffer prior to addition of laemmli buffer and western blotting analysis. For Flag-TNFα co-immunoprecipitation experiments, MEF cell extracts were incubated 4 h with anti-Flag M2-conjugated beads. For detection of endogenous NIK/RIP1 interaction, 2 mg of proteins were incubated O/N at 4 °C with 2 μg of NIK Ab. Next day, protein A/G beads were incubated 2 h with the cell lysates followed by an extensive wash with the lysis buffer prior to western blotting analyses of RIP1.
For co-immunoprecipitation of NIK and RIP1 into HEK293T cells, the latter were lysed in 150 mM NaCl, 20 mM Tris-HCl pH 7.4, 1% Triton, 10% glycerol and 1 mM EDTA. Flag-NIK and RIP1-V5 were immunoprecipitated using 20 μl of EZview Red anti-Flag M2 affinity gel (F2426) during 4 h at 4 °C. For peptide competition, 75 μg of Flag peptide (F3290, Sigma) were added to the immunoprecipitation reaction.
Plasmids, transient transfections and retrovirus production
Dominant-negative D138N and truncated mutants ΔKD (kinase domain), ΔID (intermediate domain) and ΔDD (death domain) mouse RIP1-V5 expression vectors were provided by Dr. Tom Van den Berghe (University of Ghent, Ghent, Belgium) and RIP3-GFP expression vector by Dr. Francis Chan (University of Massachusetts, MA, USA).21, 62 FL Flag-NIK expression vector was previously described.40 First, FL Flag-NIK was subcloned into pIRES2-EGFP (BD Biosciences) and used as template to generate the kinase dead version (KK429/430AA) by site-directed mutagenesis (Quick Change II, Agilent Technologies, Santa Clara, CA, USA). Primers sequences are available upon request. Transient transfections were performed using Fugene HD (Roche) with 4 μg of total DNA per 10 cm plate of HEK293T cells. For the complementation of NIK KO MEFs, pBabe control, Flag NIK WT or NIK KD were used to produce retroviral particles as previously described.40
Cell culture and flow cytometry
All cell lines were maintained in Dulbecco’s modified Eagle medium supplemented with 10% FBS, 2 mM L-glutamine and 200 μg/ml penicillin/streptomycin and grown at 37 °C in 5% CO2. For flow cytometry analysis, MEFs were seeded in 24-well plates and 200 ng/ml of Fc-Tweak and/or 200 U/ml of TNFα were added for 24 h prior to Annexin V/PI staining (11988549001, Roche, Mannheim, Germany). Experiments were performed in duplicate, and 10 000 events were measured per sample.
Abbreviations
- NIK:
-
NF-κB-inducing kinase
- c-IAP1/2:
-
cellular inhibitor of apoptosis 1/2
- NF-κB:
-
nuclear factor-kappa B
- RIP1:
-
receptor interacting protein kinase 1
- TNFα:
-
tumor necrosis factor alpha
- TNFR1:
-
TNF receptor 1
- LTβR:
-
lymphotoxin-beta receptor
- TRADD:
-
TNF receptor-associated death domain
- FADD:
-
Fas-associated protein with death domain
- LUBAC:
-
linear ubiquitin chain assembly complex
- TAB1:
-
TAK1 binding protein 1
- IKK:
-
inhibitor of nuclear factor kappa B kinase
- CHX:
-
cycloheximide
- SM:
-
Smac mimetic
- Nec-1:
-
necrostatin-1
- z-VAD-fmk:
-
Z-Val-Ala-DL-Asp-fluoromethylketone
References
Green DR, Oberst A, Dillon CP, Weinlich R, Salvesen GS . RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts. Mol Cell 2011; 44: 9–16.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Walczak H . TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol Rev 2011; 244: 9–28.
Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011; 471: 591–596.
Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 2009; 36: 831–844.
Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 2011; 471: 637–641.
Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature 2011; 471: 633–636.
Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol 2009; 11: 123–132.
Bertrand MJ, Lippens S, Staes A, Gilbert B, Roelandt R, De Medts J et al. cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1-4). PLoS One 2011; 6: e22356.
Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30: 689–700.
Blackwell K, Zhang L, Workman LM, Ting AT, Iwai K, Habelhah H . Two coordinated mechanisms underlie tumor necrosis factor alpha-induced immediate and delayed IkappaB kinase activation. Mol Cell Biol 2013; 33: 1901–1915.
Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael-Tomasevic A, Phu L et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J 2010; 29: 4198–4209.
Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ . Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22: 245–257.
Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell 2004; 15: 535–548.
Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD . Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat Cell Biol 2006; 8: 398–406.
Wang L, Du F, Wang X . TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008; 133: 693–703.
Wilson NS, Dixit V, Ashkenazi A . Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol 2009; 10: 348–355.
Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ 2013; 20: 1381–1392.
Legarda-Addison D, Hase H, O'Donnell MA, Ting AT . NEMO/IKKgamma regulates an early NF-kappaB-independent cell-death checkpoint during TNF signaling. Cell Death Differ 2009; 16: 1279–1288.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137: 1112–1123.
Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008; 4: 313–321.
Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011; 35: 908–918.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009; 325: 332–336.
Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 2007; 131: 682–693.
Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J et al. Autocrine TNFalpha signaling renders human cancer cells susceptible to Smac-mimetic-induced apoptosis. Cancer Cell 2007; 12: 445–456.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669–681.
Ganeff C, Remouchamps C, Boutaffala L, Benezech C, Galopin G, Vandepaer S et al. Induction of the alternative NF-{kappa}B pathway by lymphotoxin {alpha}{beta} (LT{alpha}{beta}) relies on internalization of LT{beta} receptor. Mol Cell Biol 2011; 31: 4319–4334.
Sanjo H, Zajonc DM, Braden R, Norris PS, Ware CF . Allosteric regulation of the ubiquitin:NIK and ubiquitin:TRAF3 E3 ligases by the lymphotoxin-beta receptor. J Biol Chem 2010; 285: 17148–17155.
Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat Immunol 2008; 9: 1364–1370.
Varfolomeev E, Goncharov T, Maecker H, Zobel K, Komuves LG, Deshayes K et al. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci Signal 2012; 5: ra22.
Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol 2008; 182: 171–184.
Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008; 9: 1371–1378.
Dejardin E . The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol 2006; 72: 1161–1179.
Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001; 293: 1495–1499.
Xiao G, Harhaj EW, Sun SC . NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell 2001; 7: 401–409.
Novack DV . Role of NF-kappaB in the skeleton. Cell Res 2011; 21: 169–182.
Sun SC . The noncanonical NF-kappaB pathway. Immunol Rev 2012; 246: 125–140.
Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 2002; 17: 525–535.
Biton S, Ashkenazi A . NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell 2011; 145: 92–103.
Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM . Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 2002; 277: 9505–9511.
de Leon-Boenig G, Bowman KK, Feng JA, Crawford T, Everett C, Franke Y et al. The crystal structure of the catalytic domain of the NF-kappaB inducing kinase reveals a narrow but flexible active site. Structure 2012; 20: 1704–1714.
Heikenwalder M, Prinz M, Zeller N, Lang KS, Junt T, Rossi S et al. Overexpression of lymphotoxin in T cells induces fulminant thymic involution. Am J Pathol 2008; 172: 1555–1570.
Liepinsh DJ, Kruglov AA, Galimov AR, Shakhov AN, Shebzukhov YV, Kuchmiy AA et al. Accelerated thymic atrophy as a result of elevated homeostatic expression of the genes encoded by the TNF/lymphotoxin cytokine locus. Eur J Immunol 2009; 39: 2906–2915.
Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M et al. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet 1999; 22: 74–77.
Wohlleber D, Kashkar H, Gartner K, Frings MK, Odenthal M, Hegenbarth S et al. TNF-induced target cell killing by CTL activated through cross-presentation. Cell Rep 2012; 2: 478–487.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43: 449–463.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43: 432–448.
Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity 2008; 29: 423–437.
Boehm T, Scheu S, Pfeffer K, Bleul CC . Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR. J Exp Med 2003; 198: 757–769.
Derbinski J, Kyewski B . Linking signalling pathways, thymic stroma integrity and autoimmunity. Trends Immunol 2005; 26: 503–506.
Kajiura F, Sun S, Nomura T, Izumi K, Ueno T, Bando Y et al. NF-kappa B-inducing kinase establishes self-tolerance in a thymic stroma-dependent manner. J Immunol 2004; 172: 2067–2075.
Kinoshita D, Hirota F, Kaisho T, Kasai M, Izumi K, Bando Y et al. Essential role of IkappaB kinase alpha in thymic organogenesis required for the establishment of self-tolerance. J Immunol 2006; 176: 3995–4002.
Lomada D, Liu B, Coghlan L, Hu Y, Richie ER . Thymus medulla formation and central tolerance are restored in IKKalpha−/− mice that express an IKKalpha transgene in keratin 5+ thymic epithelial cells. J Immunol 2007; 178: 829–837.
Zhang X, Wang H, Claudio E, Brown K, Siebenlist U . A role for the IkappaB family member Bcl-3 in the control of central immunologic tolerance. Immunity 2007; 27: 438–452.
Akiyama T, Shinzawa M, Akiyama N . TNF receptor family signaling in the development and functions of medullary thymic epithelial cells. Front Immunol 2012; 3: 278.
Jenkinson SR, Williams JA, Jeon H, Zhang J, Nitta T, Ohigashi I et al. TRAF3 enforces the requirement for T cell cross-talk in thymic medullary epithelial development. Proc Natl Acad Sci USA 2013; 110: 21107–21112.
Hacker H, Chi L, Rehg JE, Redecke V . NIK prevents the development of hypereosinophilic syndrome-like disease in mice independent of IKKalpha activation. J Immunol 2012; 188: 4602–4610.
Mills DM, Bonizzi G, Karin M, Rickert RC . Regulation of late B cell differentiation by intrinsic IKKalpha-dependent signals. Proc Natl Acad Sci USA 2007; 104: 6359–6364.
Yamada T, Mitani T, Yorita K, Uchida D, Matsushima A, Iwamasa K et al. Abnormal immune function of hemopoietic cells from alymphoplasia (aly) mice, a natural strain with mutant NF-kappa B-inducing kinase. J Immunol 2000; 165: 804–812.
Van de Craen M, Van Loo G, Declercq W, Schotte P, Van den brande I, Mandruzzato S et al. Molecular cloning and identification of murine caspase-8. J Mol Biol 1998; 284: 1017–1026.
Acknowledgements
We are grateful to Dr. Robert Schreiber (Washington University, USA) and Dr. Genghong Cheng (University of Los Angeles, USA) for the gift of the different clones of WT and NIK-deficient MEFs. We also thank Dr. Genghong Cheng for providing the pBabe-Flag-NIK WT and NIK KD. The RIP3-GFP expression vector was provided by Dr. Francis Chan (University of Massachusetts, MA, USA). We thank Monika Wolf, Ruth Hillermann, Daniel Kull and Jay Tracy for technical assistance for the in vivo work. We thank GIGA Cell Imaging and Flow Cytometry for the access to the platform. Research in Dejardin’s group is supported by Belgian grants from the ‘Fondation belge Contre le Cancer, FCC’, from the Plan Cancer Action 29, from the Télévie and from the Centre Anticancéreux of the University of Liège (Belgium). MH was supported by the Swiss National Foundations (SNF), the Helmholtz foundation, an ERC Starting grant (Liver Cancer Mechanism), the Peter Hans Hofschneider foundation, Stiftung experimentelle Biomedizin (Peter Hans Hofschneider Stiftung) and the PCCC (preclinical comprehensive cancer center). JC’s group was supported by European Union FP7 (INFLA-CARE) and by a BBSRC grant. PV holds a European grant (Euregional PACT II) and a Methusalem grant (BOF09/01M00709) from the Flemish Government. MB has a tenure track position in the Multidisciplinary Research Program of Ghent University (GROUP-ID). Research in the ED, JP, PV and MB groups are also co-financed by a Federal grant from the Belgian government (Interuniversity Attraction Poles, IAP7/32). LB and CR are supported by a fellowship from the Télévie (Belgium). MF was supported by a fellowship from the Portuguese Foundation for Science and Technology (FCT, SFRH/BD/75137/2010). ED and JP are Research Associate and Research Director at the FNRS (Belgium), respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Boutaffala, L., Bertrand, M., Remouchamps, C. et al. NIK promotes tissue destruction independently of the alternative NF-κB pathway through TNFR1/RIP1-induced apoptosis. Cell Death Differ 22, 2020–2033 (2015). https://doi.org/10.1038/cdd.2015.69
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2015.69
This article is cited by
-
Different types of cell death and their shift in shaping disease
Cell Death Discovery (2023)
-
Inhibition of LTβR signalling activates WNT-induced regeneration in lung
Nature (2020)
-
Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation
Nature Communications (2019)
-
The non-canonical NF-κB pathway in immunity and inflammation
Nature Reviews Immunology (2017)
-
Regulating the balance between necroptosis, apoptosis and inflammation by inhibitors of apoptosis proteins
Immunology & Cell Biology (2017)
